

Abstract
Burkholderia pseudomallei is the causative agent of melioidosis and is a major mediator of sepsis in its endemic areas. Because of the low LD50 via aerosols and resistance to multiple antibiotics, it is considered a Tier 1 select agent by the CDC and APHIS. B. pseudomallei is an encapsulated bacterium that can infect, multiply, and persist within a variety of host cell types. In vivo studies suggest that macrophages and neutrophils are important for controlling B. pseudomallei infections, however few details are known regarding how neutrophils respond to these bacteria. Our goal is to describe the capacity of human neutrophils to control highly virulent B. pseudomallei compared to the relatively avirulent, acapsular B. thailandensis using in vitro analyses. B. thailandensis was more readily phagocytosed than B. pseudomallei, but both displayed similar rates of persistence within neutrophils, indicating they possess similar inherent abilities to escape neutrophil clearance. Serum opsonization studies showed that both were resistant to direct killing by complement, although B. thailandensis acquired significantly more C3 on its surface than B. pseudomallei, whose polysaccharide capsule significantly decreased the levels of complement deposition on the bacterial surface. Both Burkholderia species showed significantly enhanced uptake and killing by neutrophils after critical levels of C3 were deposited. Serum-opsonized Burkholderia induced a significant respiratory burst by neutrophils compared to unopsonized bacteria, and neutrophil killing was prevented by inhibiting NADPH-oxidase. In summary, neutrophils can efficiently kill B. pseudomallei and B. thailandensis that possess a critical threshold of complement deposition, and the relative differences in their ability to resist surface opsonization may contribute to the distinct virulence phenotypes observed in vivo.
Introduction
The causative agent of melioidosis, Burkholderia pseudomallei, is a saprophytic bacterium that is endemic throughout Southeast Asia and northern Australia [1], [2], [3]. This organism is a leading cause of pneumonia and septicemia in endemic areas [1], [2], [4], [5], [6]. In the Western hemisphere, B. pseudomallei have been documented sporadically in northern South America, Central America, and certain Caribbean islands, including Puerto Rico [7], [8], [9], [10], [11], [12], [13], and melioidosis cases are becoming increasingly more widespread in these and other tropical/sub-tropical areas worldwide [14], [15]. While infection can be established in healthy individuals through skin abrasions, ingestion, or inhalation, the incidence of melioidosis is more common in individuals with certain predisposing conditions, the primary one being diabetes mellitus [4], [16], [17]. Infection with B. pseudomallei can produce widely varying clinical symptoms which often confounds accurate diagnosis. Acute melioidosis is a serious condition that can rapidly become fatal, and is commonly characterized by abscess formation in lungs, liver, and/or spleen, as well as bacteremia. Latent melioidosis is characterized by a persistent infection that can recrudesce at varying times after the initial infection to cause disease, with the longest confirmed report being 62 years post-infection [18]. Notably, B. pseudomallei are extremely virulent via aerosol exposure, with an estimated LD50 between 5–100 organisms depending on the model [19], [20], [21]. Because of these characteristics, B. pseudomallei has recently been elevated to Tier 1 status by the CDC and APHIS [22]. B. pseudomallei is inherently resistant to many classes of antibiotics, and even treatment with proven antibiotics is often unsuccessful, with mortality rates for acute melioidosis ranging from 40–90% [2], [4]. No vaccine is currently available for preventing melioidosis, and there is great interest in identifying immune mechanisms that can promote efficient clearance of these infections.
While B. pseudomallei can be readily isolated as a free-living organism in moist tropical environments, it is also particularly efficient at infecting and persisting within both non-phagocytic and phagocytic host cell types. While not extensively studied, a number of potential virulence factors have been identified that may enhance their ability to persist intracellularly. These include type III and VI secretion systems which promote cell entry and rapid escape from endosomal compartments, as well as actin-based motility which allows for intercellular spread between adjacent cells without exposure to the extracellular milieu [23], [24], [25], [26], [27], [28], [29]. Capsule production is also known to be important for persistence in animal models of infection, although the specific virulence properties it provides is not well-established [30], [31]. One tool used to address the importance of putative virulence mechanisms are comparative studies using the closely-related, but relatively avirulent B. thailandensis. This sequenced bacterium does not produce the type I mannoheptose polysaccharide capsule expressed by B. pseudomallei, as well as lacks the ability to assimilate arabinose and a few additional genes for which no known virulence properties have been described [32], [33], [34], [35]. However, B. thailandensis does display an ability to escape the endosome, replicate, and persist in the cytoplasm in certain cell types in vitro [36], [37], [38]. While we still do not have a complete understanding of B. pseudomallei virulence mechanisms, it is evident that these bacteria are well-adapted to survive and persist within host cells, but our knowledge of which immune cells are critical for protection is limited.
Historically, the interaction between B. pseudomallei and macrophages has been a primary research focus, as macrophages are believed to be a major reservoir for both the replication and dissemination of these bacteria as well as for controlling these infections (reviewed in [39], [40]). However, recent in vivo findings suggest neutrophils may also play a critical role in controlling B. pseudomallei infection, including the following: i. selective depletion of neutrophils in a mouse model leads to enhanced susceptibility to fatal melioidosis [41], ii. neutrophils are recruited to and interact with B. pseudomallei in infected lung tissues [41], [42], iii. mice lacking NADPH oxidase, an important enzyme in the generation of the microbicidal respiratory burst primarily utilized by neutrophils, are more susceptible to B. pseudomallei infection [43], iv. diabetes mellitus, which is the primary predisposing condition for melioidosis, is associated with impaired neutrophil function [44], [45], [46], [47], v. neutropenic individuals are more susceptible to B. pseudomallei infection and development of fatal disease [48], [49] and, vi. granulocyte colony-stimulating factor (G-CSF), which stimulates neutrophil differentiation, prolongs the survival of melioidosis patients, though a direct link to enhanced neutrophil function has not been proven [50], [51], [52], [53], [54].
Although in vivo studies suggest that neutrophils are important for controlling B. pseudomallei infection, a limited number of in vitro studies have provided conflicting findings on the ability of these phagocytes to directly clear B. pseudomallei [45], [55], [56], [57]. These reports have all varied as to neutrophil efficiency in phagocytosing and killing B. pseudomallei, their abilities to elicit an oxidative burst, and whether serum components provide any opsonizing properties for enhancing bacterial killing. Our current goal is to determine the ability of human primary neutrophils to clear the highly virulent B. pseudomallei compared to the relatively avirulent B. thailandensis, as well as delineate the mechanism(s) important for bacterial killing. Our findings are the first to demonstrate that neutrophils can effectively kill both B. pseudomallei and B. thailandensis in vitro, but only if sufficient complement deposition has occurred on the bacterial surface to activate an appropriate respiratory burst.
Materials and Methods
Bacterial culture and preparation
B. pseudomallei 1026b [58], B. pseudomallei DD503 [59], and B. thailandensis E264 [34] were a gift from Don Woods (University of Calgary). B. pseudomallei DD503 ΔLPS (BP2683) [60] and B. pseudomallei DD503 ΔCPS (SZ210) [30] were provided by Paul Brett and Mary Burtnick (University of Southern Alabama). Escherichia coli strain K12 substrain W3110 was used as a control in specific experiments. For these studies, all bacterial strains were cultured aerobically for 18 hours at 37°C on tryptic soy agar (TSA) (Neogen) plates. Bacteria were recovered by scraping from TSA plates into phosphate buffered saline (PBS) and initially enumerated using a spectrophotometer, which was confirmed by dilution plating. All studies utilizing live B. pseudomallei were conducted in a CDC select agent-certified BSL3 laboratory.
Serum opsonization of bacteria
For experiments involving serum opsonization, bacteria (1×108 CFU) were incubated with the described concentrations of pooled normal human serum (NHS) (Complement Technology) or heat-inactivated (HI) serum in PBS containing 0.25 µM CaCl2 and 1 µM MgCl2 at 37°C for 30 min. HI serum was prepared by incubating the pooled human serum at 56°C for 30 min prior to addition to bacteria. To evaluate the relative contributions of the classical, lectin and alternative complement pathways to bacterial opsonization, either 10 mM ethylenediaminetetraacetic acid (EDTA) (blocks classical, lectin and alternative pathways by chelating both calcium and magnesium) or a combination of 5 mM magnesium chloride and 5 mM ethylene glycol tetraacetic acid (MgEGTA) (only blocks classical and lectin pathways by preferentially chelating calcium over magnesium) final were added, respectively.
Quantification of complement deposition and antibody binding on bacterial surfaces
All bacteria were opsonized with NHS as described above. Opsonized bacteria were then washed with PBS to remove any unbound components and fixed with 1% paraformaldehyde. Fixed bacteria were washed and labeled with goat anti-human C3 polyclonal IgG-FITC conjugated (MP Biomedicals) at 1∶400, washed, and analyzed by flow cytometry using a BD FACSCalibur (Becton Dickinson). To assess the presence of bacteria-specific antibodies, bacteria opsonized with NHS were subsequently incubated with both APC-labeled donkey anti-human IgM (1∶100) and R-PE-labeled donkey anti-human IgG (1∶100) (Jackson ImmunoResearch), washed, and analyzed by flow cytometry. Results are reported as mean fluorescence intensity (MFI).
Serum-mediated killing of bacteria
All B. pseudomallei strains, B. thailandensis, and E. coli (serum-sensitive) were incubated with 0, 20, 40 or 80% NHS and 40% HI at a concentration of 106 bacteria/ml under the same conditions as described for bacterial opsonization assays. At 0, 2, and 4 h post-incubation, an aliquot of each sample was serially diluted and plated on TSA to determine CFU/ml.
Neutrophil isolation from whole human blood
All studies involving human samples were in accordance with and approved by the University of Toledo Biomedical Institutional Review Board (IRB). Neutrophils were isolated from whole venous blood obtained from healthy human volunteers, as previously described [61]. Briefly, heparinized blood was combined with equal parts 3% dextran at room temperature for 20 min to sediment erythrocytes. The leukocyte suspension was centrifuged at 500×g for 10 min, the cells were resuspended in PBS and underlayed with equal volumes Ficoll-sodium metrizoate solution (density 1.077 g/ml) (MP Biomedicals), and centrifuged for 40 min at 400×g at room temperature with no brake. The erythrocyte/granulocyte-rich pellet was resuspended in sterile water for 25 s to allow hypotonic lysis of erythrocytes, and tonicity was restored by addition of PBS. The remaining granulocytes were >95% neutrophils and >95% cell viability as determined by Wright-Giemsa and trypan blue staining, respectively.
Bacterial killing by neutrophils
Human neutrophils were seeded at 1×106 per well in 24 well plates containing 0.5 ml RPMI 1640 with HL-1 supplement (Lonza) and Glutamax (Invitrogen)(i.e. “complete RPMI”). Neutrophils were incubated at 37°C in 5% CO2 for 20 min before addition of B. pseudomallei or B. thailandensis at an MOI = 1. In specific experiments, diphenyleneiodonium (DPI; a NADPH-oxidase inhibitor) (10 µM final) or a DMSO vehicle control was added during this incubation. The plates were centrifuged at 250×g for 5 min to synchronize infection and allowed to incubate at 37°C in 5% CO2 for 10 min before washing 3x with RPMI to remove extracellular bacteria. No antibiotics were used to kill any remaining extracellular bacteria because they have been demonstrated to enter phagocytes and kill intracellular bacteria [62], [63], [64], [65]. The neutrophils in these parallel co-cultures were lysed at either 10 min (i.e. to assess uptake) or 2 h after infection (i.e. to assess clearance) with a 0.5% saponin solution, and intracellular bacteria were enumerated by serial dilution on TSA.
Quantification of neutrophil respiratory burst
Human neutrophils were seeded at 2×105 per well in 96 well plates in PBS containing calcium and magnesium in the presence or absence of DPI (10 µM final). Neutrophils were pretreated with luminol (50 µM 3-aminophthalhydrazide in 0.1 M NaOH) at 37°C in 5% CO2 for 20 min before addition of B. pseudomallei or B. thailandensis at an MOI = 1. Co-cultures were centrifuged at 500×g for 1 min to initiate bacteria:neutrophil contact and immediately analyzed on a FLUOstar Omega plate reader (BMG Labtech), with luminescence detection every min for 20 min.
Statistical analysis
Graphpad Instat (La Jolla, CA) was used for all statistical analyses. Statistical differences were determined by either performing a two-way T-test or one-way ANOVA followed by a Tukey's post-hoc test (P≤0.05).
Results
Phagocytosis and clearance of unopsonized Burkholderia species by human neutrophils
Neutrophils are observed to be rapidly recruited to the site of infection during experimental respiratory melioidosis and blocking this influx results in greater B. pseudomallei numbers and mortality [41], [42]. However, previous in vitro studies have been conflicted on the ability of neutrophils to directly clear B. pseudomallei [45], [55], [56], [57]. We sought to clarify this by evaluating the ability of and mechanism(s) necessary for human neutrophils to phagocytose and kill highly virulent B. pseudomallei. We also sought to determine if there was differential killing of B. pseudomallei by neutrophils compared to that of the closely-related, but relatively avirulent B. thailandensis.
The inherent capability of neutrophils to internalize and clear B. pseudomallei and B. thailandensis was assessed in Figure 1. Human neutrophils were able to internalize both unopsonized B. pseudomallei and B. thailandensis, although the uptake of B. thailandensis (16.5%) by neutrophils was significantly greater than B. pseudomallei (9.1%) (Figure 1A). Subsequently, there was no change in bacterial viability of either species after 2 h incubation with neutrophils compared to the initial uptake (Figure 1B). Thus, though human neutrophils inherently internalize B. thailandensis more efficiently than B. pseudomallei, they are subsequently unable to clear either bacterial species.
Figure 1. B. pseudomallei and B. thailandensis are inherently resistant to killing by human neutrophils.
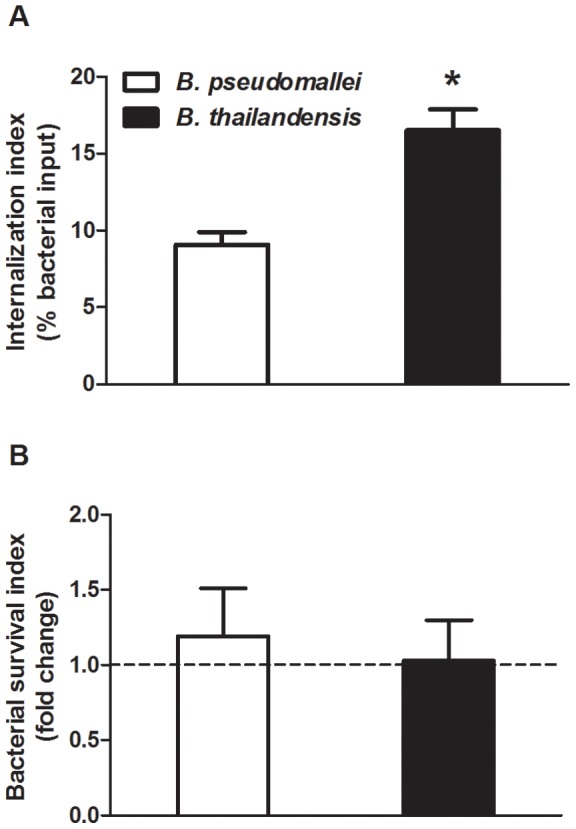
(A) B. pseudomallei and B. thailandensis were either incubated with neutrophils for 10 min to measure bacterial uptake (% bacterial input) or (B) for 2 h to measure bacterial survival (fold change from respective uptake values) as described in the Materials and Methods. The bars represent the mean±SEM of three separate experiments using neutrophils from different donors, each performed in duplicate. * indicates a statistically significant difference (P≤0.05) between B. pseudomallei and B. thailandensis values.
Complement deposition on the surface of B. pseudomallei and B. thailandensis
Before assessing the effects of serum opsonization on neutrophil responses to B. pseudomallei or B. thailandensis, the relative levels of complement deposition on the surface of these bacteria were measured in the presence of different concentrations of normal human serum (NHS), using flow cytometry (Figure 2A). After incubation with 5%, 10% and 20% NHS, significant levels of complement component C3 were detected on the surface of both Burkholderia species compared to unopsonized bacteria, whereas 1% NHS opsonization did not promote significant C3 deposition. In general, increased serum concentrations correlated with increased C3 deposition in both species. However, B. thailandensis did acquire significantly greater levels of C3 deposition at 5%, 10% and 20% NHS compared to B. pseudomallei. Bacteria incubated with heat-inactivated (HI) serum did not acquire substantial C3 on their surfaces. These data indicate that B. pseudomallei are more resistant to C3 deposition compared to B. thailandensis.
Figure 2. B. pseudomallei acquire less complement C3 on its surface than B. thailandensis.
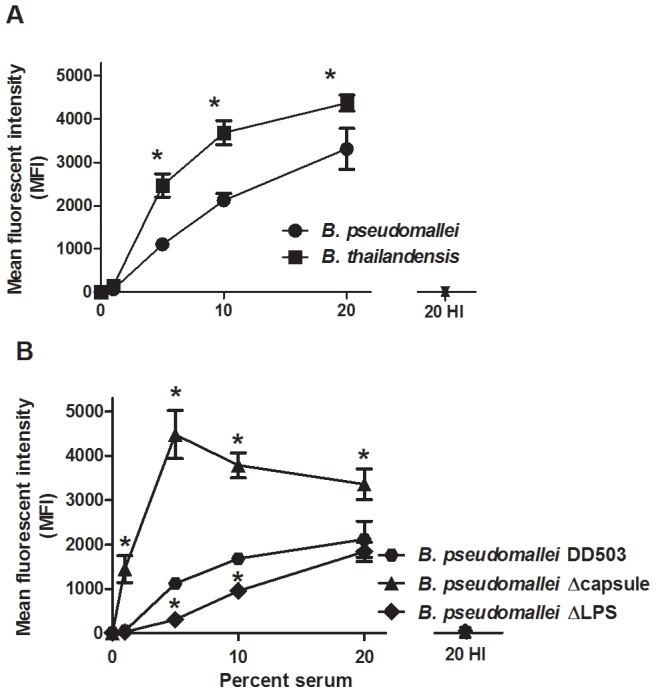
(A) B. pseudomallei 1026b and B. thailandensis E264, or (B) B. pseudomallei DD503, B. pseudomallei ΔCPS, and B. pseudomallei ΔLPS were opsonized for 30 min at the indicated serum concentrations, washed, and stained with an anti-human C3 FITC-conjugated antibody. Samples were analyzed by flow cytometry and the data reported as mean fluorescence intensity (MFI). The bars represent the mean±SEM of three separate experiments performed with duplicate samples. (A) * indicates a statistically significant difference (P≤0.05) between B. pseudomallei and B. thailandensis values; ns = not significant. (B) * indicates a statistically significant difference (P≤0.05) compared to B. pseudomallei DD503 (wild-type).
Two B. pseudomallei factors reported to be involved in serum resistance are the polysaccharide capsule and lipopolysaccharide (LPS), respectively [30], [66]. To determine if either of these factors may explain the increased C3 deposition on B. thailandensis compared to B. pseudomallei, similar experiments were performed using both capsule-deficient (ΔCPS) and LPS-deficient (ΔLPS) B. pseudomallei mutants (Figure 2B). The ΔCPS mutant showed significantly increased C3 deposition on its surface compared to the wild-type control (DD503) at all serum concentrations measured. The ΔLPS mutant showed the opposite trend, with C3 deposition being significantly decreased compared to the control at 5% and 10% NHS. These data suggest that the B. pseudomallei capsule is responsible for the differences in C3 deposition observed between B. pseudomallei and B. thailandensis.
To address which activation pathway(s) were responsible for complement deposition on B. pseudomallei and B. thailandensis, both bacterial species were incubated with 5% and 20% NHS in the presence or absence of EDTA (prevents activation of all complement pathways) or MgEGTA (allows only alternative pathway activation), and C3 deposition analyzed by flow cytometry (Figure 3). Addition of EDTA to NHS-opsonized samples reduced C3 deposition levels to that of unopsonized bacteria for both B. pseudomallei and B. thailandensis, as expected. MgEGTA addition to the 5% NHS samples allowed minimal C3 surface deposition on both bacteria, suggesting that complement deposition at 5% NHS is largely dependent on the classical or lectin pathways and that both are resistant to alternative pathway-activation at that serum concentration. After incubation in 20% NHS with MgEGTA, C3 binding to B. pseudomallei was significantly reduced compared to in the absence of MgEGTA. This decrease in C3 deposition was not observed for B. thailandensis opsonized in 20% NHS and MgEGTA, demonstrating efficient alternative pathway activation. These results suggest that B. pseudomallei are resistant to alternative pathway activation even at high serum concentrations, whereas B. thailandensis is highly susceptible at these serum levels. Surprisingly, the substantial complement deposition that occurs in NHS for both Burkholderia species appears largely due to the classical or lectin pathway. To attempt to differentiate between these pathways, we measured the levels of IgG and IgM in NHS that could bind B. pseudomallei and B. thailandensis by flow cytometry. The endogenous levels of B. pseudomallei and B. thailandensis-reactive antibodies in NHS were not significantly different from unopsonized bacteria, whereas IgG and IgM specific for a commensal bacterium (i.e. E. coli) were significantly elevated in 20% NHS (Figure 4). It is currently unclear if these very low endogenous levels of Burkholderia-reactive IgG or IgM in NHS are enough to activate the classical pathway versus the contribution of innate immune mediators that activate via the classical or lectin pathway.
Figure 3. B. pseudomallei are resistant to C3 deposition by the alternative pathway of complement.
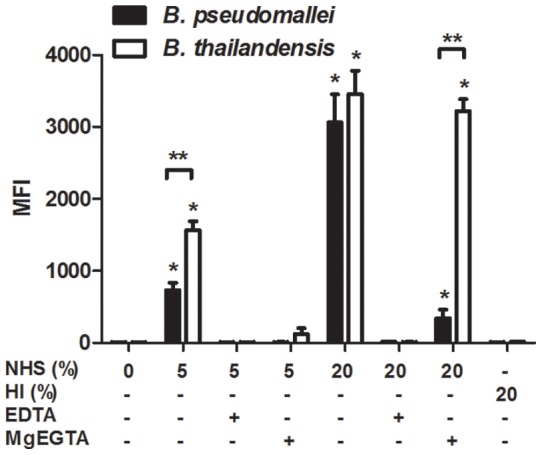
B. pseudomallei and B. thailandensis were incubated with the indicated serum concentrations in the presence or absence of MgEGTA or EDTA, washed, and stained with an anti-human C3 FITC-conjugated antibody. Samples were analyzed by flow cytometry and the data reported as mean fluorescence intensity (MFI). The bars represent the mean±SEM of three separate experiments performed with duplicate samples. The single asterisk (*) indicates a statistically significant increase (P≤0.05) over unopsonized bacteria. The double asterisks (**) indicate a statistically significant difference (P≤0.05) between B. pseudomallei and B. thailandensis values.
Figure 4. Levels of endogenous Burkholderia-reactive antibodies present in normal human serum.
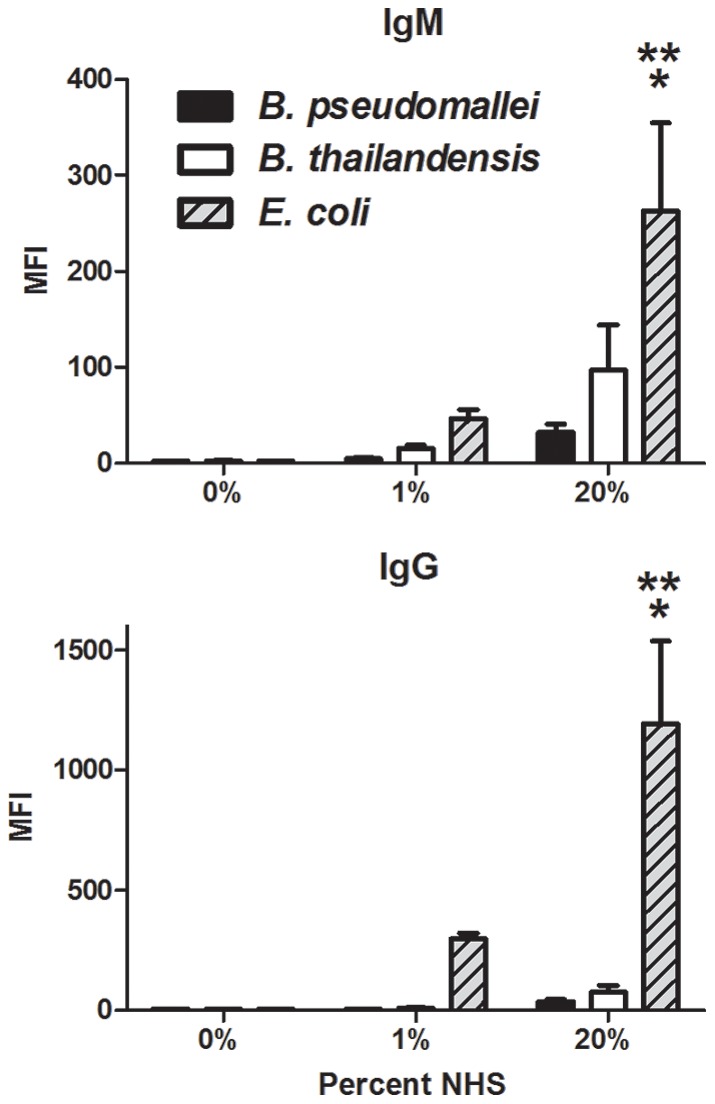
B. pseudomallei, B. thailandensis, and E. coli were incubated with the indicated serum concentrations, washed, and stained with both an APC-labeled anti-human IgM antibody and R-PE-labeled anti-human IgG antibody. Samples were analyzed by flow cytometry and the data reported as mean fluorescence intensity (MFI). The bars represent the mean±SEM of two separate experiments performed with duplicate samples. The single asterisk (*) indicates a statistically significant increase (P≤0.05) over unopsonized bacteria. The double asterisks (**) indicate a statistically significant difference (P≤0.05) over 20% NHS-opsonized B. pseudomallei and B. thailandensis values.
Resistance of Burkholderia species to direct killing by serum
To determine whether the observed differences in surface C3 deposition might directly affect survival of B. pseudomallei or B. thailandensis, the bacteria were incubated in NHS and enumerated at different times post-incubation to assess the direct killing effect. B. pseudomallei viability was unaltered up to 4 h post-incubation in 20%, 40%, and 80% NHS, confirming it is resistant to serum bactericidal activity (Figure 5) [30], [55], [66], [67]. Although, B. thailandensis was shown to have much higher levels of C3 deposition (Figure 2), it demonstrated similar resistance to direct killing as B. pseudomallei at all serum concentrations, as has been previously suggested [66]. The ΔCPS B. pseudomallei was as serum resistant as wild-type B. pseudomallei and B. thailandensis, indicating the capsule is not required for survival in serum. However, the ΔLPS B. pseudomallei showed significant death in 20, 40 and 80% serum, even though it had very low levels of C3 deposition (Figure 2). The results suggest that LPS attracts C3 deposition, but does not allow MAC formation capable of disrupting the bacterial outer membrane. As a control, no viable E. coli (serum-sensitive) were detected after a 2 h incubation in ≥20% NHS, demonstrating the NHS possessed bactericidal activity. Direct killing of E. coli was not observed in 40% HI serum, indicating the mechanism was complement-mediated. These findings suggest that, even though there are significant differences in surface C3 deposition between B. pseudomallei and B. thailandensis, both possess a similar resistance to direct killing by complement. Additionally, while LPS seems to be necessary for the survival of B. pseudomallei in serum, the capsule is not involved in resistance to serum-mediated direct killing.
Figure 5. B. pseudomallei and B. thailandensis are resistant to direct complement-mediated killing.

B. pseudomallei 1026b, B. thailandensis, B. pseudomallei DD503, B. pseudomallei ΔCPS, B. pseudomallei ΔLPS and E. coli were incubated at the indicated serum concentrations at 37°C. At the indicated times, an aliquot of each sample was serially diluted and plated on TSA to determine CFU/ml. Each condition was assessed as duplicate samples in all experiments. * indicates a significant difference (P≤0.05) between E. coli and both Burkholderia species.
Serum opsonization of B. pseudomallei and B. thailandensis results in increased uptake and killing by human neutrophils
Although neutrophils could not kill unopsonized B. pseudomallei or B. thailandensis, it is possible that surface C3 deposition could promote phagocytosis and/or killing by neutrophils. Initial studies measuring bacterial uptake by neutrophils at 10 min post-infection showed equal or decreased numbers of internalized serum-opsonized B. pseudomallei and B. thailandensis, respectively, compared to unopsonized bacteria (Figure 6). When the bacteria were opsonized with HI serum, the number of internalized B. pseudomallei and B. thailandensis appeared to increase over that of their respective unopsonized numbers. However, a significant decrease in uptake was observed between bacteria opsonized with 20% NHS compared to 20% HI for both species, indicating a role for a heat-labile serum factor(s), most likely a component of the complement cascade. However, these findings were counter-intuitive and were not consistent with any trends reported for similar intracellular bacteria (reviewed in [68], [69]).
Figure 6. Effects of serum opsonization on B. pseudomallei and B. thailandensis uptake by neutrophils.
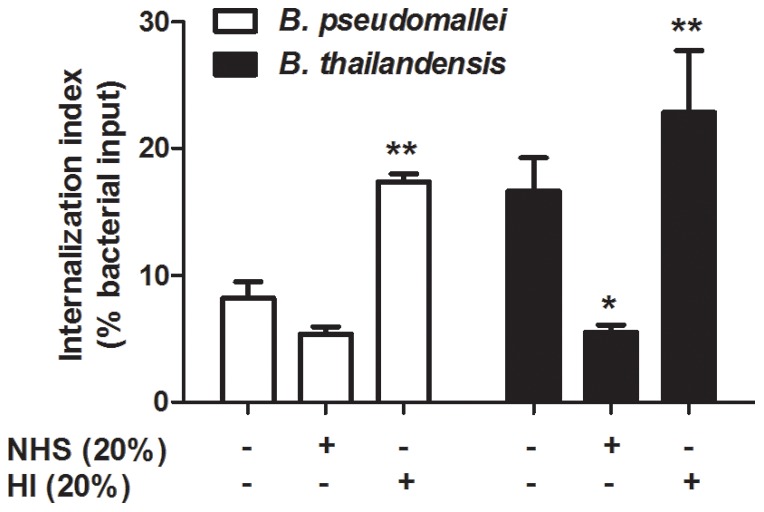
Bacteria were incubated in the presence or absence of the indicated sera prior to co-culture with neutrophils. Bacterial uptake was measured at 10 min post-infection. The bars represent the mean±SEM of three separate experiments using neutrophils from different donors and performed in duplicate. The single asterisk (*) indicates a significant difference (P≤0.05) between unopsonized and 20% NHS opsonized B. thailandensis values. The double asterisks (**) indicate a statistically significant difference (P≤0.05) between 20% NHS and 20% HI opsonized bacteria.
One possibility for these misleading results is that serum-opsonized B. pseudomallei and B. thailandensis were being rapidly killed by neutrophils, producing an artificially low internalization rate. To address this, the internalization studies were repeated in the presence or absence of diphenyleneiodonium (DPI), a NADPH-oxidase inhibitor that blocks the respiratory burst. Internalization of unopsonized B. thailandensis by neutrophils at 10 min post-infection was again significantly greater than for unopsonized B. pseudomallei, and this was unaltered by the addition of DPI (Figure 7A). B. pseudomallei opsonized in 1% NHS showed significantly greater internalization by neutrophils compared to unopsonized bacteria, both in the absence and presence of DPI. B. thailandensis opsonized with 1% NHS showed no change in internalization by neutrophils compared to unopsonized bacteria, but DPI-treated neutrophils had significantly more internalized B. thailandensis. Internalization of B. pseudomallei opsonized in 5%, 10%, and 20% NHS-opsonized B. pseudomallei was low in the absence of DPI; however, neutrophils treated with DPI showed significantly enhanced uptake of B. pseudomallei opsonized at the same serum concentrations. B. thailandensis opsonized with 5%, 10%, and 20% NHS were taken up at significantly lower numbers in the absence of DPI than unopsonized bacteria. However, in the presence of DPI, B. thailandensis opsonized at the same NHS concentrations showed significantly enhanced internalization by neutrophils, and these levels were similar to that seen for B. pseudomallei. Opsonization of both B. pseudomallei and B. thailandensis in 20% HI serum had no effect on internalization compared to unopsonized bacteria. These data suggest that serum opsonization increases the ability of neutrophils to internalize both B. pseudomallei and B. thailandensis in a complement-dependent manner. Additionally, bacterial opsonization in ≥5% serum elicits a rapid reduction in numbers of viable bacteria, which is associated with the respiratory burst (i.e. DPI-sensitive). Interestingly, B. pseudomallei opsonized with 1% NHS demonstrated increased internalization by neutrophils but did not stimulate bactericidal activity, whereas B. thailandensis showed increased uptake and rapid killing at this serum concentration, suggesting there are differences in the amount of reactive oxygen species (ROS) produced in response to these species.
Figure 7. Serum opsonization enhances internalization and clearance of B. pseudomallei and B. thailandensis by neutrophils.

Isolated human neutrophils were treated with DPI or DMSO (vehicle control) for 20 min prior to bacterial infection. Neutrophils were incubated with B. pseudomallei and B. thailandensis for (A) 10 min to measure bacterial uptake or for (B) 2 h to measure bacterial survival. The bars represent the mean±SEM of three separate experiments using neutrophils from different donors and performed in duplicate. The single asterisk (*) indicates a significant difference (P≤0.05) compared to unopsonized bacteria. The double asterisks (**) indicate a significant difference (P≤0.05) between B. pseudomallei and B. thailandensis. The triple asterisks (***) indicate a significant difference (P≤0.05) between 20% NHS opsonized Burkholderia species and all other conditions.
The ability of B. pseudomallei and B. thailandensis to subsequently survive after internalization was measured in the absence and presence of DPI for 2 h post-infection. The numbers of unopsonized B. pseudomallei and B. thailandensis within neutrophils were not reduced during the 2 h assessment period (Figure 7B). Opsonization of B. pseudomallei and B. thailandensis in 1% serum produced some reduction in bacterial numbers, but these values were not significant. Opsonization of both species in 5% and 10% serum elicited a significant reduction in intracellular numbers, and increasing the serum concentration to 20% produced a further significant reduction in bacterial numbers compared to the lower serum concentrations. Notably, opsonization of both species in 20% HI serum produced no reduction in bacterial numbers, and neutrophils treated with DPI were unable to reduce the numbers of either B. pseudomallei or B. thailandensis regardless of the serum concentration used for opsonization. The results demonstrate that opsonization of both B. pseudomallei and B. thailandensis with ≥5% NHS elicits significant activation and bactericidal activity by human neutrophils, and this is dependent on induction of a respiratory burst.
Bacterial opsonization with serum is required for rapid induction of the neutrophil respiratory burst
To further delineate the affects of serum opsonization on eliciting neutrophil killing, a luminol-based chemiluminescence assay was utilized to quantify the kinetics and magnitude of the neutrophil respiratory burst induced by B. pseudomallei and B. thailandensis. Neutrophils co-cultured with unopsonized bacteria did not induce a respiratory burst and appeared similar to uninfected cells (Figure 8A). Neutrophils co-cultured with B. pseudomallei and B. thailandensis opsonized in 5%, 10%, or 20% NHS induced a rapid and substantial respiratory burst compared to unopsonized bacteria, whereas bacteria opsonized in 1% serum produced a more intermediate response. The maximum respiratory burst for B. pseudomallei and B. thailandensis opsonized with ≥5% serum was reached within 2–3 min after inoculation. These values were all significantly greater than neutrophils exposed to unopsonized bacteria, which produced little to no ROS (Figure 8B). Neutrophils infected with either bacteria opsonized with 1% serum did show some increase in ROS activity, but these levels were not significantly different from background values. It is noteworthy that we also did not observe significant neutrophil killing of B. pseudomallei or B. thailandensis opsonized with 1% NHS (Figure 7B). Neutrophils infected with 20% HI opsonized B. pseudomallei or B. thailandensis produced baseline levels of ROS, reiterating the importance of complement deposition in inducing the respiratory burst against these Burkholderia species.
Figure 8. Correlation of serum-opsonization and induction of respiratory burst by B. pseudomallei and B. thailandensis.

(A) Kinetics of reactive oxygen species produced by neutrophils following bacterial infection. Neutrophils were pre-incubated in the presence of luminol before addition of B. pseudomallei or B. thailandensis at an MOI = 1 with or without DPI. Co-cultures were centrifuged briefly to initiate bacteria:neutrophil contact and immediately analyzed on a plate reader with luminescence detection continuously for a total of 20 min. The data represents the combined results of three separate experiments using neutrophils from different donors and performed in triplicate. (B) Chemiluminescence levels measured at t = 2 min post-infection. The bars represent the mean±SEM of three separate experiments performed in triplicate. * indicates a statistically significant difference (P≤0.05) compared to neutrophils stimulated with unopsonized B. pseudomallei or B. thailandensis.
Discussion
Melioidosis can be a highly lethal disease if not appropriately diagnosed and treated, particularly with the development of pneumonia and bacteremia that frequently leads to involvement of multiple organs. A major issue in controlling these infections is that B. pseudomallei are highly efficient at infecting and persisting within multiple non-immune and immune cell types. Within susceptible cell types, these bacteria can quickly escape endosomal compartments and subsequently utilize actin polymerization to efficiently invade adjacent cells without being exposed to the extracellular environment, thus limiting their exposure to antibodies and other soluble immune effectors. Therefore, it is important to identify immune cells involved in the cellular response that are best able to control these infections, as well as the mechanisms that promote bacterial clearance. Neutrophils are important for controlling systemic infections caused by numerous bacterial species, including many that are associated with pneumonia and bacteremia [70]. While neutrophils have been demonstrated to be critical for controlling melioidosis in vivo directly through depletion studies, it is still unclear if the neutrophils have a direct effect on B. pseudomallei clearance or the cells have an indirect role through modulation of other cell types [41]. A recent study conversely indicated that neutrophil recruitment during melioidosis may be detrimental in controlling bacterial numbers and host survival, and suggested monocytes may be important to limit B. pseudomallei infection [71]. There is indirect evidence that neutrophils may play a role in controlling melioidosis infection through correlative findings based on cellular recruitment to the infection site, predisposing conditions, and adjunctive therapies; however any protective properties have not been clearly confirmed to be attributable to neutrophils [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54]. Notably, in vitro studies to delineate the relative abilities of neutrophils to kill B. pseudomallei have provided conflicting results [45], [55], [56], [57]. In our current study, we sought to determine the requirements that allow human neutrophils to kill B. pseudomallei, the mechanisms needed for this process, and whether there are differences in killing efficiencies between the relatively avirulent B. thailandensis and highly virulent B. pseudomallei that correlate with their contrasting pathogenesis in vivo.
When neutrophils were assessed for their inherent abilities to neutralize these bacteria, both species were recognized and phagocytosed within 10 min of co-incubation, although B. thailandensis was taken up at twice the rate as B. pseudomallei. Regardless, both B. pseudomallei and B. thailandensis showed similar abilities to subsequently resist killing and persist within neutrophils. One explanation for the differences in uptake could be the capsule produced by B. pseudomallei makes it more difficult for neutrophils to recognize and/or internalize compared to the acapsular B. thailandensis. Such anti-phagocytic properties have been described for a number of Gram-positive and Gram-negative bacteria possessing a carbohydrate capsule, including Streptococcus pneumoniae, Staphylococcus aureus, Neisseria meningitis, Haemophilus influenzae, Klebsiella pneumoniae, and Escherichia coli (reviewed in [72], [73], [74], [75]). However, possession of this capsule alone does not correlate with their ability to evade neutrophil clearance, as acapsular B. thailandensis displayed a similar ability to resist neutrophil killing as B. pseudomallei. Thus, both bacteria appear able to inherently evade neutrophil clearance and additional immune mechanisms must be involved if neutrophils are able to control these infections in vivo.
Antibodies and other serum opsonins are known to be crucial for neutrophils to recognize and kill certain bacteria, and particularly those that possess a capsule (reviewed in [72], [73], [74], [75]). We chose to focus on complement as a critical opsonin to promote efficient killing of these Burkholderia species, particularly since these innate components would be present early during infection and before the development of Burkholderia-specific antibodies. Quantification of C3 deposition on the bacterial surface indicated that B. thailandensis acquired significantly more C3 on its surface compared to B. pseudomallei. Parallel studies using the ΔCPS B. pseudomallei mutant suggested that this capsular material is largely responsible for the reduced C3 levels deposited on B. pseudomallei, and titration studies indicated that this protection was most apparent in low levels of serum/complement, which likely reflects the levels encountered in most host tissues. When serum levels were relatively low (≤5%), components of the classical or lectin pathways were necessary for complement activation on both bacterial species; however when the serum concentration was increased to 20% NHS, activation through the alternative pathway contributed to the majority of C3 on B. thailandensis, but not for B. pseudomallei. Although, B. thailandensis acquired more surface C3 than B. pseudomallei, both Burkholderia species were equally resistant to complement-mediated direct killing. The mechanism(s) that Burkholderia species use to resist direct killing by complement is not known. While B. pseudomallei LPS is known to be involved in serum resistance [66], and our findings indicate it is absolutely required for the complete complement resistance observed by B. pseudomallei, it has not been determined how it mediates this effect and what host or other bacterial factors are involved in this process. The length of the LPS O-antigen has been associated with serum resistance in some Gram-negative bacteria [76], and the structure of B. pseudomallei and B. thailandensis O-antigen are similar, suggesting this could represent a common serum-resistance mechanism between these closely-related bacterial species [34], [77], [78]. Since the B. pseudomallei ΔLPS mutant had less C3 deposition than wild-type B. pseudomallei but still resulted in direct bacterial killing, this indicated that C3 is directly deposited onto LPS and may thus prevent the assembly of the MAC on the bacterial outer membrane. Multiple bacterial species including Haemophilus influenzae, Neisseria meningitidis and N. gonorrhoeae, Borrelia burgdorferi, Streptococcus pyogenes, and Moraxella catarrhalis bind negative regulators of the complement system as a means to avoid direct killing by complement (reviewed in [79]), particularly via the alternative pathway. However, there have been no reports that B. pseudomallei or B. thailandensis can similarly bind complement regulatory proteins to avoid direct killing. Our data also indicate that B. pseudomallei are resistant to activation of the complement system by the alternative pathway. This finding goes against previous studies showing the alternative pathway is the predominant pathway of complement activation on B. pseudomallei [55], [66]. This discrepancy may be explained by differences in serum concentration and incubation times between these studies. While our studies clearly show that activation via the classical or lectin pathway is initiated in response to B. pseudomallei and B. thailandensis in NHS, it is still unclear which molecule(s) present in NHS is initiating these pathways for both Burkholderia species, and future studies will address this finding. There is evidence that NHS contains both IgG and IgM “natural” antibodies that can inherently bind to many bacterial surfaces and activate the classical pathway of complement [80], [81], although the levels of Burkholderia-specific antibodies we detected in NHS for our studies was extremely low (Figure 4). Alternatively, the pentraxins serum amyloid P (SAP) and C-reactive protein (CRP) are known to bind certain sugars present on the surfaces of bacteria to directly activate the classical pathway, and CRP can also interact with ficolin to activate the lectin-mediated pathway [82], [83]. The identification of the innate mediators responsible for this complement activation may provide innovative targets for Burkholderia-targeted innate immunotherapies.
Because of the significant differences in surface C3 deposition observed between B. pseudomallei and B. thailandensis, particularly at lower serum concentrations, we hypothesized that neutrophils would internalize and kill opsonized B. thailandensis more effectively than B. pseudomallei. Serum opsonization did elicit a significant increase in phagocytosis and killing of both Burkholderia species by neutrophils, and these activities did correlate with the relative levels of C3 surface deposition on these bacteria. B. pseudomallei and B. thailandensis opsonized with ≥5% NHS were both internalized more efficiently than unopsonized bacteria, and subsequently were cleared at a significantly greater rate within 2 h of co-culture. Increasing the NHS levels to 20% did not change the uptake rate, but did significantly enhance the clearance levels of both strains over the lower serum concentrations. Interestingly, when B. pseudomallei were opsonized with 1% NHS, an increased uptake by neutrophils was observed both in the presence and absence of the NADPH-oxidase inhibitor, whereas 1% serum-opsonized B. thailandensis only showed increased uptake in the presence of the inhibitor. These results suggest that the different degrees of C3 deposition observed in 1% serum represent the threshold levels required for enhanced bacterial uptake (observed in both strains opsonized in 1% NHS) versus the levels required to initiate the respiratory burst; i.e. the uptake levels for B. pseudomallei opsonized in 1% NHS were not increased in the presence of DPI. This suggests the C3 levels on B. pseudomallei were not sufficient to initiate the ROS production that was observed for B. thailandensis in 1% NHS, where the enhanced uptake could only be visualized after adding the NADPH-oxidase inhibitor. These observations also correlate with our direct in vitro analyses demonstrating that certain serum levels are required to elicit neutrophil NADPH oxidase activity to clear B. pseudomallei. Bacteria opsonized in ≥5% NHS induce significant ROS generation by neutrophils compared to unopsonized bacteria, with maximum levels acquired within 2–4 minutes of bacterial contact. The rapidity of the ROS generation is likely critical for B. pseudomallei clearance, as these bacteria are known to escape from phagosomal/endosomal compartments within 15–20 minutes of uptake [26], [84]. Bacteria opsonized in 1% NHS elicited a greatly reduced ROS response, which corresponds to the inefficient clearance observed for these bacteria. Interestingly, bacteria opsonized with 20% NHS elicited similar ROS levels as bacteria opsonized in 5% and 10% NHS (Figure 8), however the 20% opsonized bacteria were cleared at a significantly higher rate than the 5–10% opsonized bacteria (Figure 7). This suggests that other neutrophil-related mechanisms besides the respiratory burst may become activated in response to these higher levels of C3 deposition and contribute to killing of these Burkholderia species B. pseudomallei are susceptible to the bactericidal action of certain antimicrobial peptides that are produced by neutrophils, specifically the cathelicidin peptide, LL-37 [85], [86], [87]. In addition, neutrophils contain a wide variety of antimicrobial molecules including additional antimicrobial peptides or defensins, myeloperoxidase, neutrophil extracellular traps (NETs), and serine proteases which could work in concert with ROS generation [88], [89], [90], [91], [92], [93]. NETs were recently demonstrated to be antibacterial against B. pseudomallei, and NET release in response to B. pseudomallei was NADPH-oxidase dependent, as has been demonstrated previously with other agonists [94], [95], [96]. Since those studies were performed with unopsonized B. pseudomallei, our findings indicate that NET release may occur at a much faster rate with serum-opsonized bacteria in coordination with the rapid induction of the respiratory burst. Altogether, our data demonstrate a clear requirement for complement opsonization to allow for efficient uptake and killing of B. pseudomallei by neutrophils, and this bacterial clearance is dependent on achieving a rapid respiratory burst that elicits a threshold level of ROS generation.
Although melioidosis could historically be considered a neglected tropical disease, the global interest in these infections has increased dramatically in the past decade. The regions where B. pseudomallei has been recovered from soils has expanded beyond southeast Asia and northern Australia, and now includes large areas of the Middle East and South/Central America [reviewed in [14], [15]]. Concurrently, an increase in melioidosis cases has been observed in many of these regions. Diabetes is a major risk factor for acquiring these infections, as well as for developing severe disease. Because diabetes rates are rapidly increasing worldwide, it is likely that the number of melioidosis cases will also rise substantially. Together with the reports that B. pseudomallei possesses virulence attributes that make it attractive for misuse in bioterrorism-related releases has generated great interest in better understanding how this pathogen can so efficiently evade our immune defenses [97], [98]. Neutrophils have recently been reported to be prominently associated with B. pseudomallei infections in vivo and possess many qualities that would suggest they are capable of promoting host clearance [42]. However, the previous in vitro studies have reported disparate findings as to abilities of neutrophils to control these infections [55], [56], [57]. While our findings strongly indicate that neutrophils can efficiently clear B. pseudomallei, these activities appear to be dependent on the presence of critical C3 levels deposited on the surface of this pathogen. Thus, the differences observed between our findings and those of some previous publications may be partly attributed to differences in the serum concentration used, in the experimental set-up for neutrophil analyses, or even the handling of the bacteria and neutrophils [55], [56], [57]. However, based on the experimental methods described in these publications, there is no obvious indication for the discrepancies in the reported results. Our findings that neutrophil-generated NADPH-oxidase activity is important for B. pseudomallei clearance also strongly correlates with current in vivo studies reporting the significance of this pathway in melioidosis development [43], as well as the large number of recent reports associating the presence of neutrophil with improved outcome in B. pseudomallei infections [45], [55], [56], [57]. Several recent studies have also reported that neutrophils possess previously unappreciated abilities to interact directly and indirectly with other immune cells including macrophages, dendritic cells, NK cells, and T cells, and modulate their functions to further enhance disease resolution [reviewed in [99], [100]]; this suggests that neutrophils may possess additional important roles in resolution of melioidosis.
In conclusion, it is apparent that human neutrophils are able to effectively internalize and kill B. pseudomallei that have been opsonized with complement. The deposition of sufficient levels of complement components on the bacterial surface is critical for these processes. The mechanism of killing can largely be attributed to ROS production by these appropriately activated neutrophils. The primary predisposing condition for melioidosis, diabetes, is known to reduce chemotactic activity, phagocytosis, and microbicidal activities including decreased production of reactive oxygen species (reviewed in [44] and [45], [46]). Therefore, therapeutic strategies to promote recruitment of neutrophils to sites of infection and restore their phagocytic and bactericidal activities may allow diabetic individuals to control and limit the systemic spread of B. pseudomallei infections. Furthermore, modulation of neutrophil recruitment and function and/or complement deposition on the surface of B. pseudomallei may enhance the resolution of melioidosis regardless of predisposing condition.
Acknowledgments
We would like to thank Don Woods (University of Calgary) for providing the B. pseudomallei and B. thailandensis strains, and for many helpful discussions and support. We also acknowledge Sean Linkes for his assistance with the flow cytometry analyses. Additionally, we thank Viviana Ferreira, Claudio Cortes, J.P. Lavik, Minal Mulye, and Michael Bechill for their technical assistance and helpful comments.
Funding Statement
This work was supported by American Heart Association Postdoctoral Fellowship 11POST7320010 (MEW), Public Health Service grant R01-AI073452 (RMW) from the National Institute of Allergy and Infectious Disease, and Public Health Service grant U01-AI077764 (RMW) from the National Institute of Allergy and Infectious Disease. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chaowagul W, White NJ, Dance DA, Wattanagoon Y, Naigowit P, et al. (1989) Melioidosis: a major cause of community-acquired septicemia in northeastern Thailand. J Infect Dis 159: 890–899. [DOI] [PubMed] [Google Scholar]
- 2. Currie BJ, Fisher DA, Howard DM, Burrow JN, Lo D, et al. (2000) Endemic melioidosis in tropical northern Australia: a 10-year prospective study and review of the literature. Clin Infect Dis 31: 981–986. [DOI] [PubMed] [Google Scholar]
- 3. Currie BJ, Fisher DA, Howard DM, Burrow JN, Selvanayagam S, et al. (2000) The epidemiology of melioidosis in Australia and Papua New Guinea. Acta Trop 74: 121–127. [DOI] [PubMed] [Google Scholar]
- 4. White NJ (2003) Melioidosis. Lancet 361: 1715–1722. [DOI] [PubMed] [Google Scholar]
- 5. Leelarasamee A (2000) Melioidosis in Southeast Asia. Acta Trop 74: 129–132. [DOI] [PubMed] [Google Scholar]
- 6. Cheng AC, Currie BJ (2005) Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev 18: 383–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corral DM, Coates AL, Yau YC, Tellier R, Glass M, et al. (2008) Burkholderia pseudomallei infection in a cystic fibrosis patient from the Caribbean: a case report. Can Respir J 15: 237–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pérez JM, Petiot A, Adjidé C, Gerry F, Goursaud R, et al. (1997) First case report of melioidosis in Guadeloupe, a French West Indies archipelago. Clin Infect Dis 25: 164–165. [DOI] [PubMed] [Google Scholar]
- 9. Rolim DB, Vilar DC, Sousa AQ, Miralles IS, de Oliveira DC, et al. (2005) Melioidosis, northeastern Brazil. Emerg Infect Dis 11: 1458–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dorman SE, Gill VJ, Gallin JI, Holland SM (1998) Burkholderia pseudomallei infection in a Puerto Rican patient with chronic granulomatous disease: case report and review of occurrences in the Americas. Clin Infect Dis 26: 889–894. [DOI] [PubMed] [Google Scholar]
- 11. Inglis TJ, Rolim DB, Q SA (2006) Melioidosis in the Americas. Am J Trop Med Hyg 75: 947–954. [PubMed] [Google Scholar]
- 12. Miralles IS, Maciel Mdo C, Angelo MR, Gondini MM, Frota LH, et al. (2004) Burkholderia pseudomallei: a case report of a human infection in Ceará, Brazil. Rev Inst Med Trop Sao Paulo 46: 51–54. [DOI] [PubMed] [Google Scholar]
- 13. Christenson B, Fuxench Z, Morales JA, Suarez-Villamil RA, Souchet LM (2003) Severe community-acquired pneumonia and sepsis caused by Burkholderia pseudomallei associated with flooding in Puerto Rico. Bol Asoc Med P R 95: 17–20. [PubMed] [Google Scholar]
- 14. Dance DA (2000) Melioidosis as an emerging global problem. Acta Trop 74: 115–119. [DOI] [PubMed] [Google Scholar]
- 15. Currie BJ, Dance DA, Cheng AC (2008) The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans R Soc Trop Med Hyg 102: 1–4. [DOI] [PubMed] [Google Scholar]
- 16. Currie BJ, Ward L, Cheng AC (2010) The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis 4: e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suputtamongkol Y, Chaowagul W, Chetchotisakd P, Lertpatanasuwun N, Intaranongpai S, et al. (1999) Risk factors for melioidosis and bacteremic melioidosis. Clin Infect Dis 29: 408–413. [DOI] [PubMed] [Google Scholar]
- 18. Ngauy V, Lemeshev Y, Sadkowski L, Crawford G (2005) Cutaneous melioidosis in a man who was taken as a prisoner of war by the Japanese during World War II. J Clin Microbiol 43: 970–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeddeloh JA, Fritz DL, Waag DM, Hartings JM, Andrews GP (2003) Biodefense-driven murine model of pneumonic melioidosis. Infect Immun 71: 584–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vietri NJ, DeShazer D (2007) Melioidosis. Medical Aspects of Biological Warfare. Washington, DC.: Borden Institute, Walter Reed Army Medical Center. 147–166.
- 21.Titball RW, Russell P, Cuccui J, Easton A, Haque A, et al. (2008) Burkholderia pseudomallei: animal models of infection. Trans R Soc Trop Med Hyg 102. [DOI] [PubMed]
- 22.FESAP (2011) Recommendations concerning the select agent program.
- 23. Pilatz S, Breitbach K, Hein N, Fehlhaber B, Schulze J, et al. (2006) Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect Immun 74: 3576–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stevens MP, Haque A, Atkins T, Hill J, Wood MW, et al. (2004) Attenuated virulence and protective efficacy of a Burkholderia pseudomallei bsa type III secretion mutant in murine models of melioidosis. Microbiology 150: 2669–2676. [DOI] [PubMed] [Google Scholar]
- 25. Stevens MP, Stevens JM, Jeng RL, Taylor LA, Wood MW, et al. (2005) Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei . Mol Microbiol 56: 40–53. [DOI] [PubMed] [Google Scholar]
- 26. Stevens MP, Wood MW, Taylor LA, Monaghan P, Hawes P, et al. (2002 ) An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol Microbiol 46: 649–659. [DOI] [PubMed] [Google Scholar]
- 27. Warawa J, Woods DE (2005) Type III secretion system cluster 3 is required for maximal virulence of Burkholderia pseudomallei in a hamster infection model. FEMS Microbiol Lett 242: 101–108. [DOI] [PubMed] [Google Scholar]
- 28. Kespichayawattana W, Rattanachetkul S, Wanun T, Utaisincharoen P, Sirisinha S (2000) Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect Immun 68: 5377–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, et al. (2011) The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei . Infect Immun 79: 1512–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reckseidler-Zenteno SL, DeVinney R, Woods DE (2005) The capsular polysaccharide of Burkholderia pseudomallei contributes to survival in serum by reducing complement factor C3b deposition. Infect Immun 73: 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Warawa JM, Long D, Rosenke R, Gardner D, Gherardini FC (2009) Role for the Burkholderia pseudomallei capsular polysaccharide encoded by the wcb operon in acute disseminated melioidosis. Infect Immun 77: 5252–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reckseidler SL, DeShazer D, Sokol PA, Woods DE (2001) Detection of bacterial virulence genes by subtractive hybridization: identification of capsular polysaccharide of Burkholderia pseudomallei as a major virulence determinant. Infect Immun 69: 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brett PJ, Deshazer D, Woods DE (1997) Characterization of Burkholderia pseudomallei and Burkholderia pseudomallei-like strains. Epidemiol Infect 118: 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brett PJ, DeShazer D, Woods DE (1998) Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int J Syst Bacteriol 48: 317–320. [DOI] [PubMed] [Google Scholar]
- 35. Smith MD, Angus BJ, Wuthiekanun V, White NJ (1997) Arabinose assimilation defines a nonvirulent biotype of Burkholderia pseudomallei . Infect Immun 65: 4319–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Charoensap J, Utaisincharoen P, Engering A, Sirisinha S (2009) Differential intracellular fate of Burkholderia pseudomallei 844 and Burkholderia thailandensis UE5 in human monocyte-derived dendritic cells and macrophages. BMC Immunol 10: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kespichayawattana W, Intachote P, Utaisincharoen P, Sirisinha S (2004) Virulent Burkholderia pseudomallei is more efficient than avirulent Burkholderia thailandensis in invasion of and adherence to cultured human epithelial cells. Microb Pathog 36: 287–292. [DOI] [PubMed] [Google Scholar]
- 38. French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, et al. (2011) Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc Natl Acad Sci USA 108: 12095–12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brett PJ, Woods DE (2000) Pathogenesis of and immunity to melioidosis. Acta Trop 74: 201–210. [DOI] [PubMed] [Google Scholar]
- 40. Lazar Adler NR, Govan B, Cullinane M, Harper M, Adler B, et al. (2009) The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS Microbiol Rev 33: 1079–1099. [DOI] [PubMed] [Google Scholar]
- 41. Easton A, Haque A, Chu K, Lukaszewski R, Bancroft GJ (2007) A critical role for neutrophils in resistance to experimental infection with Burkholderia pseudomallei . J Infect Dis 195: 99–107. [DOI] [PubMed] [Google Scholar]
- 42. Laws TR, Smither SJ, Lukaszewski RA, Atkins HS (2011) Neutrophils are the predominant cell-type to associate with Burkholderia pseudomallei in a BALB/c mouse model of respiratory melioidosis. Microb Pathog 51: 471–475. [DOI] [PubMed] [Google Scholar]
- 43. Breitbach K, Klocke S, Tschernig T, van Rooijen N, Baumann U, et al. (2006) Role of inducible nitric oxide synthase and NADPH oxidase in early control of Burkholderia pseudomallei infection in mice. Infect Immun 74: 6300–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alba-Loureiro TC, Munhoz CD, Martins JO, Cerchiaro GA, Scavone C, et al. (2007) Neutrophil function and metabolism in individuals with diabetes mellitus. Braz J Med Biol Res 40: 1037–1044. [DOI] [PubMed] [Google Scholar]
- 45. Chanchamroen S, Kewcharoenwong C, Susaengrat W, Ato M, Lertmemongkolchai G (2009) Human polymorphonuclear neutrophil responses to Burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun 77: 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, et al. (1997) Impaired leucocyte functions in diabetic patients. Diabet Med 14: 29–34. [DOI] [PubMed] [Google Scholar]
- 47. Naghibi M, Smith RP, Baltch AL, Gates SA, Wu DH, et al. (1987) The effect of diabetes mellitus on chemotactic and bactericidal activity of human polymorphonuclear leukocytes. Diabetes Res Clin Pract 4: 27–35. [DOI] [PubMed] [Google Scholar]
- 48. Healey T, Selva-Nayagam S (2001) Retrospective review of febrile neutropenia in the Royal Darwin Hospital, 1994–99. Intern Med J 31: 406–412. [DOI] [PubMed] [Google Scholar]
- 49. Mukhopadhyay C, Chawla K, Vandana KE, Krishna S, Saravu K (2010) Pulmonary melioidosis in febrile neutropenia: the rare and deadly duet. Trop Doct 40: 165–166. [DOI] [PubMed] [Google Scholar]
- 50. Powell K, Ulett G, Hirst R, Norton R (2003) G-CSF immunotherapy for treatment of acute disseminated murine melioidosis. FEMS Microbiol Lett 224: 315–318. [DOI] [PubMed] [Google Scholar]
- 51. Cheng AC, Dasari P, Currie BJ (2004) Granulocyte colony-stimulating factor and an in vitro whole blood model of melioidosis. Eur J Clin Microbiol Infect Dis 23: 205–207. [DOI] [PubMed] [Google Scholar]
- 52. Cheng AC, Limmathurotsakul D, Chierakul W, Getchalarat N, Wuthiekanun V, et al. (2007) A randomized controlled trial of granulocyte colony-stimulating factor for the treatment of severe sepsis due to melioidosis in Thailand. Clin Infect Dis 45: 308–314. [DOI] [PubMed] [Google Scholar]
- 53. Cheng AC, Stephens DP, Anstey NM, Currie BJ (2004) Adjunctive granulocyte colony-stimulating factor for treatment of septic shock due to melioidosis. Clin Infect Dis 38: 32–37. [DOI] [PubMed] [Google Scholar]
- 54. Stephens DP, Fisher DA, Currie BJ (2002) An audit of the use of granulocyte colony-stimulating factor in septic shock. Intern Med J 32: 143–148. [DOI] [PubMed] [Google Scholar]
- 55. Egan AM, Gordon DL (1996) Burkholderia pseudomallei activates complement and is ingested but not killed by polymorphonuclear leukocytes. Infect Immun 64: 4952–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Razak N, Ismail G (1982) Interaction of human polymorphonuclear leukocytes with Pseudomonas pseudomallei . J Gen Appl Microbiol 28: 509–518. [Google Scholar]
- 57. Jones AL, Beveridge TJ, Woods DE (1996) Intracellular survival of Burkholderia pseudomallei . Infect Immun 64: 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeShazer D, Brett PJ, Carlyon R, Woods DE (1997) Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J Bacteriol 179: 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moore RA, DeShazer D, Reckseidler S, Weissman A, Woods D (1999) E (1999) Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei . Antimicrob Agents Chemother 43: 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heiss C, Burtnick MN, Wang Z, Azadi P, Brett PJ (2012) Structural analysis of capsular polysaccharides expressed by Burkholderia mallei and Burkholderia pseudomallei . Carbohydr Res 15: 90–94. [DOI] [PubMed] [Google Scholar]
- 61.Nauseef W (2007) Isolation of human neutrophils from venous blood. In: Quinn MT, DeLeo FR, Bokoch GM, editors. Methods in Molecular Biology. Totowa, NJ: Human Press Inc. 15–20. [DOI] [PubMed]
- 62. Drevets DA, Canono BP, Leenen PJ, Campbell PA (1994) Gentamicin kills intracellular Listeria monocytogenes . Infect Immun 62: 2222–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van den Broek PJ (1989) Antimicrobial drugs, microorganisms, and phagocytes. Rev Infect Dis 11: 213–245. [DOI] [PubMed] [Google Scholar]
- 64. Mandell GL (2005) Uptake, transport, delivery, and intracellular activity of antimicrobial agents. Pharmacotherapy 25: 130–133. [DOI] [PubMed] [Google Scholar]
- 65. Prokesch RC, Hand WL (1982) Antibiotic entry into human polymorphonuclear leukocytes. Antimicrob Agents Chemother 21: 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DeShazer D, Brett PJ, Woods DE (1998) The type II O-antigenic polysaccharide moiety of Burkholderia pseudomallei lipopolysaccharide is required for serum resistance and virulence. Mol Microbiol 30: 1081–1100. [DOI] [PubMed] [Google Scholar]
- 67. Ismail G, Razak N, Mohamed R, Embi N, Omar O (1988) Resistance of Pseudomonas pseudomallei to normal human serum bactericidal action. Microbiol Immunol 32: 645–652. [DOI] [PubMed] [Google Scholar]
- 68. Underhill DM, Ozinsky A (2002) Phagocytosis of microbes: complexity in action. Annu Rev Immunol 20: 825–852. [DOI] [PubMed] [Google Scholar]
- 69. Brown EJ (1991) Complement receptors and phagocytosis. Curr Opin Immunol 3: 76–82. [DOI] [PubMed] [Google Scholar]
- 70. Conlan JW (1997) Critical role of neutrophils in host defense against experimental systemic infections of mice by Listeria monocytogenes, Salmonella typhimurium, and Yersinia enterocolitica . Infect Immun 65: 630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F (2011) Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1β is deleterious. PLoS Pathog 7. [DOI] [PMC free article] [PubMed]
- 72. Corbett D, Roberts IS (2009) The role of microbial polysaccharides in host-pathogen interaction. F1000 Biol Rep 1: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Roberts I, S., Saunders FK, Boulnois GJ (1989) Bacterial capsules and interactions with complement and phagocytes. Biochem Soc Trans 17: 462–464. [DOI] [PubMed] [Google Scholar]
- 74. Taylor CM, Roberts IS (2005) Capsular polysaccharides and their role in virulence. Contrib Microbiol 12: 55–66. [DOI] [PubMed] [Google Scholar]
- 75.Merino S, Tomas JM (2010) Bacterial capsules and evasion of immune responses. Encylopedia of Life Sciences. Hoboken, NJ: John Wiley and Sons, Ltd. 1–9.
- 76. Grossman N, Schmetz MA, Foulds J, Klima EN, Jimenez-Lucho VE, et al. (1987) Lipopolysaccharide size and distribution determine serum resistance in Salmonella montevideo . J Bacteriol 169: 856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brett PJ, Burtnick MN, Woods DE (2003) The wbiA locus is required for the 2-O-acetylation of lipopolysaccharides expressed by Burkholderia pseudomallei and Burkholderia thailandensis . FEMS Microbiol Lett 218: 323–328. [DOI] [PubMed] [Google Scholar]
- 78. Ngugi SA, Ventura VV, Qazi O, Harding SV, Kitto GB, et al. (2010) Lipopolysaccharide from Burkholderia thailandensis E264 provides protection in a murine model of melioidosis. Vaccine 28: 7551–7555. [DOI] [PubMed] [Google Scholar]
- 79. Blom AM, Hallstrom T, Riesbeck K (2009) Complement evasion strategies of pathogens–Acquisition of inhibitors and beyond. Mol Immunol 46: 2808–2817. [DOI] [PubMed] [Google Scholar]
- 80. Balagopal A, MacFarlane AS, Mohapatra N, Soni S, Gunn JS, et al. (2006) Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect Immun 74: 5114–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schlesinger LS, Horwitz MA (1994) A role for natural antibody in the pathogenesis of leprosy: antibody in nonimmune serum mediates C3 fixation to the Mycobacterium leprae surface and hence phagocytosis by human mononuclear phagocytes. Infect Immun 62: 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yuste J, Botto M, Bottoms SE, Brown JS (2007) Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae . PLoS Pathog 3: 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ng PM, Le Saux A, Lee CM, Tan NS, Lu J, et al. (2007) C-reactive protein collaborates with plasma lectins to boost immune response against bacteria. EMBO J 26: 3431–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Harley VS, Dance DA, Tovey G, McCrossan MV, Drasar BS (1998) An ultrastructural study of the phagocytosis of Burkholderia pseudomallei . Microbios 94: 35–45. [PubMed] [Google Scholar]
- 85. Kanthawong S, Bolscher JG, Veerman EC, van Marle J, de Soet HJ, et al. (2012) Antimicrobial and antibiofilm activity of LL-37 and its truncated variants against Burkholderia pseudomallei . Int J Antimicrob Agents 39: 39–44. [DOI] [PubMed] [Google Scholar]
- 86. Kanthawong S, Nazmi K, Wongratanacheewin S, Bolscher JG, Wuthiekanun V, et al. (2009) In vitro susceptibility of Burkholderia pseudomallei to antimicrobial peptides. Int J Antimicrobial Agents 34: 309–314. [DOI] [PubMed] [Google Scholar]
- 87. Sim SH, Liu Y, Tan J, Thong TW, Wang D, et al. (2011) Antimicrobial activity of cathelicidin peptides against Burkholderia pseudomallei, the causative agent of melioidosis. Int J Antimicrob Agents 38: 270–271. [DOI] [PubMed] [Google Scholar]
- 88. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, et al. (2004) Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 89. Standish AJ, Weiser JN (2009) Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol 183: 2602–2609. [DOI] [PubMed] [Google Scholar]
- 90. Nauseef WM (2007) How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219: 88–102. [DOI] [PubMed] [Google Scholar]
- 91. Parry MF, Root RK, Metcalf JA, Delaney KK, Kaplow LS, et al. (1981) Myeloperoxidase deficiency: prevalence and clinical significance. Ann Intern Med 95: 293–301. [DOI] [PubMed] [Google Scholar]
- 92. Chapman ALP, Hampton MB, Senthilmohan R, Winterbourn CC, Kettle AJ (2002) Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus . J Biol Chem 277: 9757–9762. [DOI] [PubMed] [Google Scholar]
- 93.Mayer-Scholl A, Hurwitz R, Brinkmann V, Schmid M, Jungblut P, et al. (2005) Human neutrophils kill Bacillus anthracis. PLoS Pathog 1. [DOI] [PMC free article] [PubMed]
- 94.Riyapa D, Buddhisa S, Korbsrisate S, Cuccui J, Wren BW, et al. (2012) Neutrophil extracellular traps exhibit antibacterial activity against Burkholderia pseudomallei and are influenced by bacterial and host factors. Infect Immun: In press. [DOI] [PMC free article] [PubMed]
- 95. Ermert D, Urban CF, Laube B, Goosmann C, Zychlinsky A, et al. (2009) Mouse neutrophil extracellular traps in microbial infections. J Innate Immun 1: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, et al. (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dance DA (2005) Melioidosis and glanders as possible biological weapons. In: Fong, Alibek, editors. Bioterrorism and Infectious Agents. New York: Springer Science. 99–145.
- 98. Galyov EE, Brett PJ, Deshazer D (2010) Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol 64: 495–517. [DOI] [PubMed] [Google Scholar]
- 99. Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11: 519–531. [DOI] [PubMed] [Google Scholar]
- 100. Costantini C, Cassatella MA (2011) The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. J Leukoc Biol 89: 221–233. [DOI] [PubMed] [Google Scholar]