

Abstract
Ca2+ signaling is of vital importance to cardiac cell function and plays an important role in heart failure. It is based on sarcolemmal, sarcoplasmic reticulum and mitochondrial Ca2+ cyclings. While the first two are well characterized, the latter remains unclear, controversial and technically challenging.
In mammalian cardiac myocytes, Ca2+ influx through L-type calcium channels in the sarcolemmal membrane triggers Ca2+ release from the nearby junctional sarcoplasmic reticulum to produce Ca2+ sparks. When this triggering is synchronized by the cardiac action potential, a global [Ca2+]i transient arises from the coordinated Ca2+ release events. The ends of intermyofibrillar mitochondria are located within 20 nm of the junctional sarcoplasmic reticulum and thereby experience the high local [Ca2+] during the Ca2+ release process. Both local and global Ca2+ signals may thus influence calcium signaling in mitochondria and, reciprocally, mitochondria may contribute to local control of calcium signaling. In addition to the intermyofibrillar mitochondria, morphologically distinct mitochondria are also located in the perinuclear and subsarcolemmal regions of the cardiomyocyte and thus experience different local [Ca2+].
Here we review the literature in regard to several issues of broad interest: (1) the ultrastructural basis for mitochondrion - sarcoplasmic reticulum cross-signaling; (2) mechanisms of sarcoplasmic reticulum signaling; (3) mitochondrial calcium signaling; and (4) the possible interplay of calcium signaling between the sarcoplasmic reticulum and adjacent mitochondria.
Finally, this review discusses experimental findings and mathematical models of cardiac calcium signaling between the sarcoplasmic reticulum and mitochondria, identifies weaknesses in these models, and suggests strategies and approaches for future investigations.
Keywords: mitochondria, sarcoplasmic reticulum, ventricular cell, calcium cycling, heart
1. Introduction
The release of Ca2+ from the sarcoplasmic reticulum (SR) is central to the normal physiology of cardiac myocytes and the cellular and subcellular control of intracellular calcium concentration ([Ca2+]i) is the focus of much current research. The major mechanism by which the cardiac action potential (AP) triggers Ca2+ release from the SR is by Ca2+-induced Ca2+ release (Bers, 2001, 2002a, 2002b; Cannell et al., 1995; Fabiato, 1985, 1992). The released Ca2+, after having activating contractile elements, is pumped back into the SR by SR Ca2+-ATPase (SERCA2a). The global [Ca2+]i elevation is restored to the low diastolic [Ca2+]i when Ca2+ is removed from the cytoplasm by sarcolemmal Ca2+-ATPase and the Na+-Ca2+-exchanger. Some of the elevated [Ca2+]i may also be buffered by the mitochondria, with entry into the matrix permitted down an electrochemical gradient through a channel known as the mitochondrial Ca2+ uniporter (MCU). In the complete cycle of Ca2+ elevation and reduction, the Ca2+ levels in each compartment at the end of the cycle must be restored to the pre-release state. Thus the amount of Ca2+ released from the SR (triggered by ICa, the sarcolemmal voltage-dependent Ca2+ channel current) must be equal to the amount of Ca2+ taken up by the SR in the steady-state. This requirement for steady-state Ca2+ flux balance applies not only to SR Ca2+ cycling (Bers, 2001; Eisner et al., 2000), but also for uptake and release of Ca2+ across the sarcolemma and Ca2+ entry and exit for the mitochondria.
This cellular and subcellular Ca2+ cycling is of vital importance to cardiac cell function and plays an important role in ventricular dysfunctions such as heart failure. The regulation of this system in vivo is an area of active investigation by many laboratories. An important clue to understanding these regulatory mechanisms may come from the recognition that the control of the Ca2+ cycling, and therefore signal transduction, occurs in spatially discrete sub-domains, as suggested earlier for Ca2+-induced Ca2+ release (Izu and Balke, 2002; Niggli and Lederer, 1990; Santana et al., 1996; Stern, 1992; Stern et al., 1999; Wier et al., 1994). For example, when local control mechanisms dominate, the triggering of SR Ca2+ release channels (type 2 ryanodine receptors, RyR2s) is governed not by the global, cell averaged [Ca2+], but instead by the Ca2+ microdomain surrounding each cluster of RyR2s at the junctional SR (jSR) due initially to the influx of Ca2+ from sarcolemmal L-type Ca2+ channels that are near to the jSR. The complex of L-type Ca2+ channels (located in sarcolemma) and the jSR (with its cluster of about 100 RyR2s (Franzini-Armstrong et al., 1999; Soeller et al., 2007) constitute the couplon (Franzini-Armstrong et al., 1999; Stern, 1992). The local-control theory and our current understanding of local Ca2+ dynamics increase the importance of knowing about the location, density, and regulation of intracellular ultrastructures (channels, pumps, regulatory proteins, membrane structures, etc.) involved in SR Ca2+ cycling.
Intermyofibrillar mitochondria (IMFMs; Fig. 1) span the sarcomere from the couplon at one Z-disk to the couplon at the next Z-disk and are thus “bookended” by the jSR. They are surrounded by the network (“free”) SR (nSR) which forms a thin intricate network (rete) from one jSR to another jSR (while interconnected with the entire SR within the cell and to the ER and nuclear envelope (Wu and Bers, 2006). Additionally, these IMFMs are packed between the nearby myofibrils of the sarcomere that contract with each [Ca2+]i transient (i.e. global calcium release). The IMFMs are the intracellular organelles (other than the SR) that are positioned closest to the microdomains of elevated local [Ca2+] during each Ca2+ spark, the localized calcium signal from a single jSR (Cheng et al., 1993), or during each [Ca2+]i transient (Ramesh et al., 1998; Sharma et al., 2000). The major role for the mitochondria is to provide ATP needed for cellular function including contraction and for SERCA2a Ca2+ pumping (Chen et al., 1996, 1998; Maack et al., 2008; Yang and Steele, 2000, 2001). Because of its location and the specific features of its biology and function, another possible mitochondrial function is in the regulation of SR Ca2+ cycling. For example, mitochondria appear to play a role in the synthesis of an activator of Ca2+ uptake into SR, cyclic ADPR (Lukyanenko et al., 2001a). ADPR cyclase (also known as CD38) which produces two potent Ca2+ messengers, cyclic ADPR and NAADP from β-NAD+, was found to be bound to mitochondrial membranes in a variety of cells including cardiac myocytes (Chini and Dousa, 1995; Franco et al., 1998; Guse, 2000; Mészáros et al., 1997; Mojzisova et al., 2001; Munshi et al., 2000; Lee, 2001; Lee et al., 1997; Liang et al., 1999; Okamoto et al., 2000; Yusufi et al., 2001; Ziegler et al., 1997). Under some conditions, Ca2+ release from the SR could be modulated by mitochondrial reactive oxygen species (ROS) (Akar et al., 2005; Wang et al., 2008; Yan et al., 2008; Zorov et al., 2006); however, the most intriguing effect of mitochondria on local Ca2+ signaling could be from the possible involvement of mitochondria in the uptake and release of Ca2+, a process we will call “mitochondrial Ca2+ cycling”. Reports of dynamic fluctuations of mitochondrial Ca2+ ([Ca2+]m) vary with respect to the extent and speed of both uptake and release (Brandes and Bers, 2002; Dedkova and Blatter, 2008; Maack et al., 2006; O’Rourke, 2007; Robert et al., 2001; Sedova et al., 2006). Certainly the existence of a favorable electrochemical gradient for passive Ca2+ accumulation by the mitochondria (matrix potential is about −180 mV with respect to the cytosol), a Ca2+ permeable channel (the so-called uniporter or MCU) and a mechanism for extrusion (the mitochondrial Na+/Ca2+ exchanger along with the mitochondrial Na+-proton exchanger) lay the foundation for such possibilities. These possibilities are certainly intriguing and suggest that mitochondrial Ca2+ cycling may be important in normal and pathological conditions, but this assessment depend critically on the speed and extent of Ca2+ movement across the mitochondria. In this review we discuss data and hypotheses concerning mitochondrial Ca2+ movement in ventricular myocytes and review approaches for productive future work.
Figure 1. Location of mitochondria in rat ventricular myocyte.
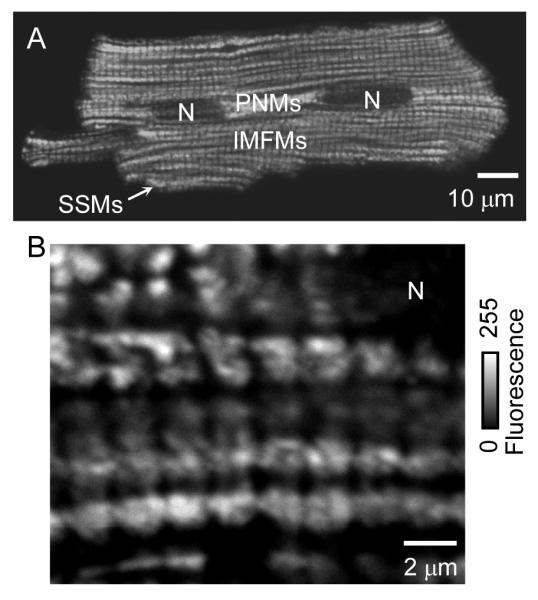
I Immunofluorescent labeling of VDAC as a mitochondrial marker. Two representative cells are shown with lower (A) and higher (B) resolution. IMFMs, intermyofibrillar mitochondria; N, nucleus; PNMs, perinuclear mitochondria; SSMs, subsarcolemmal mitochondria. Adapted from Parfenov et al., 2006.
2. Ultrastructural basis
It has been suggested that Ca2+, released from the endoplasmic reticulum of nonmuscle cells, may be accumulated to some extent by local mitochondria because of the close anatomical relationship between the two organelles (Csordas et al., 2002; Frey and Mannella, 2000; Hajnoczky et al., 2000; Pacher et al., 2002; Rizzuto et al., 1993; Spät et al., 2008; Szalai et al., 2000). Similarly, Ohata et al. (1998) suggested that mitochondrial accumulation of Ca2+ released from the cardiac SR could be mediated by the structural association between IMFM and the SR. The details of ultrastructural links, however, remain under investigation.
Cardiac mitochondria
Three subpopulations of mitochondria are often discussed: IMFMs, subsarcolemmal mitochondria (SSMs) and perinuclear mitochondria (PNMs). The three types are distinguishable by their location, morphology, or by method of isolation. SSMs are readily discernable in skeletal muscle fibers (Rambourg and Segretain, 1980). Originally, the name “subsarcolemmal” implied all mitochondria that could be easily isolated by polytron treatment of the tissue (Chemnitius et al., 1993; Matlib et al., 1978). In contrast, IMFMs are tightly packed within the cell as described earlier, and can only be isolated after nagarse treatment (Matlib et al., 1978). It was shown that these two groups of cardiac mitochondria (SSMs and IMFMs) are very distinct morphologically and, probably, biochemically from each other (Matlib et al., 1978; Palmer et al., 1977).
Cardiac mitochondria are also classified into PNMs and IMFMs (Fawcett and McNutt, 1969; McNutt and Fawcett, 1969; Segretain et al., 1981). Immunofluorescent labeling (Fig. 1) shows the difference in their shape and size. IMFMs are much larger and have a complicated shape in comparison to the smaller and more rounded PNMs. Fig. 2A shows a micrograph from an ultrathin section through the central part of a cardiac cell. Dissimilarities in shapes, matrix electron densities, and internal membrane extensions between the two mitochondria populations are readily observed. The differences in matrix electron density and in internal membrane extension have been suggested to reflect differences in functional activity between the two types of mitochondria (Fawcett, 1966; Hackenbrock, 1968; Kononova, 1982). PNMs usually appear circular on electron micrographs, while IMFMs appear oval and occupy the entire space between Z-lines (Fawcett and McNutt, 1969). Fig. 2B shows that the tightly packed IMFMs are located in very close proximity to known Ca2+ release site, the jSR, which is located between the IMFM and sarcolemma of T-tubule (TT).
Figure 2. Types of cardiac mitochondria in rat ventricular myocyte.

Electron micrograph; longitudinal ultrathin section, conventional fixation. Longitudinal ultrathin section. (A) Perinuclear and intermyofibrillar mitochondria. (B, C) Intermyofibrillar mitochondria. (D) Perinuclear mitochondria. (E) Intermyofibrillar and slender mitochondria. jSR, junctional sarcoplasmic reticulum; Z, Z-line; IMFM, intermyofibrillar mitochondrion; N, nucleus; PNM, perinuclear mitochondrion; SM, slender mitochondrion; TT, T-tubule; black arrows show regions where 4 membranes are clearly seen.
Questions regarding the shape and size of cardiac mitochondria are direct (Aon et al., 2006). Quenching of ethylrhodamine fluorescence by a narrow laser beam revealed several mitochondrial clusters within one cardiomyocyte (Amchenkova et al., 1988), each of which could represent a single branched mitochondrion. Indeed, cardiac mitochondria were shown to branch, bend back upon themselves, or have two lobes connected by narrower segments (Fawcett and McNutt, 1969; Segretain et al., 1981). Figs. 2C and D show that IMFM (Fig. 2C) could span four sarcomeres (8 μm) in length, and that PNM could be of shapes far from round (Fig. 2D).
Although the morphology of the three mitochondria groups is dynamic and varied (Rube and van der Bliek, 2004), there are also two rarely observed groups of cardiac mitochondria: slender mitochondria (SM) and nuclear mitochondria. SM are very long mitochondria or mitochondrial branches, though both extremities of the mitochondrion have not yet been imaged. A typical SM is shown in Fig. 2E. Arrowheads indicate regions where four membranes are clearly visible. The continuity of these mitochondria makes them ideal candidates for what was described in classical studies by Fawcett and McNutt (1969) as Nuclear mitochondria in cardiac myocytes were described recently (Skulachev et al., 2004), but only in end-stage cardiac failure.
During heart failure, mitochondria undergo changes and IMFMs may lose their connections to the cytoskeleton and SR (Jones et al., 1975; Su et al., 2000). Fig. 3 shows a significant reduction in the density of IMFMs and abnormally small mitochondria in a ventricular cell from rat suffering from heart failure (compare with Fig. 2). Recently, we demonstrated that under normal (i.e. physiological) conditions the mitochondrial outer membrane is very close to the surrounding structures, including the SR and other mitochondria (Fig. 4) (Lukyanenko et al., 2007; Parfenov et al., 2006; Salnikov et al., 2007). The packing was largely inaccessible even to 3 nm particles, and the molecular details of this organization remain largely unknown. Some data suggest a role for desmin and tubulin in maintaining structural integrity (Watkins et al., 1987). Milner et al. (1999, 2000) showed that desmin-null (desmin -/-) cardiac myocytes have subsarcolemmal mitochondrial clumping and reduced IMFMs, although these observations require further quantitative and physiological investigation. The manner in which desmin could be connected to the outer mitochondrial membrane (OMM) is unclear; however it could be similar to adhesive structures described for bacteria (Knight et al., 2000; Mootha et al., 2003; Paschen et al., 2003; Sauer et al., 2000).
Figure 3. Ultrastructure of ventricular cell from rat suffering from heart failure.

Heart failure was induced with isoproterenol (3rd week; 0.3 mg/kg injections). Note mitochondrial regressive changes and abnormal I bands. Left ventricle; conventional microwave fixation. M, mitochondrion; Z, Z line. Adapted from Lukyanenko, 2007.
Figure 4. Mitochondrial contacts in rat ventricular myocyte.

(A,B) The electron micrographs show areas of contact between a mitochondrion and T-tubules. (C). Intermitochondrial contact. Inset shows the ultrastructural organization of the contact. Arrowheads show mitochondrial cristae. IMFM, intermyofibrillar mitochondrion; jSR, junctional SR; M, mitochondrion; TT, T-tubules; zSR, z-tubules of the SR; Z, Z line (Z disk); black arrows show RyRs; white arrows show structures connecting IMFM and jSR. Adapted from Parfenov et al., 2006.
Tubulin was reported to establish tight contacts to the voltage-dependent anion channel (VDAC) (Carré et al., 2002; Monge et al., 2008; Rostovtseva and Bezrukov, 2008; Rostovtseva et al., 2008). In addition to tubulin, another connective candidate is mitofusin. Mitofusin was shown to tether the endoplasmic reticulum to mitochondria in mouse embryonic fibroblasts and HeLa cells (Brito and Scorrano, 2008). Most recently, García-Pérez et al. (2008) reported very specific physical coupling between the OMM and SR in cardiac cells. This hypothesis was based on the existence of direct Ca2+ channeling from the SR to the mitochondrial matrix. The physiological or pathophysiological context of this hypothesis is yet to be elucidated.
Are IMFMs different from other cardiac mitochondria?
Cardiac IMFMs have been isolated from a number of animal species (Hoppel et al., 1982; Matlib et al., 1978; McMillin-Wood et al., 1980; Ohata et al., 1998; Palmer et al., 1977; Weinstein et al., 1985, 1986). The abundance of fibrillar material in the heart, coupled with the tight packing of this mitochondria between the Z-disks make isolation of IMFMs very difficult. Electron microscopy of the corresponding pellets showed that the polytron preparation was practically unable to extract IMFMs. Therefore, to release IMFMs, a nagarse preparation was used. Once isolated, IMFMs were shown to have different biochemical properties than other cardiac mitochondria types. Compared to the polytron-isolated mitochondria, IMFMs have: (1) up to three-times higher rates of Ca2+ uptake and up to two-times lower Km values; (2) 50% higher rates of oxidative phosphorylation, and (3) significantly higher cytochrome content (Matlib et al., 1978; McMillin-Wood et al., 1980; Palmer et al., 1977). IMFMs were shown to be less vulnerable to global ischemia (Weinstein et al., 1985), while cardiomyopathy lead to mitochondrial oxidative defects confined wholly to the IMFMs (Hoppel et al., 1982), though possible damage to IMFMs by nagarse may invalidate these data.
These dissimilarities between cardiac mitochondrial subpopulations were confirmed, to some extent, in intact mitochondria by Kononova (1982). In these experiments, hypoxia was followed by quantitative analysis of changes in mitochondrial ultrastructure. Hypoxia resulted in the swelling of all cardiac mitochondria. However, after a one day period, only IMFMs and PNMs showed a significant increase in area and number of cristae, and only PNMs significantly (almost double) increased in number, thereby suggesting different biochemical properties in the studied mitochondrial subpopulations.
Reports presented by Isenberg et al. (1993) and Gallitelli et al. (1999) supported this hypothesis. They described experiments in which isolated guinea-pig ventricular myocytes were instantly frozen with supercooled propane (−196°C) during stimulation with paired voltage or current clamp depolarizing pulses. Cell contact with the coolant induced a negative spike followed by a large positive current. The position of this artifact enables temporal resolution of the freezing incident. Using electron beam microanalysis it was determined that during systole, the Ca2+ increase in peripheral mitochondria was at least 3 times that of the central mitochondria. However, although the analyzing beam had a diameter of 16 nm (Isenberg et al., 1993), it was uncertain whether it was specifically focused on the mitochondrial matrix, given that the ultrastructure of the mitochondria, more specifically the deeply infolded cristae, does not permit distinction between Ca2+ inside the matrix, and Ca2+ in the mitochondrial intermembrane space. Therefore, the data may reflect changes in Ca2+ within both the mitochondrial intermembrane space and in the matrix. This provides a plausible explanation as to why Ho et al. (2003) did not detect any increase in IMFM Ca2+ during contractions. It should be noted, however, that the approach used by Ho et al. (2003) was less precise than that of Isenberg and co-authors.
Junctional Sarcoplasmic Reticulum
SR Ca2+ cycling is a balance between Ca2+ release and uptake (Eisner et al., 2000) and under steady-state conditions these fluxes must be equal. Electron microscopy and immunolabeling are common visualization tools used to study the localization and membrane structures of Ca2+ transport proteins. The intracellular complexes primarily responsible for Ca2+ release and uptake during the cardiac cell contractile cycle are the couplons (Frank, 1990; Franzini-Armstrong et al., 1998, 1999; Gathercole et al., 2000; Jorgensen et al., 1982-1993; Yang et al., 2002). The jSR located in close proximity to the TT is seen as a pancake that is wrapped around the TT with wispy connections to the network SR (Brochet et al., 2005). A cross sectional view of the jSR reveals two membrane surfaces (Figs. 3B and C): one facing the TT sarcolemma studded with “feet” (i.e. RyR2 homotetramers), and the other facing the mitochondrial outer membrane rich in SERCA2a (Jorgensen and Jones, 1987; Jorgensen et al., 1982). The jSR is equidistant (~15 nm) from the sarcolemma and the IMFM outer membrane. These jSR membranes were shown to contain major structural components of the SR Ca2+ cycling system (Jorgensen and Jones, 1987; Jorgensen et al., 1982; Ozawa et al., 1976; Sommer and Spach, 1964).
Recently, we have developed a practical approach to measure the functional distances between membranes in vivo (Lukyanenko, 2007; Parfenov et al., 2006; Salnikov et al., 2007). Our experiments revealed that even during contraction, gold nanoparticles as small as 3 nm in diameter could not enter the space between the jSR and the membranes (Parfenov et al., 2006). The full molecular and biophysical explanation for this observation involves the consideration of the many proteins and molecular structures that may fill those spaces.
The distribution of sarcolemmal Ca2+ channels, exchangers, and pumps is critical to the understanding of Ca2+ signaling. DHPRs are located in the TT and face the “subspace” (or junctional cleft), which separates the TT and the jSR. The sarcolemmal Na+-Ca2+ exchanger proteins are found in both the exterior sarcolemma and the TT sarcolemma but not in the junctional cleft (Frank et al., 1992; Kieval et al., 1992; Scriven et al., 2005). Little is known about the function of the IMFM outer membrane, facing the Ca2+ uptake proteins of the jSR, located in close proximity to the Z-line network SR which is thought to be rich in SERCA2a (Jorgensen et al., 1982; Ozawa et al., 1976; Prestle et al., 2003).
3. Mechanisms of Ca2+ cycling
Our purpose here is to focus on the interplay between mitochondria and SR Ca2+ signaling. Therefore, other aspects of membrane (sarcolemmal) Ca2+ cycling will not be discussed. See D.M. Bers (2001) for detailed review on this topic.
Sarcoplasmic reticulum Ca2+ cycling
In mammalian cardiac myocytes, the SR serves as the intracellular Ca2+ store. It amplifies the “trigger” Ca2+ that enters across the sarcolemma to produce the [Ca2+]i transient which underlies cardiac contraction (Bers, 2001, 2002a, 2002b; Chiesi et al., 1994; Cannell et al., 1995; Feher and Fabiato, 1990). The amount of Ca2+ in the SR lumen ([Ca2+]SR) depends on the functional state of SR Ca2+ uptake and Ca2+ release mechanisms. Cytoplasmic Ca2+ itself is the main regulator of Ca2+ release from the SR in cardiac myocytes (Bers, 2001, 2002a, 2002b; Fabiato, 1985, 1992; Feher and Fabiato, 1990). Ca2+ release and re-uptake are highly coordinated through changes in [Ca2+]SR and also depend on the [Ca2+] gradient across the SR membrane (Bhogal and Colyer, 1998; Ching et al., 2000; Fabiato, 1992; Györke and Györke, 1998; Ikemoto and Yamamoto, 2000; Lukyanenko et al., 1996, 1998-2001b; Saiki and Ikemoto, 1999; Sitsapesan and Williams, 1995). SERCA2a, the SR Ca2+ pump, maintains the Ca2+ gradient between the cytosol and the SR lumen ([Ca2+]SR/[Ca2+]cyt=~15,000), using the free energy available from hydrolysis of ATP (ΔGATP~−60 kJ/mol). There is a tight coupling between the SR Ca2+ gradient and the ΔGATP (Chen et al., 1996, 1998). As Ca2+ accumulates in the lumen the off-rate of Ca2+ from SERCA2a may become the rate-limiting step, and Ca2+ pumping decreases through “back-inhibition” (Pozzan et al., 1994). The details of these events clearly depend on the efficiency of transport, its stoichiometry (how many Ca2+ ions per ATP consumed) and other features of the pump. Intraluminal Ca2+ can also modulate the activity of SERCA2a by modulating the activity of protein kinases that interact with the luminal complex of SERCA2a (Bhogal and Colyer, 1998). The efficiency of transport by SERCA2a in both physiological and pathophysiological conditions (such as ischemia) may be at 75-85% of the theoretical thermodynamic limit on the basis of the ΔGATP, leaving the possibility of kinetic and further thermodynamic regulation (Chen et al., 1998; Feher and Fabiato, 1990). Overall, the activity of SERCA2a is not only dependent on the energy state of the cell but can also be kinetically regulated by SR proteins, protein kinases, and by phospholamban (PLN) (Bers, 2001). For instance, removing the kinetic limitation of PLN on the activity of the SERCA2a allows the SR Ca2+ gradient to move closer to its thermodynamic limit (Chen et al., 1998). Exactly what the thermodynamic limit is, however, is not precisely known.
Failure in the control mechanisms of SR Ca2+ cycling leads to a variety of cardiac dysfunctions. Spontaneous Ca2+ release and increased SR Ca2+ “leak” has been implicated in cardiac dysfunctions such as genetic and acquired triggered arrhythmias and the initiation of ventricular fibrillation during postischemic reflow (Bellinger et a. al., 2008; Carmeliet, 1999; Ferrier, 1976; Ishide, 1996; Janse, 1999; Kihara and Morgan, 1991; Lakatta, 1992; Lehnart et al., 2006, 2008; Marks, 2001; Pogwizd and Bers, 2002). Defective SR Ca2+ cycling was found to be responsible for defective excitation-contraction coupling in heart failure (Currie and Smith, 1999,Haghighi et al., 2001; Hasenfuss and Pieske, 2002;; Hobai and O’Rourke, 2001; Kirchhefer et al., 1999; Lehnart et al., 2006, 2008; Maier and Bers, 2007; Schmidt et al., 1998;).
Ca2+ sparks
A Ca2+ spark is a fluorescent signal corresponding to the localized release of Ca2+ from a jSR cluster of RyR2s. The Ca2+ spark has a time to peak of about 10 ms with a size (full-width at half of the maximum level) of about 2 μm and a volume at that time of about 10 fl. It is thought to represent the efflux of Ca2+ from a RyR2 cluster (average size is about 100 RyR2s (Franzini-Armstrong et al., 1999; Soeller et al., 2007)). While the exact number of RyR2s that are involved in the Ca2+ spark is not precisely known, there is reason to believe that it could involve all of the channels in the cluster, but via a variable fraction could also be involved with little difference in Ca2+ spark characteristics (Cheng and Lederer, 2008; Sobie et al., 2002). The Ca2+ spark is the elementary event of SR Ca2+ release (Cheng et al., 1993; Györke et al., 1997; Guatimosim et al., 2002; Lopez-Lopez et al., 1995; Lukyanenko et al., 2000, 2007). Ca2+ sparks can occur spontaneously, or can be evoked by the activation of sarcolemmal L-type Ca2+ channels (Cannell et al., 1994; Lopez-Lopez et al., 1995; Wang et al., 2001). Under normal conditions, nearly all of the spontaneous or diastolic Ca2+ sparks remain localized and do not activate nearby (1 μm away) Ca2+ spark sites (jSR) (Cheng et al., 1993, 1996; Lukyanenko and Györke, 1999; Lukyanenko et al., 1996, 1999). Under conditions of increased SR Ca2+ load, Ca2+ sparks increase in amplitude and frequency and become initiation sites of propagating Ca2+ waves (Cheng et al., 1993, 1996; Izu et al., 2001; Lukyanenko and Györke, 1999; Lukyanenko et al., 1996). With respect to our discussion of mitochondria, Ca2+ sparks are of comparable size to IMFM and originate in close proximity to IMFM. Ca2+ sparks can be readily influenced by changes in local [Ca2+]i and are clearly an excellent tool to use in the study of crosstalk between the IMFM and the SR.
Ca2+ sparks have been recorded in close proximity to IMFMs and PNMs (Cheng et al., 1996; Lukyanenko et al., 2007; Shacklock et al., 1995; Yang and Steele, 2005). Recently we showed that the spatio-temporal characteristics of sparks found around PNMs and around IMFMs are very similar with respect to many of the parameters. However, Ca2+ sparks from the PNM zones were of significantly longer in duration (Lukyanenko et al., 2007) than the usual diastolic Ca2+ sparks. Our data suggested that the RyR2 clusters which produce stereotyped Ca2+ sparks are likely to be similar in structure. We speculate that the differences in Ca2+ spark duration could be due to the functional differences in the nearby mitochondria, but more experiments are needed to verify the observation and better characterize and account for it.
Mitochondrial Ca2+ cycling
In adult ventricular myocytes, mitochondria occupy 30 to 40% of the intracellular volume, presumably reflecting the huge demands of the contractile machinery for ATP production (Maack et al., 2008). Under normal conditions, the very negative inner mitochondrial membrane (IMM) potential (~−180 mV relative to the cytosol) provides a strong electrochemical driving force for Ca2+ to enter the mitochondrial matrix from the cytosol (Fig. 5). It is thought that the pathway by which Ca2+ crosses the IMM is the MCU (Dedkova and Blatter, 2008; Gunter and Pfeiffer, 1990; O’Rourke, 2007; Robert et al., 2001; Sedova et al., 2006). The molecular identity of the uniporter remains uncertain (Kirichok et al., 2004) and hence, the characteristics of its conductance, kinetics and regulation remain largely unknown and untested. Even more obscure is a faster mode of mitochondrial Ca2+ uptake known as rapid uptake mode (RaM), which has been described as a rapid self inhibitory Ca2+ uptake with recovery period of ~60 s (Buntinas et al., 2001; Sparagna et al., 1995).
Figure 5. Mitochondrial Ca2+ cycling in ventricular myocytes.

Schematic of local Ca2+ cycling in ventricular cardiac myocytes: sarcolemmal, SR, and mitochondrial Ca2+ cycling. This schematic is a conceptual representation of the location of structures involved in the local interplay of IMFM, SR, and sarcolemmal Ca2+ cycling. ADP, adenosine diphosphate; ANT, adenine nucleotide translocase; ATP, adenosine trisphosphate; junctional sarcoplasmic reticulum, MCU, electrogenic mitochondrial Ca2+ uniporter; mNCX, Na+/Ca2+ exchanger; NHE, Na+/H+ exchanger; nSR, network SR; PTP, permeability transition pore; RaM, rapid mode of Ca2+ uptake; TT, Transverse tubule; VDAC, voltage-dependent anion channel.
At steady state, the Ca2+ influx into the mitochondria must be balanced by an efflux. However, as noted above the mitochondrial Ca2+ dynamics are still both uncertain and controversial. The efflux of Ca2+ is believed to depend on mitochondrial Na+-Ca2+ exchanger (mNCX) and Na+-proton exchanger (NHE). It has been suggested that the mNCX could extrude Ca2+ from the matrix as long as the Na+ that enters the matrix also has a way to exit. This has been thought possible by the NHE with K0.5~4–8 mM (Bers et al., 2003; Cox and Matlib, 1993; Dash and Beard, 2008; Fry et al., 1984b; Saotome et al., 2005). Other possible contributors to the mitochondrial Ca2+ flux include the permeability transition pore (PTP, the molecular and functional characteristics of which are also uncertain and controversial) and a putative H+-Ca2+ exchanger (Hüser and Blatter, 1999; Kang et al., 2007; Nicholls and Chalmers, 2004; Rizzuto et al., 2000), but the details remain murky. As [Ca2+] in mitochondria ([Ca2+]mito) increases with time, Chalmers and Nicholls (2003) suggest that there may be three phases of mitochondrial Ca2+ accumulation. First, the modest increase in [Ca2+]mito influences enzyme function (e.g. matrix dehydrogenase). Second, as [Ca2+]mito increases further, the mitochondria may serve to buffer [Ca2+]i. Third, when [Ca2+]mito becomes even higher, PTP may be activated. Exactly how these elements may interact with physiological Ca2+ extrusion or with the hypothesized mitochondrial Ca2+-activated K+ channels (mBKCa) (Kang et al., 2007) will be model dependent and the answer awaits critical new experiments. There continues to be related vexing questions about many aspects of mitochondrial Ca2+ regulation.
Due to its dependency on the mitochondrial inner membrane potential, mitochondrial Ca2+ entry and exit should be affected by everything that may affect the potential across the mitochondrial inner membrane. This should included pH, Pi, ADP, ATP, [Na+]i and [Ca2+]i (Cortassa et al., 2003; Dedkova and Blatter, 2008; Nicholls and Crompton, 1980; Oliveira and Kowaltowski, 2004). Under normal conditions, with the simple models put forward so far, the extrusion of Ca2+ from the mitochondria should depend primarily on [Na+]mito but also on [Na+]i. Thus, mNCX is thought to be the primary Ca2+ release mechanism under physiological conditions (Gunter et al., 1994; Rizzuto et al., 2000), however its capacity and kinetics remain uncertain. The mNCX is pharmacologically and molecularly distinct from sarcolemmal Na+/Ca2+ exchanger. For example, the diltiazem analogue, benzothiazepine CGP 37157, has been reported to inhibit mNCX (Baron and Thayer, 1997; Cox and Matlib, 1993; Cox et al., 1993; White and Reynolds, 1997), while diltiazem itself inhibits Ca2+ channels and not the sarcolemmal Na+/Ca2+ exchange (Kuo et al., 2002; Watano et al., 1999).
The PTP, a large non-selective conductance pore, appears to be regulated by [Ca2+]mito (Bernardi et al., 1994; Haworth and Hunter, 1979; Kroemer et al., 2007; Szabadkai and Duchen, 2008; Zoratti and Szabo, 1995). Recently such a Ca2+-induced permeability transition was demonstrated in cardiac mitochondria (Kang et al., 2007; Salnikov et al., 2007), but the totality of the data are not compelling. Activation of the putative PTP collapses the membrane potential and can release Ca2+ through the pore itself and/or may allow Ca2+ efflux via reversal of the MCU (Fiskum and Cockrell, 1985; Kang et al., 2007; Pacher and Hajnoczky, 2001), but these possibility remain largely speculative. The most potent inhibitor of the PTP in cardiac cells is cyclosporin A (CsA) (Rizzuto et al., 2000).
The PTP (as we understand it to exist) permits molecules as large as 1500 Da to pass. The exact molecular composition of the PTP remains uncertain, however. It is argued that the PTP has a component in the OMM (e.g. the voltage-dependent anion channel, VDAC) and another component in the IMM (e.g. ANT, the adenine nucleotide transporter), and appears to be regulated by cyclophilin D in the IMM. The outer mitochondrial membrane is a minimal barrier for small molecules because it contains VDAC, a channel that permits both anions and cations as well as uncharged substances to pass; VDAC allows non-electrolytes up to ~ 5000 MW to permeate, and has been referred to as “mitochondrial porin” (Colombini, 1987, 2004; Crompton, 1999; Gincel et al., 2001; Kroemer et al., 2007; Murphy and Steenbergen, 2007; Rostovtseva et al., 2002a, 2002b, 2005; Szabadkai and Duchen, 2008). For comparison, VDAC is much larger than cardiac gap junctions (CX43) which permit organic molecules as big as 900 Da to pass (Veenstra et al 2005). The VDAC pore does not prevent passage of calcium ions even in the closed state, while passage of molecules as big as ATP is inhibited (Rostovtseva et al., 2005). The involvement of VDAC in mitochondrial Ca2+ signaling is complex. For example, despite its low selectivity, VDAC was reported to exhibit Ca2+-dependent regulation (Gincel et al., 2001; Shoshan-Bormatz et al., 2003; but see Rostovtseva et al., 2005).
Other mitochondrial Ca2+ permeation paths have also been reported. Shey-Shing Sheu’s group found RyRs type one (RyR1) in the mitochondrial membrane (Beutner et al., 2001; Sharma et al., 2000). The existence of RyR1s in the mitochondrial inner membrane was recently supported by additional work from this group (Altschafl et al., 2007). However, due to the existence of Ca2+ microdomains at the ends of the intermyofibrillar mitochondria described above, there is high probability of SR membranes may contaminate mitochondrial membranes preparations despite the great care used in their preparation (Taylor et al., 2003). It was, however, RyR1 that was found not the normal cardiac type 2 isoform. This issue was recently reexamined by Spät et al. (2008) who concluded that mitochondria in the rat heart are highly resistant to purification from SR membranes. Our own experiments involving the use of anti-bodies to all types of RyRs (Salnikov et al., 2005) failed to reveal any RyRs at the center of mitochondria as expected, assuming that the entire IMM is available to the RyR1. This work however, did not rule out the possibility that they could be located at the connections between the inner and outer mitochondrial membranes and therefore undetectable by immunogold labeling, which with two IgGs, can have an error margin of up to 20 nm.
Recently, reports of “Ca2+ channeling” from the SR to the mitochondrial matrix have emerged (García-Pérez et al., 2008; Spät et al., 2008). These reports suggest that in cardiac myocytes, the SR RyR2s could be located in close proximity to the OMM, and could provide a sufficient Ca2+ trigger for induction of mitochondrial membrane permeabilization allowing the transfer of Ca2+ into the cardiac mitochondria (García-Pérez et al., 2008). García-Pérez et al. (2008) also suggested that mitochondrial type 1 RyRs reported earlier (Altschafl et al., 2007) could be involved in coupling between the SR and OMM. To date no compelling functional linkage between either SR or ER and mitochondria has been provided. In 2001 Kaasik et al. showed the possible existence of direct adenine nucleotide channeling between the cardiac jSR and IMFM. The provocative hypothesis that an array of “direct connections” between the SR/ER and the mitochondrial matrix cannot be readily disproved and the data to date are, at best, suggestive. Additional unambiguous experiments are needed to test the hypothesis. Considering the highly restricted space between the SR and IMFM (Lukyanenko et al., 2007; Parfenov et al., 2006; Salnikov et al., 2007), it can be concluded that regions of close apposition or contact between mitochondria and the SR/ER are likely to be important. However, direct evidence of the involvement of mitochondria in cardiac Ca2+ signaling remains largely absent. The role(s) of mitochondrial [Ca2+]mito fluctuations with the local Ca2+ sparks or the cell-wide [Ca2+]i transients remains intriguing and provocative (Maack et al., 2006; O’Rourke, 2007).
The dynamics of the reported SR-IMFM contacts and the time-dependent changes in quantitative morphometry must be carried out to provide support for putative SR-IMFM crosstalk. At this point we can only speculate that the systolic increase in [Ca2+]i (that could be higher locally) may be important. Many questions are raised regarding these matters. If present, could crosstalk affect IMFM dependent apoptosis (Pan et al., 2001)? How do local and global [Ca2+]i affect changes in cytochrome C and relocation of Bax (a Bcl-2 family member) from the cytoplasm to the IMFM outer membrane (Heiskanen et al., 1999; Pan et al., 2001)? Can these proteins form aggregates with VDAC in the mitochondrial outer membrane (Godlewski et al., 2002; Kluck et al., 1999; Kuwana et al., 2002)? If so, what are the consequences?
4. Interplay between mitochondrial and sarcoplasmic reticulum Ca2+ signals
Experimental data
There are two aspects of SR Ca2+ signaling that could be influenced by the mitochondria under normal physiological conditions. During systole the mitochondria are bathed by the very high [Ca2+]i at their ends near the jSR and the global [Ca2+]i in the middle. They compete (albeit poorly) with the SR for uptake of Ca2+. The efflux of Ca2+ from the mitochondria during diastole must equal the influx acquired during systole (on average and in the steady-state). To the extent that there is Ca2+ influx during systole, the mitochondria should produce an efflux during diastole. Depending on the amount and rate of Ca2+ efflux from the mitochondria, there may be a measurable effect on [Ca2+]i. If the efflux is high and focused near the jSR, it may “bias” the local jSR Ca2+ signal and influence the probability of the RyR2s to be triggered by the L-Type Ca2+ channel current influx. If, however, the efflux is low and spread out in space and time, it may have no significant effect. Until the end of the 1970s, mitochondria were considered an important structure in the control of Ca2+ homeostasis (Bers, 2001; Pozzan et al., 1994). However, later it was shown that the MCU only became activated to appreciable levels when [Ca2+] rose above 0.5 μM (Fry et al., 1984a; Pozzan et al., 1994; Sedova et al., 2006) with K0.5~4-10 μM (Bassani et al., 1998; Sedova et al., 2006). The cellular [Ca2+]i peaks at about 1 μM, although some regions of each mitochondrion presumably experience a higher concentration. Importantly the conductance of the MCU (i.e. the effective turnover rate) is thought to be quite low (Kirichok et al., 2004), and the density of the uniporters in the IMM is unknown. If the local [Ca2+]i at the jSR end of the IMFM were to reach 3 μM the nearby MCUs would be significantly activated (García-Pérez et al., 2008). However, the MCUs in the middle of the IMFM would be bathed with a lower [Ca2+]i. Experiments with cardiomyocytes (Bassani et al., 1992, 1993; Bowser et al., 1998; Brandes and Bers, 2002; Duchen, 1999, 2000; García-Pérez et al., 2008; Isenberg et al., 1993; Martin et al., 1998; Ohata et al., 1998; Pacher et al., 2000; Pitter et al., 2002; Robert et al., 2001; Sedova et al., 2006; Sharma et al., 2000; Sheu and Sharma, 1999; Szalai et al., 2000; Territo et al., 2001a, 2001b) and other cells types (Arnaudeau et al., 2001; Connor, 1993; Haak et al., 2002; Isaeva and Shirokova, 2003; Isaeva et al., 2005; Jouaville et al., 1995; Maack and O’Rourke, 2008; Spät et al., 2008; Wang and Thayer, 2002) do suggest that the [Ca2+]mito changes with time and reflects the sarcomeric [Ca2+]i gradient, but this [Ca2+]mito is not calibrated. For example, it was reported that free [Ca2+] in mitochondria under physiological conditions is ~100 nM (Miyata et al., 1991). This is a level that is about the same as the measured [Ca2+]I. However it was also suggested that free [Ca2+]mito may increase during systole to ~700 nM (Brandes and Bers, 2002; Miyata et al., 1991; Ohata et al., 1998). The actual normal change in total mitochondrial Ca2+ content under physiological conditions is not known but could readily exceed the 1 mM Ca2+ measured by Isenberg et al. (1993).
Recently, the role played by Na+ in mitochondrial Ca2+ regulation was demonstrated in vascular endothelial cells (Sedova and Blatter, 2000), cortex neurons (Raiteri et al., 2002) and cardiac myocytes (Bers et al., 2003; Maack and O’Rourke, 2008; Maack et al., 2006; Sedova et al., 2006). In the cardiac cell, during an action potential, the [Na+] in the region closest to the membrane was estimated to increase to as high as 80 mM within milliseconds (Gallitelli et al., 1999). While unverified, if the Gallitelli estimate of this increase in [Na+] during INa in a narrow subcellular microdomain were supported and applied to the mitochondrion, it may be sufficient to power significant Ca2+ efflux from the mitochondria (Piacentino et al., 2003). For many reasons, however, this number is unreasonably high (Lederer et al., 1990). The pressing questions are how fast Ca2+ can be released from the IMFM and how much? Using patch clamp and electron probe microanalysis, Isenberg and coworkers (Isenberg et al., 1993) reported that the peak total [Ca2+] in mitochondria could diminish from 1.0 mM to ~0.5 mM in just 50 ms. As an isolated event, this Ca2+ efflux from a mitochondrion would produce a 36 nM elevation in a 10 fl volume (assuming a rectangular shaped mitochondrion of 0.2 × 0.2 × 1.8 microns if the cytosolic Ca2+ buffering power were 100). This would be visible using our current methods, unless it were blurred by overlapping [Ca2+]i signals such as Ca2+ sparks. Such Ca2+ release events, if they did contribute, would not add any net Ca2+ to the signal, instead they would alter the kinetics of the [Ca2+]i transient. Do note that the condition that triggers SR Ca2+ release (the AP) and thus underlie high mitochondrial Ca2+ uptake is the same condition that favors mitochondrial Ca2+ extrusion (high local [Na+]). Therefore, there are many details that must be addressed both experimentally and with respect to mitochondrial Ca2+ modeling before any firm conclusions can be drawn.
Mathematical models
As noted above, the precise details of SR and mitochondrial Ca2+ interplay remain obscure experimentally and theoretically. Since the experimental findings are inconsistent, there is much room for speculation. Mathematical models provide us with the ability to study Ca2+ transport in each system in isolation and enable us to refine our experiments or the analysis associated with them. Mathematical models have been used to gain insights into the regulation of energy metabolism in the mitochondria (Jafri et al., 2001; Lambeth and Kushmerick, 2002; Magnus and Keizer,1998a, 1998b; Nguyen et al., 2007; Tornheim, 1979), Ca2+ cycling in the SR (Greenstein et al., 2006; Greenstein and Winslow, 2002; Shannon et al., 2000, 2002, 2004), and the “supply meets demand” phenomena in cardiomyocytes (Cortassa et al., 2006; Nguyen and Jafri, 2005). These models are constructed in a modular fashion, where each module is a detailed kinetic model of the individual elements (i.e. enzymes, other proteins) that constitute the system. While models of Ca2+ cycling in the SR and cytosol are abundant, models of Ca2+ cycling in the mitochondria are scarce. The few models that have been developed are constrained by parameters largely derived from experiments conducted on isolated mitochondria preparations. In this section we briefly discuss several of these models along with their respective contributions.
One of the most extensive efforts to model mitochondrial Ca2+ handling, and its effect on energy metabolism, was made by Magnus and Keizer in pancreatic β-cells (Keizer and Magnus, 1989; Magnus and Keizer, 1997, 1998a, 1998b). Their first model included six transport mechanisms in the inner mitochondrial membrane: proton pumping via respiration, proton uptake by way of the F1F0-ATPase, a proton leak, adenine nucleotide exchange, Ca2+ uptake via the MCU, and extrusion via the Na+/Ca2+ exchanger. The kinetic models of each mechanism were developed separately and shown to successfully reproduce the rates of transport measured experimentally. When combined, these mechanisms were used to describe resting mitochondria and phosphorylating mitochondria, by fixing NADH and Ca2+ concentrations. Under these conditions, variation of mitochondrial Ca2+ concentration was then used to describe mitochondrial Ca2+ handling.
Using this minimal model, Magnus and Keizer predicted a very sharp increase in the mitochondrial ability to take up Ca2+ at normal cytosolic Ca2+ concentrations (i.e. 0.4 – 0.5 μM), in agreement with experimental observations (Saavedra-Molina et al., 1990). By extending the model to include a more physiological formulation of energy metabolism, which included the Ca2+ dependence of mitochondrial dehydrogenases, as well as a dynamic formulation of plasma membrane currents, Magnus and Keizer (1997, 1998) showed that in phosphorylating mitochondria, when NADH levels are constant, the depolarizing influence of Ca2+ influx via the MCU would decrease phosphorylation and increase oxidation. Although they predicted this effect to be quite large at cytosolic Ca2+ concentrations exceeding 1 – 2 μM, they showed that this significantly affects ATP production, enough to provoke adverse reactions from ATP-dependent plasma membrane ionic channels, even at lower, more physiological concentrations.
Elements of the Magnus and Keizer models were used by Cortassa and co-workers to develop a model of isolated cardiac mitochondria (Cortassa et al., 2003). For better qualitative approximation of the system, extensive modifications, including the addition of NADH as a dynamic state variable dependent upon the activity of the tricarboxylic citric acid (TCA) cycle, were applied. The resulting model provided the first mathematical means of studying the dynamic regulation of energy metabolism by Ca2+ cycling in cardiac mitochondria. Model simulations suggested that increases in cytosolic [Ca2+] had two opposite effects on mitochondria: a dissipative effect on the inner mitochondrial membrane due to the shuttling of the divalent cation through the MCU, and a stimulatory effect on the activity of the TCA cycle dehydrogenases resulting from higher intramitochondrial [Ca2+]. They concluded that increase in ATP production by Ca2+ can only be achieved when the extent of NADH production exceeds the depolarizing effect of Ca2+ influx on the change in membrane potential.
By embedding a similar formulation of mitochondria energetics into a whole-cell model of excitation-contraction coupling in the ventricular myocyte, Nguyen and Jafri (2005) were able to study the effects of cytosolic Ca2+ transients on Ca2+ cycling in the mitochondria, and on energy metabolism. The model predicted that in addition to activation of the TCA cycle dehydrogenases, Ca2+ - dependent activation of the F1F0-ATPase is necessary in order to achieve significant increases in ATP production. They predicted that mitochondria exposed to the small Ca2+ transients in the bulk myoplasm undergo a mild inner membrane depolarization (~10%) in response to such transients, while IMFM located in close proximity to the Ca2+ release sites (see Cheng et al., 1993, 1996) undergo depolarization large enough to cause a decline in ATP production that recovers quickly, minimizing its impact on the overall ATP production. The latter provided insight into the self inhibitory mechanism of mitochondrial Ca2+ uptake characterized experimentally as the RaM (Buntinas et al., 2001; Sparagna et al., 1995). This mechanism describes intramitochondrial free [Ca2+] concentration regulation of both Ca2+ uptake, and Ca2+ extrusion, through changes in membrane potential. The Nguyen and Jafri model also reproduced intramitochondrial Ca2+ oscillations in response to pacing, with Ca2+ rising to approximately 1/3 of the cytosolic Ca2+ concentration (at 1Hz frequency), in agreement with experimental measurements by Trollinger and co-workers (Trollinger et al., 2000).
In a more comprehensive model of the ventricular myocyte, Cortassa and coworkers (2006) integrated all major cellular ATP consuming processes, thereby providing an all-inclusive platform for studying the relationship between cardiac energy supply and demand, and through it the interplay between Ca2+ cycling in the SR and in the mitochondria. Researchers have used this platform to study cellular energetics duringphysiological and pathophysiological excitation-contraction coupling (Korzeniewsky, 2007; Maack and O’Rourke, 2007; Mack et al., 2006; O’Rourke and Maack, 2007; Plank et al., 2008). There have been other notable modeling attempts to link electrophysiology, ion homeostasis, Ca2+ handling, ATP consumption, and mitochondrial energetics. One model developed by Matsuoka and co-workers (2004) used Ca2+ as the sole signaling molecule to study the interaction between the cytoplasmic and mitochondrial spaces. Despite being based on sound electrophysiology, the interrelationship between changes in excitation-contraction coupling and bioenergetics was deemed incomplete due to the lack of significant respiratory control in the mitochondrial component of the model (Korzeniewski and Mazat, 1996). Others have elegantly modeled the energetic processes in the mitochondria but do not incorporate the electrophysiological components that regulate cytosolic Ca2+ cycling (Saks et al., 2001, 2004; Vendelin et al., 2000, 2004).
Current mathematical models of mitochondrial bioenergetics support intrinsic interactions between SR and mitochondrial Ca2+ cycling by way of regulating energy metabolism in the mitochondria. These models are fitted to data obtained from experiments conducted on isolated mitochondria, and validated by their ability to reproduce results obtained in vitro. Moreover, the scarcity of experimental data on intact mitochondria, particularly on the spatial geometry of the inner mitochondrial matrix, limits accurate modeling of phenomena such as RaM and spontaneous local depolarization events. For this reason, these models provide only a semiqualitative means of studying bioenergetics in intact mitochondria. In all, the predictive accuracy of mathematical models is restricted by the paucity of detailed spatially and temporally resolved experimental evidence.
5. Future prospects
Mathematical modeling
The limitations of current mathematical models of mitochondria and how they interact with the cellular and subcellular environment are many. One of the key deficiencies is that the models do not fully include spatially and temporally resolved relationships between Ca2+ cycling in the cytosol, in the SR and in the mitochondria. In current models, both SR and mitochondria are each largely modeled as individual homogeneous (“lumped”) compartments. Experimental evidence discussed earlier suggests that the ultrastructure of both mitochondria and SR play a critical role in the regulation of Ca2+ cycling within these compartments. The conceptual differences in the lumped versus spatially resolved models have not yet been convincingly articulated. Our inability to account for the existence of metabolic compartments (and subcompartments), which is supported by the heterogeneous distribution of ATP within the cell (Saks et al., 1996), was acknowledge by Cortassa and coworkers (2006) to negatively affect the accuracy of the models. Additionally, Ca2+ sparks and the rapid uptake mode (RaM) in mitochondria, which both suggest local intracellular compartments of rapid Ca2+ dynamics, are not considered. A starting point in addressing these limitations is a more physiological formulation of SR Ca2+ release. This would require inclusion of Ca2+ release units (Jafri et al., 1998) that would generate Ca2+ sparks (Sobie et al., 2002), such that the cytosolic Ca2+ transient is a summation of these individual release events. A partitioning of the mitochondrial compartment such that some regions are exposed to the larger local [Ca2+] near the jSR end of the IMFM, while other mitochondrial compartments interact with [Ca2+] in the bulk cytoplasm, is also important. The jSR regions may then be the equivalent to the microdomains and possible sites of RaM. Although spatially resolved structures and functions of virtually all critical mitochondrial elements are unknown, one may be able to take steps to approach this goal. For example, parameters could be derived through parameter fitting, where known biophysical properties of the system are used as boundary conditions. Such approaches have been successfully employed in mathematical models of excitation contraction coupling, and to model Ca2+ dynamics in the junctional cleft (Cannell et al., 2006; Jafri et al., 1998; Shannon et al., 2004; Soeller and Cannell, 2004). The resulting models would provide a more detailed and physiologically realistic estimate of cardiac cellular energetics and Ca2+ signaling with respect to both the mitochondria and SR and their interactions under the modeled conditions.
Effects of mitochondrial agents on the sarcoplasmic reticulum Ca2+ cycling
Table 1 summarizes information about the direct targets of the major mitochondrial Ca2+ cycling inhibitors and their concentrations used by different authors in vivo and in vitro, in different cells. One of the most significant restrictions in studies of IMFM-SR crosstalk is the lack of data on the effects of mitochondrial agents on SR Ca2+ cycling mechanisms. The table shows that such effects are either unknown or questionable. Moreover, some data for SR Ca2+ release were obtained from changes in global [Ca2+] that do not reveal direct targets because (1) inhibition of RyR2s can result in an actual increase in [Ca2+]SR] due to inhibition of Ca2+ leakage through RyR2s and (2) activation of RyR2s can decrease [Ca2+]SR due to increase in Ca2+ leakage from the SR (Lukyanenko et al., 1996, 2001b). Effects of some of these inhibitors on cardiac SR Ca2+ uptake are not well described because the methods used only permit the authors to evaluate their inhibitory effects on SR Ca2+ cycling in general (Cox et al., 1993; Matlib et al., 1998). Therefore, more work is needed to characterize the effects of these mitochondrial agents on the SR.
Table 1.
Inhibitors of mitochondrial Ca2+ cycling: their direct targets, commonly used concentrations and known effects on the cardiac SR Ca2+ cycling.
| Mitochondrial target |
IC50 (μM) |
Concentrations used (μM) |
Effect on SR Ca2+ cycling | ||
|---|---|---|---|---|---|
| Inhibitor | Release | Uptake | |||
| CGP 37157 | mNCX | 0.36 10 | 1-100 4, 5,10, 17,19,24, 26 | unknown | no?10 |
| Clonazepam | mNCX | 7.00 10 | 5-100 7,23 | unknown | no?10 |
| Ru360 | MCU | 0.184 16 | 0.1-10 11,19,22,26 | no?16 | no?16 |
| Rotenone | H+ transport | <1? 14,20 | 1-50 4,8,14,27,29 | unknown | unknown |
| Cyclosporin A | PTP | 10? 12, <0.1? 25 | 0.01-25 1-3,5,9,13, 5,21,25-27,29 | no? 9; yes? 6,18 | unknown |
MCU, electrogenic mitochondrial Ca2+ uniporter; mNCX, mitochondrial Na+/Ca2+ exchanger; NHE, Na+/H+ exchanger; nSR, network SR; PTP, permeability transition pore; “?” – questionable
Development of novel approaches to measure mitochondrial Ca2+ cycling in vivo
Recently a number of novel and powerful approaches were developed and used in cardiac cells that should help to resolve some of the issues noted above. These include: proteomics of Ca2+-sensing proteins in heart mitochondria (Balaban, 2006; Hopper et al., 2006; Taylor et al., 2003); mitochondrial matrix–targeted redox- or Ca2+-sensitive fluorescent proteins and photoactivable GFP (Gerencser et al., 2008b; Karbowski et al., 2004, 2006; Wang et al., 2008), measurement of mitochondrial swelling in situ by optimized spatial filtering (Gerencser et al., 2008a), and measurement of instantaneous velocity vectors of mitochondrial transport and bioenergetic parameters (Gerencser et al., 2008b). For example, using the latter, it was discovered that hippocampal mitochondria with a higher oxidized thiol redox status have lower membrane potentials and are smaller in size. On average, these mitochondria also have higher motility, which only slightly depended on bioenergetic parameters, but is correlated to the size of the mitochondria. This approach could help to distinguish between the three groups of cardiac mitochondria, including possible interchange in their location within the myocyte.
Recently, we developed a novel approach, confocal monitoring of fluorescence from the mitochondrial intermembrane space loaded with Ca2+ sensitive fluorescent dye (Lukyanenko et al., 2008). This approach could have a great advantage in studying local SRIMFM Ca2+ interplay. It will enable or improve: (1) recording of [Ca2+] in close proximity to IMFM, (2) characterization of the dependence of IMFM Ca2+ cycling on Na+, (3) confirmation or rejection of the functional difference between the three subpopulations of cardiac mitochondria, (4) visualization of the changes in mitochondrial size in conjunction with their effects on the SR Ca2+ cycling (Ca2+ sparks), and finally, (5) clarification of the role of mitochondrial Ca2+ cycling in the regulation of SR Ca2+ cycling. Recently, after loading the mitochondrial intermembrane space with fluo-3 pentapotassium salt (fluo-35−) in isolated cardiac mitochondria, we showed that fluo-35−: (1) enters the mitochondrial intermembrane space through VDAC, (2) emits a brighter fluorescence signal there than in the surrounding solution due to a higher apparent concentration, and,(3) shows no changes in properties in this mitochondrial low pH sub-domain (Lukyanenko et al., 2008). We concluded that fluo-35− may be used as a probe for cardiac mitochondrial research in its original membrane environment and under quasi-physiological conditions. Preliminary experiments in permeabilized ventricular myocytes support these conclusions, and in addition, suggest that during pathologies involving mitochondrial swelling, mitochondria can produce Ca2+ release, which may increase Ca2+ leakage from the SR.
Summary
Research into the role of mitochondrial Ca2+ cycling and its relationship to SR Ca2+ cycling is likely to benefit our understanding of Ca2+ dynamics in heart. Experimental investigations and parallel mathematical modeling at high spatial and temporal resolution are needed and should enable us to better investigate the molecular physiology in normal hearts and more precisely examine Ca2+ and electrical dysfunction in heart disease including arrhythmia and failure.
ACKNOWLEDGEMENTS
The authors research was supported by a network-grant from Fondation Leducq (“European North American Atrial Fibrillation Research Alliance”, to W.J.L.), by the Interdisciplinary Training Program in Muscle Biology, National Institute of Arthritis and Musculoskeletal and Skin Diseases (to A.C.), by The Maryland Stem Cell Research Fund (to W.J.L.), and by the National Heart Lung and Blood Institute (to W.J.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–35. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschafl BA, Beutner G, Sharma VK, Sheu SS, Valdivia HH. The mitochondrial ryanodine receptor in rat heart: a pharmaco-kinetic profile. Biochim Biophys Acta. 2007;1768:1784–95. doi: 10.1016/j.bbamem.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J Cell Biol. 1988;107:481–95. doi: 10.1083/jcb.107.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, O’Rourke B. The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys J. 2006;91:4317–27. doi: 10.1529/biophysj.106.087817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appaix F, Guerrero K, Rampal D, Izikki M, Kaambre T, Sikk P, et al. Bax and heart mitochondria: uncoupling and inhibition of respiration without permeability transition. Biochim Biophys Acta. 2002;1556:155–67. doi: 10.1016/s0005-2728(02)00358-4. [DOI] [PubMed] [Google Scholar]
- Arnaudeau S, Kelley WL, Walsh JV, Demaurex N. Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–9. doi: 10.1074/jbc.M103274200. [DOI] [PubMed] [Google Scholar]
- Ban K, Handa SR, Chapman A. On the mechanism of the failure of mitochondrial function in isolated guinea-pig myocytes subjected to a Ca2+ overload. Cardiovasc Res. 1999;44:556–67. doi: 10.1016/s0008-6363(99)00233-3. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A, Shin DW, Ahn JO, Kim DH. Calcineurin regulates ryanodine receptor/Ca2+-release channels in rat heart. Biochem J. 2000;352:61–70. [PMC free article] [PubMed] [Google Scholar]
- Baron KT, Thayer SA. CGP37157 modulates mitochondrial Ca2+ homeostasis in cultured rat dorsal root ganglion neurons. Eur J Pharmacol. 1992;340:295–300. doi: 10.1016/s0014-2999(97)01433-7. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992;453:591–608. doi: 10.1113/jphysiol.1992.sp019246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani JW, Bassani RA, Bers DM. Ca2+ cycling between sarcoplasmic reticulum and mitochondria in rabbit cardiac myocytes. J Physiol. 1993;460:603–21. doi: 10.1113/jphysiol.1993.sp019489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani RA, Fagian MM, Bassani JW, Vercesi AE. Changes in calcium uptake rate by rat cardiac mitochondria during postnatal development. J Mol Cell Cardiol. 1998;30:2013–23. doi: 10.1006/jmcc.1998.0762. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA. 2008;105:2198–202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–17. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002a;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-contraction coupling and cardiac contractile force. 2nd ed Kluwer Academic Publishers; Dordrecht: 2001. [Google Scholar]
- Bers DM. Sarcoplasmic reticulum Ca release in intact ventricular myocytes. Front Biosci. 2002b;7:d1697–711. doi: 10.2741/A873. [DOI] [PubMed] [Google Scholar]
- Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276:21482–8. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- Bhogal MS, Colyer J. Depletion of Ca2+ from the sarcoplasmic reticulum of cardiac muscle prompts phosphorylation of phospholamban to stimulate store refilling. Proc Natl Acad Sci USA. 1998;95:1484–9. doi: 10.1073/pnas.95.4.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes R, Bers DM. Simultaneous measurements of mitochondrial NADH and Ca2+ during increased work in intact rat heart trabeculae. Biophys J. 2002;83:587–604. doi: 10.1016/S0006-3495(02)75194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–10. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci USA. 2005;102:3099–104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser DN, Minamikawa T, Nagley P, Williams DA. Role of mitochondria in calcium regulation of spontaneously contracting cardiac muscle cells. Biophys J. 1998;75:2004–14. doi: 10.1016/S0006-3495(98)77642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996;66:403–11. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Buntinas L, Gunter KK, Sparagna GC, Gunter TE. The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta. 2001;1504:248–61. doi: 10.1016/s0005-2728(00)00254-1. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–56. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–9. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Crossman DJ, Soeller C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J Muscle Res Cell Motil. 2006;27:297–306. doi: 10.1007/s10974-006-9089-y. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- Carré M, André N, Carles G, Borghi H, Brichese L, Briand C, Braguer D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage - dependent anion channel. J Biol Chem. 2002;277:33664–9. doi: 10.1074/jbc.M203834200. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278:19062–70. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Chemnitius JM, Manglitz T, Kloeppel M, Doenst T, Schwartz P, Kreuzer H, Zech R. Rapid preparation of subsarcolemmal and interfibrillar mitochondrial subpopulations from cardiac muscle. Int J Biochem. 1993;25:589–96. doi: 10.1016/0020-711x(93)90668-5. [DOI] [PubMed] [Google Scholar]
- Chen W, London R, Murphy E, Steenbergen C. Regulation of the Ca2+ gradient across the sarcoplasmic reticulum in perfused rabbit heart. A 19F nuclear magnetic resonance study. Circ Res. 1998;83:898–907. doi: 10.1161/01.res.83.9.898. [DOI] [PubMed] [Google Scholar]
- Chen W, Steenbergen C, Levy LA, Vance J, London RE, Murphy E. Measurement of free Ca2+ in sarcoplasmic reticulum in perfused rabbit heart loaded with 1,2-bis(2-amino-5,6-difluorophenoxy)ethane-N,N,N’,N’-tetraacetic acid by 19F NMR. J Biol Chem. 1996;271:7398–403. [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer W, Cannell M. Calcium Sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–4. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270:C148–59. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- Chiesi M, Wrzosek A, Grueninger S. The role of the sarcoplasmic reticulum in various types of cardiomyocytes. Mol Cell Biochem. 1994;130:159–71. doi: 10.1007/BF01457397. [DOI] [PubMed] [Google Scholar]
- Ching LL, Williams AJ, Sitsapesan R. Evidence for Ca2+ activation and inactivation sites on the lumenal side of the cardiac ryanodine receptor complex. Circ Res. 2000;87:201–6. doi: 10.1161/01.res.87.3.201. [DOI] [PubMed] [Google Scholar]
- Chini EN, Dousa TP. Enzymatic synthesis and degradation of nicotinate adenine dinucleotide phosphate (NAADP), a Ca2+-releasing agonist, in rat tissues. Biochem. Biophys Res Commun. 1995;209:167–74. doi: 10.1006/bbrc.1995.1485. [DOI] [PubMed] [Google Scholar]
- Colombini M. Regulation of the mitochondrial outer membrane channel, VDAC. J Bioenerg Biomembr. 1987;19:309–20. doi: 10.1007/BF00768534. [DOI] [PubMed] [Google Scholar]
- Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell Biochem. 2004;256-257:107–15. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- Connor JA. Intracellular calcium mobilization by inositol 1,4,5-trisphosphate: intracellular movements and compartmentalization. Cell Calcium. 1993;14:185–200. doi: 10.1016/0143-4160(93)90066-f. [DOI] [PubMed] [Google Scholar]
- Cortassa S, Aon MA, Marban E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003;84:2734–55. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortassa S, Aon MA, O’Rourke B, Jacques R, Tseng HJ, Marban E, Winslow RL. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J. 2006;91:1564–89. doi: 10.1529/biophysj.105.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DA, Conforti L, Sperelakis N, Matlib MA. Selectivity of inhibition of Na+-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol. 1993;21:595–9. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- Cox DA, Matlib MA. Modulation of intramitochondrial free Ca2+ concentration by antagonists of Na+-Ca2+ exchange. Trends Pharmacol Sci. 1993;14:408–13. doi: 10.1016/0165-6147(93)90063-P. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–49. [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Madesh M, Antonsson B, Hajnoczky G. tcBid promotes Ca2+ signal propagation to the mitochondria: control of Ca2+ permeation through the outer mitochondrial membrane. EMBO J. 2002;21:2198–206. doi: 10.1093/emboj/21.9.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie S, Smith GL. Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc Res. 1999;41:135–46. doi: 10.1016/s0008-6363(98)00241-7. [DOI] [PubMed] [Google Scholar]
- Dash RK, Beard DA. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J Physiol. 2008;586:3267–85. doi: 10.1113/jphysiol.2008.151977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44:77–91. doi: 10.1016/j.ceca.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–48. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Choi HS, Diaz ME, O’Neill SC, Trafford AW. Integrative analysis of calcium cycling in cardiac muscle. Circ Res. 2000;87:1087–94. doi: 10.1161/01.res.87.12.1087. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced calcium release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A. Two kinds of calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cardiac cells. In: Frank GB, editor. Excitation-contraction coupling in skeletal, cardiac and smooth muscle. Plennum Press; New York: 1992. pp. 245–62. [DOI] [PubMed] [Google Scholar]
- Fawcett DW. The cell. Saunders Company; Philadelphia: 1966. [Google Scholar]
- Fawcett DW, McNutt NS. The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J Cell Biol. 1969;42:1–45. doi: 10.1083/jcb.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher JJ, Fabiato A. Cardiac sarcoplasmic reticulum: calcium uptake and release. In: Langer GA, editor. Calcium and heart. Raven Press; New York: 1990. pp. 199–268. [Google Scholar]
- Ferrier GR. The effects of tension on acetylstrophanthidin-induced transient depolarizations and aftercontractions in canine myocardial and Purkinje tissues. Circ Res. 1976;38:156–62. doi: 10.1161/01.res.38.3.156. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Cockrell RS. Uncoupler-stimulated release of Ca2+ from Ehrlich ascites tumor cell mitochondria. Arch Biochem Biophys. 1985;240:723–33. doi: 10.1016/0003-9861(85)90081-5. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann NY Acad Sci. 1998;853:20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–39. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JS, Mottino G, Reid D, Molday RS, Philipson KD. Distribution of the Na+-Ca2+ exchange protein in mammalian cardiac myocytes: an immunofluorescence and immunocolloidal gold-labeling study. J Cell Biol. 1992;117:337–45. doi: 10.1083/jcb.117.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JS. Ultrastructure of the unfixed myocardial sarcolemma and cell surface. In: Langer GA, editor. Calcium and the heart. Raven Press; New York: 1990. pp. 1–25. [Google Scholar]
- Franco L, Guida L, Bruzzone S, Zocchi E, Usai C, De Flora A. The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP-ribose across membranes. FASEB J. 1998;12:1507–20. doi: 10.1096/fasebj.12.14.1507. [DOI] [PubMed] [Google Scholar]
- Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–24. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- Fry CH, Powell T, Twist VW, Ward JP. Net calcium exchange in adult rat ventricular myocytes: an assessment of mitochondrial calcium accumulating capacity. Proc R Soc Lond B Biol Sci. 1984a;223:223–38. doi: 10.1098/rspb.1984.0091. [DOI] [PubMed] [Google Scholar]
- Fry CH, Powell T, Twist VW, Ward JP. The effects of sodium, hydrogen and magnesium ions on mitochondrial calcium sequestration in adult rat ventricular myocytes. Proc R Soc Lond B Biol Sci. 1984b;223:239–54. doi: 10.1098/rspb.1984.0092. [DOI] [PubMed] [Google Scholar]
- Gallitelli MF, Schultz M, Isenberg G, Rudolf F. Twitch-potentiation increases calcium in peripheral more than in central mitochondria of guinea-pig ventricular myocytes. J Physiol. 1999;518:433–447. doi: 10.1111/j.1469-7793.1999.0433p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Pérez C, Hajnóczky G, Csordás G. Physical coupling supports the local Ca2+ transfer between SR subdomains and the mitochondria in heart muscle. J Biol Chem. 2008;283:32771–80. doi: 10.1074/jbc.M803385200. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathercole DV, Colling DJ, Skepper JN, Takagishi Y, Levi AJ, Severs NJ. Immunogold-labeled L-type calcium channels are clustered in the surface plasma membrane overlying junctional sarcoplasmic reticulum in guinea-pig myocytes-implications for excitation-contraction coupling in cardiac muscle. J Mol Cell Cardiol. 2000;32:1981–94. doi: 10.1006/jmcc.2000.1230. [DOI] [PubMed] [Google Scholar]
- Chini EN, Dousa TP. Enzymatic synthesis and degradation of nicotinate adenine dinucleotide phosphate (NAADP), a Ca2+-releasing agonist, in rat tissues. Biochem Biophys Res Commun. 1995;209:167–74. doi: 10.1006/bbrc.1995.1485. [DOI] [PubMed] [Google Scholar]
- Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358:147–55. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski MM, Gajkowska B, Lamparska-Przybysz M, Motyl T. Colocalization of BAX with BID and VDAC-1 in nimesulide-induced apoptosis of human colon adenocarcinoma COLO 205 cells. Anticancer Drugs. 2002;13:1017–29. doi: 10.1097/00001813-200211000-00006. [DOI] [PubMed] [Google Scholar]
- Gerencser AA, Doczi J, Töröcsik B, Bossy-Wetzel E, Adam-Vizi V. Mitochondrial swelling measurement in situ by optimized spatial filtering: astrocyte-neuron differences. Biophys J. 2008a;95:2583–98. doi: 10.1529/biophysj.107.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Nicholls DG. Measurement of instantaneous velocity vectors of organelle transport: mitochondrial transport and bioenergetics in hippocampal neurons. Biophys J. 2008b;95:3079–99. doi: 10.1529/biophysj.108.135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J. 2006;90:77–91. doi: 10.1529/biophysj.105.065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J. 2002;83:2918–45. doi: 10.1016/S0006-3495(02)75301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatimosim S, Dilly K, Santana LF, Jafri MS, Sobie EA, Lederer WJ. Local Ca2+ signaling and EC coupling in heart: Ca2+ sparks and the regulation of the [Ca2+]i transient. J Mol Cell Cardiol. 2002;34:941–50. doi: 10.1006/jmcc.2002.2032. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–39. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–86. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Guse AH. Cyclic ADP-ribose. J Mol Med. 2000;78:26–35. doi: 10.1007/s001090000076. [DOI] [PubMed] [Google Scholar]
- Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–10. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke S, Lukyanenko V, Györke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol. 1997;500:297–309. doi: 10.1113/jphysiol.1997.sp022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak LL, Grimaldi M, Smaili SS, Russell JT. Mitochondria regulate Ca2+ wave initiation and inositol trisphosphate signal transduction in oligodendrocyte progenitors. J Neurochem. 2002;80:405–15. doi: 10.1046/j.0022-3042.2001.00727.x. [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J Cell Biol. 1968;37:345–69. doi: 10.1083/jcb.37.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi K, Schmidt AG, Hoit BD, Brittsan AG, Yatani A, Lester JW, et al. Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. J Biol Chem. 2001;276:24145–52. doi: 10.1074/jbc.M102403200. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Krishnamurthy R, Szalai G. Mitochondrial calcium signaling driven by the IP3 receptor. J Bioenerg Biomembr. 2000;32:15–25. doi: 10.1023/a:1005504210587. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Call Cardiol. 2002;34:951–69. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195:460–7. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- Heiskanen KM, Bhat MB, Wang HW, Ma J, Nieminen AL. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J Biol Chem. 1999;274:5654–6568. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- Ho R, Fan D, Somlyo AV, Somlyo AP. Calcium content of peripheral and central mitochondria in the guinea pig myocardium: electron probe analysis. Cell Calcium. 2003;33:247–56. doi: 10.1016/s0143-4160(02)00238-5. [DOI] [PubMed] [Google Scholar]
- Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–84. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- Hoppel CL, Tandler B, Parland W, Turkaly JS, Albers LD. Hamster cardiomyopathy. A defect in oxidative phosphorylation in the cardiac interfibrillar mitochondria. J Biol Chem. 1982;257:1540–8. [PubMed] [Google Scholar]
- Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, et al. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45:2524–36. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999;343:311–7. [PMC free article] [PubMed] [Google Scholar]
- Ikemoto N, Yamamoto T. The luminal Ca2+ transient controls Ca2+ release/re-uptake of sarcoplasmic reticulum. Biochem Biophys Res Commun. 2000;279:858–63. doi: 10.1006/bbrc.2000.4031. [DOI] [PubMed] [Google Scholar]
- Isaeva EV, Shirokova N. Metabolic regulation of Ca2+ release in permeabilized mammalian skeletal muscle fibres. J Physiol. 2003;547:453–62. doi: 10.1113/jphysiol.2002.036129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaeva EV, Shkryl VM, Shirokova N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J Physiol. 2005;565:855–72. doi: 10.1113/jphysiol.2005.086280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg G, Han S, Schiefer A, Wendt-Gallitelli MF. Changes in mitochondrial calcium concentration during the cardiac contraction cycle. Cardiovasc Res. 1993;27:1800–9. doi: 10.1093/cvr/27.10.1800. [DOI] [PubMed] [Google Scholar]
- Ishide N. Intracellular calcium modulators for cardiac muscle in pathological conditions. Jap Heart J. 1996;37:1–17. doi: 10.1536/ihj.37.1. [DOI] [PubMed] [Google Scholar]
- Izu L, Balke CW. The Ca2+ synapse redo: a matter of location, location, location. Circ Res. 2002;91:276–7. doi: 10.1161/01.res.0000032008.49697.e3. [DOI] [PubMed] [Google Scholar]
- Izu LT, Wier WG, Balke CW. Evolution of cardiac calcium waves from stochastic calcium sparks. Biophys J. 2001;80:103–20. doi: 10.1016/S0006-3495(01)75998-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri MS, Dudycha SJ, O’Rourke B. Cardiac energy metabolism: models of cellular respiration. Ann Rev Biomed Eng. 2001;3:57–81. doi: 10.1146/annurev.bioeng.3.1.57. [DOI] [PubMed] [Google Scholar]
- Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J. 1998;74:1149–68. doi: 10.1016/S0006-3495(98)77832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse MJ. Electrophysiology of arrhythmias. Arch Mal Coeur Vaiss. 1999;92:9–16. [PubMed] [Google Scholar]
- Jones M, Ferrans VJ, Morrow AG, Roberts WC. Ultrastructure of crista supraventricularis muscle in patients with congenital heart diseases associated with right ventricular outflow tract obstruction. Circulation. 1975;51:39–67. doi: 10.1161/01.cir.51.1.39. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–41. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Jordani MC, Santos AC, Prado IM, Uyemura SA, Curti C. Flufenamic acid as an inducer of mitochondrial permeability transition. Mol Cell Biochem. 2000;210:153–8. doi: 10.1023/a:1007185825101. [DOI] [PubMed] [Google Scholar]
- Jorgensen AO, Jones LR. Immunoelectron microscopical localization of phospholamban in adult canine ventricular muscle. J Cell Biol. 1987;104:1343–52. doi: 10.1083/jcb.104.5.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen AO, Shen AC, Arnold W, McPherson PS, Campbell KP. The Ca2+-release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J Cell Biol. 1993;120:969–80. doi: 10.1083/jcb.120.4.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen AO, Shen AC, Daly P, MacLennan DH. Localization of Ca2+ + Mg2+-ATPase of the sarcoplasmic reticulum in adult rat papillary muscle. J Cell Biol. 1982;93:883–92. doi: 10.1083/jcb.93.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res. 2001;89:153–9. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- Kang SH, Park WS, Kim N, Youm JB, Warda M, Ko JH, Ko EA, Han J. Mitochondrial Ca2+ - activated K+ channels more efficiently reduce mitochondrial Ca2+ overload in rat ventricular myocytes. Am J Physiol. 2007;293:H307–13. doi: 10.1152/ajpheart.00789.2006. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164:493–9. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–62. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Kieval RS, Bloch RJ, Lindenmayer GE, Ambesi A, Lederer WJ. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am J Physiol. 1992;263:C545–50. doi: 10.1152/ajpcell.1992.263.2.C545. [DOI] [PubMed] [Google Scholar]
- Keizer J, Magnus G. ATP-sensitive potassium channel and bursting in the pancreatic beta cell. A theoretical study. Biophys J. 1989;56:229–42. doi: 10.1016/S0006-3495(89)82669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara Y, Morgan JP. Intracellular calcium and ventricular fibrillation. Studies in the aequorin-loaded isovolumic ferret heart. Circ Res. 1991;68:1378–89. doi: 10.1161/01.res.68.5.1378. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–61. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, et al. The proapoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–22. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SD, Berglund J, Choudhury D. Bacterial adhesins: structural studies reveal chaperone function and pilus biogenesis. Curr Opin Chem Biol. 2000;4:653–60. doi: 10.1016/s1367-5931(00)00144-7. [DOI] [PubMed] [Google Scholar]
- Kononova VA. Quantitative analysis of the mitochondrial ultrastructure of cardiomyocytes in rats adapting to altitude hypoxia. Biull Eksp Biol Med. 1982;94:116–8. [PubMed] [Google Scholar]
- Korzeniewski B, Mazat JP. Theoretical studies on the control of oxidative phosphorylation in muscle mitochondria: application to mitochondrial deficiencies. Biochem J. 1996;319:143–8. doi: 10.1042/bj3190143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski B. Regulation of oxidative phosphorylation through parallel activation. Biophys Chem. 1970;139:93–110. doi: 10.1016/j.bpc.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kuo TH, Zhu L, Golden K, Marsh JD, Bhattacharya SK, Liu BF. Altered Ca2+ homeostasis and impaired mitochondrial function in cardiomyopathy. Mol Cell Biochem. 2002;238:119–27. doi: 10.1023/a:1019967323419. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–42. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc Res. 1992;26:193–214. doi: 10.1093/cvr/26.3.193. [DOI] [PubMed] [Google Scholar]
- Lambeth MJ, Kushmerick MJ. A computational model for glycogenolysis in skeletal muscle. Ann Biomed Eng. 2002;30:808–27. doi: 10.1114/1.1492813. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- Lee HC. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu Rev Pharmacol Toxicol. 2001;41:317–45. doi: 10.1146/annurev.pharmtox.41.1.317. [DOI] [PubMed] [Google Scholar]
- Lee HC, Graeff RM, Walseth TF. ADP-ribosyl cyclase and CD38. Multi-functional enzymes in Ca2+ signaling. Adv Exp Med Biol. 1997;419:411–9. [PubMed] [Google Scholar]
- Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–45. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Terrenoire C, Reiken S, Wehrens XH, Song LS, Tillman EJ, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci USA. 2006;103:7906–10. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Chini EN, Cheng J, Dousa TP. Synthesis of NAADP and cADPR in mitochondria. Arch Biochem Biophys. 1999;371:317–25. doi: 10.1006/abbi.1999.1463. [DOI] [PubMed] [Google Scholar]
- Liu M, Siess M, Hoffmann PC. Inhibition of the mitochondrial respiratory chain in isolated atria-a comparison of rotenone and amytal. Biochem Pharmacol. 1970;19:197–207. doi: 10.1016/0006-2952(70)90340-0. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez J, Shacklock P, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–5. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- Loupatatzis C, Seitz G, Schonfeld P, Lang F, Siemen DA. Single-channel currents of the permeability transition pore from the inner mitochondrial membrane of rat liver and of a human hepatoma cell line. Cell Physiol Biochem. 2002;12:269–78. doi: 10.1159/000067897. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V. Delivery of nano-objects to functional sub-domains of healthy and failing cardiac myocytes. Nanomed. 2007;2:831–46. doi: 10.2217/17435889.2.6.831. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Györke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized cardiac myocytes. J Physiol. 1999;521:575–85. doi: 10.1111/j.1469-7793.1999.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Györke I, Györke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflug Arch. 1996;432:1047–54. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Györke I, Subramanian S, Smirnov A, Wiesner TF, Györke S. Inhibition of Ca2+ sparks by ruthenium red in permeabilized rat ventricular myocytes. Biophys J. 2000;79:1273–84. doi: 10.1016/S0006-3495(00)76381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Györke I, Wiesner TF, Györke S. Potentiation of Ca2+ release by cADPR in heart is mediated by enhanced Ca2+ uptake into the sarcoplasmic reticulum. Circ Res. 2001a;89:614–22. doi: 10.1161/hh1901.098066. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Lederer WJ, Salnikov VV. Role of intermyofibrillar mitochondria (IMM) in local regulation of the SR Ca2+ cycling. Biophys J. 2003;84:338a. [Google Scholar]
- Lukyanenko V, Rostovtseva TK, Lederer WJ. Fluo-3 in the mitochondrial intermembrane space. Biophys J. 2008;93:25a. [Google Scholar]
- Lukyanenko V, Subramanian S, Györke I, Wiesner TF, Györke S. The role of sarcoplasmic reticlum lumenal Ca2+ in generation of Ca2+ wave in rat ventricular myocytes. J Physiol. 1999;518:173–86. doi: 10.1111/j.1469-7793.1999.0173r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Viatchenko-Karpinski S, Smirnov A, Wiesner TF, Györke S. Dynamic regulation of the SR Ca2+ content and release by lumenal Ca2+-sensitive leak through RyRs in rat ventricular myocytes. Biophys J. 2001b;81:785–98. doi: 10.1016/S0006-3495(01)75741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Wiesner TF, Györke S. Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J Physiol. 1998;507:667–77. doi: 10.1111/j.1469-7793.1998.667bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Ziman A, Lukyanenko A, Salnikov V, Lederer WJ. Functional groups of ryanodine receptors in rat ventricular cells. J Physiol. 2007;583:251–69. doi: 10.1113/jphysiol.2007.136549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–82. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2008;102:369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus G, Keizer J. Minimal model of beta-cell mitochondrial Ca2+ handling. Am J Physiol. 1997;273:C717–33. doi: 10.1152/ajpcell.1997.273.2.C717. [DOI] [PubMed] [Google Scholar]
- Magnus G, Keizer J. Model of beta-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am J Physiol. 1998a;274:C1158–73. doi: 10.1152/ajpcell.1998.274.4.C1158. [DOI] [PubMed] [Google Scholar]
- Magnus G, Keizer J. Model of beta-cell mitochondrial calcium handling and electrical activity. II. Mitochondrial variables. Am J Physiol. 1998b;274:C1174–84. doi: 10.1152/ajpcell.1998.274.4.C1174. [DOI] [PubMed] [Google Scholar]
- Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–40. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Marks AR. Ryanodine receptors/calcium release channels in heart failure and sudden cardiac death. J Mol Cell Cardiol. 2001;33:615–24. doi: 10.1006/jmcc.2000.1343. [DOI] [PubMed] [Google Scholar]
- Martin BJ, Valdivia HH, Bunger R, Lasley RD, Mentzer RM., Jr Pyruvate augments calcium transients and cell shortening in rat ventricular myocytes. Am J Physiol. 1998;274:H8–17. doi: 10.1152/ajpheart.1998.274.1.H8. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Rouslin W, Kraft G, Berner P, Schwartz A. On the existence of two populations of mitochondria in a single organ. Respiration, calcium transport and enzyme activities. Biochem Biophys Res Commun. 1978;84:482–8. doi: 10.1016/0006-291x(78)90194-8. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, et al. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem. 1998;273:10223–31. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Sarai N, Jo H, Noma A. Simulation of ATP metabolism in cardiac excitation-contraction coupling. Prog Biophys Mol Biol. 2004;85:279–99. doi: 10.1016/j.pbiomolbio.2004.01.006. [DOI] [PubMed] [Google Scholar]
- McMillin-Wood J, Wolkowicz PE, Chu A, Tate CA, Goldstein MA, Entman ML. Calcium uptake by two preparations of mitochondria from heart. Biochim Biophys Acta. 1980;591:251–65. doi: 10.1016/0005-2728(80)90157-7. [DOI] [PubMed] [Google Scholar]
- McNutt NS, Fawcett DW. The ultrastructure of the cat myocardium. II. Atrial muscle. J Cell Biol. 1969;42:46–67. doi: 10.1083/jcb.42.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin-Wood J, Wolkowicz PE, Chu A, Tate CA, Goldstein MA, Entman ML. Calcium uptake by two preparations of mitochondria from heart. Biochim Biophys Acta. 1980;591:251–65. doi: 10.1016/0005-2728(80)90157-7. [DOI] [PubMed] [Google Scholar]
- Meszaros LG, Wrenn RW, Varadi G. Sarcoplasmic reticulum-associated and protein kinase C-regulated ADP-ribosyl cyclase in cardiac muscle. Biochem Biophys Res Commun. 1997;234:252–6. doi: 10.1006/bbrc.1997.6620. [DOI] [PubMed] [Google Scholar]
- Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–98. doi: 10.1083/jcb.150.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, et al. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol. 1999;31:2063–76. doi: 10.1006/jmcc.1999.1037. [DOI] [PubMed] [Google Scholar]
- Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol. 1991;261:H1123–34. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- Mojzisova A, Krizanova O, Zacikova L, Kominkova V, Ondrias K. Effect of nicotinic acid adenine dinucleotide phosphate on ryanodine calcium release channel in heart. Pflugers Arch. 2001;441:674–7. doi: 10.1007/s004240000465. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–40. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- Monge C, Beraud N, Kuznetsov AV, Rostovtseva T, Sackett D, Schlattner U, et al. Regulation of respiration in brain mitochondria and synaptosomes: restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol Cell Biochem. 2008;318:147–65. doi: 10.1007/s11010-008-9865-7. [DOI] [PubMed] [Google Scholar]
- Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, Lee HC. Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J Biol Chem. 2000;275:21566–71. doi: 10.1074/jbc.M909365199. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- Nguyen MH, Dudycha SJ, Jafri MS. Effect of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am J Physiol. 2007;292:C2004–20. doi: 10.1152/ajpcell.00271.2006. [DOI] [PubMed] [Google Scholar]
- Nguyen MH, Jafri MS. Mitochondrial calcium signaling and energy metabolism. Ann NY Acad Sci. 2005;1047:127–37. doi: 10.1196/annals.1341.012. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Chalmers S. The integration of mitochondrial calcium transport and storage. J Bioenerg Biomembr. 2004;36:277–81. doi: 10.1023/B:JOBB.0000041753.52832.f3. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Crompton M. Mitochondrial calcium transport. FEBS Lett. 1980;111:261–8. doi: 10.1016/0014-5793(80)80806-4. [DOI] [PubMed] [Google Scholar]
- O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli E, Lederer WJ. Voltage-independent calcium release in heart muscle. Science. 1990;250:565–8. doi: 10.1126/science.2173135. [DOI] [PubMed] [Google Scholar]
- Ohata H, Chacon E, Tesfai SA, Harper IS, Herman B, Lemasters JJ. Mitochondrial Ca2+ transients in cardiac myocytes during the excitation-contraction cycle: effects of pacing and hormonal stimulation. J Bioenerg Biomembr. 1998;30:207–22. doi: 10.1023/a:1020588618496. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Takasawa S, Nata K, Kato I, Tohgo A, Noguchi N. Physiological and pathological significance of the CD38-cyclic ADP-ribose signaling system. Chem Immunol. 2000;75:121–45. doi: 10.1159/000058766. [DOI] [PubMed] [Google Scholar]
- Oliveira GA, Kowaltowski AJ. Phosphate increases mitochondrial reactive oxygen species release. Free Radic Res. 2004;38:1113–8. doi: 10.1080/10715760400009258. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, Maack C. The role of Na dysregulation in cardiac disease and how it impacts electrophysiology. Drug Discov Today Dis Models. 2007;4:207–17. doi: 10.1016/j.ddmod.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Katagiri T, Yoshida F, Nitani H, Nakai Y. Recent advances in studies on cardiac structure and metabolism. University Park Press; Baltimore: 1976. Electron microscopic studies on ATPase activities in myocardial infarction; pp. 471–5. [PubMed] [Google Scholar]
- Pacher P, Csordas P, Schneider T, Hajnoczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol. 2000;529:553–64. doi: 10.1111/j.1469-7793.2000.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Hajnoczky G. Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 2001;20:4107–21. doi: 10.1093/emboj/20.15.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Thomas AP, Hajnoczky G. Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc Natl Acad Sci USA. 2002;99:2380–5. doi: 10.1073/pnas.032423699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–9. [PubMed] [Google Scholar]
- Pan Z, Bhat MB, Nieminen AL, Ma J. Synergistic movements of Ca2+ and Bax in cells undergoing apoptosis. J Biol Chem. 2001;276:32257–63. doi: 10.1074/jbc.M100178200. [DOI] [PubMed] [Google Scholar]
- Parfenov AS, Salnikov V, Lederer WJ, Lukyanenko V. Aqueous diffusion pathways as a part of the ventricular cell ultrastructure. Biophys J. 2008;90:1107–19. doi: 10.1529/biophysj.105.071787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Kim TK, Kim DH. Cyclosporin A treatment alters characteristics of Ca2+-release channel in cardiac sarcoplasmic reticulum. Am J Physiol. 1999;276:H865–72. doi: 10.1152/ajpheart.1999.276.3.H865. [DOI] [PubMed] [Google Scholar]
- Paschen SA, Waizenegger T, Stan T, Preuss M, Cyrklaff M, Hell K, et al. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature. 2003;426:862–6. doi: 10.1038/nature02208. [DOI] [PubMed] [Google Scholar]
- Piacentino V, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–8. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- Pitter JG, Maechler P, Wollheim CB, Spat A. Mitochondria respond to Ca2+ already in the submicromolar range: correlation with redox state. Cell Calcium. 2002;31:97–104. doi: 10.1054/ceca.2001.0264. [DOI] [PubMed] [Google Scholar]
- Plank G, Zhou L, Greenstein JL, Cortassa S, Winslow RL, O’Rourke B, Trayanova NA. From mitochondrial ion channels to arrhythmias in the heart: computational techniques to bridge the spatio-temporal scales. Philos Transact A Math Phys Eng Sci. 2008;366:3381–409. doi: 10.1098/rsta.2008.0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM. Calcium cycling in heart failure: the arrhythmia connection. J Cardiovasc Electrophysiol. 2002;13:88–91. doi: 10.1046/j.1540-8167.2002.00088.x. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Prestle J, Quinn FR, Smith GL. Ca2+-handling proteins and heart failure: novel molecular targets? Curr Med Chem. 2003;10:967–81. doi: 10.2174/0929867033457656. [DOI] [PubMed] [Google Scholar]
- Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann NY Acad Sci. 1998;853:341–4. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Stigliani S, Zedda L, Raiteri M, Bonanno G. Multiple mechanisms of transmitter release evoked by “pathologically” elevated extracellular [K+]: involvement of transporter reversal and mitochondrial calcium. J Neurochem. 2002;80:706–14. doi: 10.1046/j.0022-3042.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- Rambourg A, Segretain D. Three-dimensional electron microscopy of mitochondria and endoplasmic reticulum in the red muscle fiber of the rat diaphragm. Anat Rec. 1980;1197:33–48. doi: 10.1002/ar.1091970104. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529:37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20:4998–5007. doi: 10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Bezrukov SM. VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr. 2008;40:163–70. doi: 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Komarov A, Bezrukov SM, Colombini M. Dynamics of nucleotides in VDAC channels: structure-specific noise generation. Biophys J. 2002a;82:193–205. doi: 10.1016/S0006-3495(02)75386-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Komarov A, Bezrukov SM, Colombini M. VDAC channels differentiate between natural metabolites and synthetic molecules. J Membr Biol. 2002b;187:147–56. doi: 10.1007/s00232-001-0159-1. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–42. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci USA. 2008;105:18746–51. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands JC, Casida JE. NADH: ubiquinone oxidoreductase inhibitors block induction of ornithine decarboxylase activity in MCF-7 human breast cancer cells. Pharmacol Toxicol. 1998;83:214–9. doi: 10.1111/j.1600-0773.1998.tb01471.x. [DOI] [PubMed] [Google Scholar]
- Rube DA, van der Bliek AM. Mitochondrial morphology is dynamic and varied. Mol Cell Biochem. 2004;256-257:331–9. doi: 10.1023/b:mcbi.0000009879.01256.f6. [DOI] [PubMed] [Google Scholar]
- Saavedra-Molina A, Uribe S, Devlin TM. Control of mitochondrial matrix calcium: studies using fluo-3 as a fluorescent calcium indicator. Biochem Biophys Res Com. 1990;167:148–53. doi: 10.1016/0006-291x(90)91743-c. [DOI] [PubMed] [Google Scholar]
- Saks VA, Kaambre T, Sikk P, Eimre M, Orlova E, Paju K, et al. Intracellular energetic units in red muscle cells. Biochem J. 2001;356:643–57. doi: 10.1042/0264-6021:3560643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks VA, Kuznetsov AV, Vendelin M, Guerrero K, Kay L, Seppet EK. Functional coupling as a basic mechanism of feedback regulation of cardiac energy metabolism. Mol Cel Biochem. 2004;256:257–185. doi: 10.1023/b:mcbi.0000009868.92189.fb. [DOI] [PubMed] [Google Scholar]
- Saks VA, Ventura-Clapier R, Aliev MK. Metabolic control and metabolic capacity: two aspects of creatine kinase functioning in the cells. Biochim Biophys Acta. 1996;1274:81–8. doi: 10.1016/0005-2728(96)00011-4. [DOI] [PubMed] [Google Scholar]
- Salnikov VV, Lukyanenko YO, Frederick CA, Lederer WJ, Lukyanenko V. Probing the outer mitochondrial membrane in cardiac mitochondria with nanoparticles. Biophys J. 2007;92:1058–71. doi: 10.1529/biophysj.106.094318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikov VV, Lukyanenko A, Lederer WJ, Lukyanenko V. Spatial distribution of ryanodine receptors in rat ventricular cells. Biophys J. 2005;88:87a. [Google Scholar]
- Sanchez JA, Garcia MC, Sharma VK, Young KC, Matlib MA, Sheu SS. Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J Physiol. 2001;536:387–96. doi: 10.1111/j.1469-7793.2001.0387c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996;78:166–71. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- Saotome M, Katoh H, Satoh H, Nagasaka S, Yoshihara S, Terada H, Hayashi H. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am J Physiol. 2005;288:H1820–8. doi: 10.1152/ajpheart.00589.2004. [DOI] [PubMed] [Google Scholar]
- Sauer FG, Barnhart M, Choudhury D, Knight SD, Waksman G, Hultgren SJ. Chaperone-assisted pilus assembly and bacterial attachment. Curr Opin Struct Biol. 2000;10:548–56. doi: 10.1016/s0959-440x(00)00129-9. [DOI] [PubMed] [Google Scholar]
- Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–37. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- Scriven DR, Klimek A, Asghari P, Bellve K, Moore ED. Caveolin-3 is adjacent to a group of extradyadic ryanodine receptors. Biophys J. 2005;89:1893–901. doi: 10.1529/biophysj.105.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedova M, Dedkova EN, Blatter LA. Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am J Physiol. 2006;291:C840–50. doi: 10.1152/ajpcell.00619.2005. [DOI] [PubMed] [Google Scholar]
- Sedova M, Blatter LA. Intracellular sodium modulates mitochondrial calcium signaling in vascular endothelial cells. J Biol Chem. 2000;275:35402–7. doi: 10.1074/jbc.M006058200. [DOI] [PubMed] [Google Scholar]
- Segretain D, Rambourg A, Clermont Y. Three dimensional arrangement of mitochondria and endoplasmic reticulum in the heart muscle fiber of the rat. Anat Rec. 1981;200:139–51. doi: 10.1002/ar.1092000204. [DOI] [PubMed] [Google Scholar]
- Shacklock PS, Wier WG, Balke CW. Local Ca2+ transients (Ca2+ sparks) originate at transverse tubules in rat heart cells. J Physiol. 1995;487:601–8. doi: 10.1113/jphysiol.1995.sp020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan-Bormatz V, Gincel D, Yehezkel G, Zaid H. Divalent cation and nucleotide binding sites of VDAC: characterization and modulation of mitochondria permeability transition pore. Biophys J. 2003;84:322a. [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000;78:322–33. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–71. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr. 2000;32:97–104. doi: 10.1023/a:1005520714221. [DOI] [PubMed] [Google Scholar]
- Sheu SS, Sharma VK. A novel technique for quantitative measurement of free Ca2+ concentration in rat heart mitochondria. J Physiol. 1999;518:577–84. doi: 10.1111/j.1469-7793.1999.0577p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. The gating of the sheep skeletal sarcoplasmic reticulum Ca2+-release channel is regulated by luminal Ca2+ J Membr Biol. 1995;146:133–44. doi: 10.1007/BF00238004. [DOI] [PubMed] [Google Scholar]
- Skulachev VP, Bakeeva LE, Chernyak BV, Domnina LV, Minin AA, Pletjushkina OY, et al. Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis. Mol Cell Biochem. 2004;256-257:341–58. doi: 10.1023/b:mcbi.0000009880.94044.49. [DOI] [PubMed] [Google Scholar]
- Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeller C, Cannell MB. Analysing cardiac excitation-contraction coupling with mathematical models of local control. Prog Biophys Mol Biol. 2004;85:141–62. doi: 10.1016/j.pbiomolbio.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Soeller C, Crossman D, Gilbert R, Cannell MB. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc Natl Acad Sci USA. 2007;104:14958–63. doi: 10.1073/pnas.0703016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer JR, Spach MS. Electron microscopic demonstration of adenosinetriphosphatase in myofibrils and sarcoplasmic membranes of cardiac muscle of normal and abnormal dogs. Am J Pathol. 1964;44:491–505. [PMC free article] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270:27510–5. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Spät A, Szanda G, Csordás G, Hajnóczky G. High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling. Cell Calcium. 2008;44:51–63. doi: 10.1016/j.ceca.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, Rios E. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol. 1999;113:469–89. doi: 10.1085/jgp.113.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Sekiguchi M, Endo M. An ultrastructural study of cardiac myocytes in postmyocardial infarction ventricular aneurysm representative of chronic ischemic myocardium using semiquantitative and quantitative assessment. Cardiovasc Pathol. 2000;9:1–8. doi: 10.1016/s1054-8807(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology. 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266:3376–9. [PubMed] [Google Scholar]
- Szalai G, Csordas G, Hantash BM, Thomas AP, Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem. 2000;275:15305–13. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, et al. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–6. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- Territo PR, French SA, Balaban RS. Simulation of cardiac work transitions, in vitro: effects of simultaneous Ca2+ and ATPase additions on isolated porcine heart mitochondria. Cell Calcium. 2001a;30:19–27. doi: 10.1054/ceca.2001.0211. [DOI] [PubMed] [Google Scholar]
- Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation. J Biol Chem. 2001b;276:2586–99. doi: 10.1074/jbc.M002923200. [DOI] [PubMed] [Google Scholar]
- Tornheim K. Oscillations of the glycolytic pathway and the purine nucleotide cycle. J Theor Biol. 1979;79:491–541. doi: 10.1016/0022-5193(79)90240-6. [DOI] [PubMed] [Google Scholar]
- Trollinger DR, Cascio WE, Lemasters JJ. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca2+-indicating fluorophores. Biophys J. 2000;79:39–50. doi: 10.1016/S0006-3495(00)76272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost LC, Lemasters JJ. Role of the mitochondrial permeability transition in salicylate toxicity to cultured rat hepatocytes: implications for the pathogenesis of Reye’s syndrome. Toxicol Appl Pharmacol. 1997;147:431–41. doi: 10.1006/taap.1997.8313. [DOI] [PubMed] [Google Scholar]
- Vanden Hoek TL, Shao Z, Li C, Schumacker PT, Becker LB. Mitochondrial electron transport can become a significant source of oxidative injury in cardiomyocytes. J Mol. Cell Cardiol. 1997;29:2441–50. doi: 10.1006/jmcc.1997.0481. [DOI] [PubMed] [Google Scholar]
- Vendelin M, Kongas O, Saks V. Regulation of mitochondrial respiration in heart cells analyzed by reaction-diffusion model of energy transfer. Am J Physiol. 2000;278:C747–64. doi: 10.1152/ajpcell.2000.278.4.C747. [DOI] [PubMed] [Google Scholar]
- Vendelin M, Lemba M, Saks VA. Analysis of functional coupling: mitochondrial creatine kinase and adenine nucleotide translocase. Biophys J. 2004;87:696–713. doi: 10.1529/biophysj.103.036210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA. NMDA-induced calcium loads recycle across the mitochondrial inner membrane of hippocampal neurons in culture. J Neurophysiol. 2002;87:740–9. doi: 10.1152/jn.00345.2001. [DOI] [PubMed] [Google Scholar]
- Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–6. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watano T, Harada Y, Harada K, Nishimura N. Effect of Na+/Ca2+ exchange inhibitor, KBR7943 on ouabain-induced arrhythmias in guinea-pigs. Br J Pharmacol. 1999;127:1846–50. doi: 10.1038/sj.bjp.0702740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SC, Samuel JL, Marotte F, Bertier-Savalle B, Rappaport L. Microtubules and desmin filaments during onset of heart hypertrophy in rat: a double immunoelectron microscope study. Circ Res. 1987;60:327–36. doi: 10.1161/01.res.60.3.327. [DOI] [PubMed] [Google Scholar]
- Weinstein ES, Benson DW, Fry DE. Subpopulations of human heart mitochondria. J Surg Res. 1986;40:495–8. doi: 10.1016/0022-4804(86)90221-0. [DOI] [PubMed] [Google Scholar]
- Weinstein ES, Benson DW, Ratcliffe DJ, Maksem J, Fry DE. Experimental myocardial ischemia. Differential injury of mitochondrial subpopulations. Arch Surg. 1985;120:332–8. doi: 10.1001/archsurg.1985.01390270070012. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurons. J Physiol. 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier WG, Egan TM, Lopez-Lopez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. J Physiol. 1994;474:463–71. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bers DM. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res. 2006;99:283–91. doi: 10.1161/01.RES.0000233386.02708.72. [DOI] [PubMed] [Google Scholar]
- Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, Cheng H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. 2008;77:432–41. doi: 10.1093/cvr/cvm047. [DOI] [PubMed] [Google Scholar]
- Yang Z, Pascarel C, Steele DS, Komukai K, Brette F, Orchard CH. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ Res. 2002;91:315–22. doi: 10.1161/01.res.0000030180.06028.23. [DOI] [PubMed] [Google Scholar]
- Yang Z, Steele DS. Effects of cytosolic ATP on Ca2+ sparks and SR Ca2+ content in permeabilized cardiac myocytes. Circ Res. 2001;89:526–33. doi: 10.1161/hh1801.096264. [DOI] [PubMed] [Google Scholar]
- Yang Z, Steele DS. Effects of cytosolic ATP on spontaneous and triggered Ca2+-induced Ca2+ release in permeabilized rat ventricular myocytes. J Physiol. 523:29–44. doi: 10.1111/j.1469-7793.2000.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Steele DS. 2005 Characteristics of prolonged Ca2+ release events associated with the nuclei in adult cardiac myocytes. Circ Res. 2000;96:82–90. doi: 10.1161/01.RES.0000151841.63705.01. [DOI] [PubMed] [Google Scholar]
- Yusufi AN, Cheng J, Thompson MA, Chini EN, Grande JP. Nicotinic acid-adenine dinucleotide phosphate (NAADP) elicits specific microsomal Ca2+ release from mammalian cells. Biochem J. 2001;353:531–6. [Google Scholar]
- Ziegler M, Jorcke D, Schweiger M. Identification of bovine liver mitochondrial NAD+ glycohydrolase as ADP-ribosyl cyclase. Biochem J. 1997;S326:401–5. doi: 10.1042/bj3260401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–76. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–17. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]