

Abstract
The complexity of gene regulation has created obstacles to defining mechanisms that establish the patterns of gene expression characteristic of the different clinical phenotypes of breast cancer. TFAP2C is a transcription factor, which plays a critical role in the regulation of both estrogen receptor-alpha (ERα) and c-ErbB2/HER2 (Her2). Herein, we performed chromatin immunoprecipitation and direct sequencing (ChIP-seq) for TFAP2C in four breast cancer cell lines. Comparing the genomic binding sites for TFAP2C, we identified that glutathione peroxidase (GPX1) is regulated by TFAP2C through an AP-2 regulatory region in the promoter of the GPX1 gene. Knock down of TFAP2C, but not the related factor TFAP2A, resulted in an abrogation of GPX1 expression. Selenium-dependent GPX activity correlated with endogenous GPX1 expression and overexpression of exogenous GPX1 induced GPX activity and significantly increased resistance to tert-butyl hydroperoxide. Methylation of the CpG island encompassing the AP-2 regulatory region was identified in cell lines where TFAP2C failed to bind the GPX1 promoter and GPX1 expression was unresponsive to TFAP2C. Furthermore, in cell lines where GPX1 promoter methylation was associated with gene silencing, treatment with 5-aza-dC (an inhibitor of DNA methylation) allowed TFAP2C to bind to the GPX1 promoter resulting in activation of GPX1 RNA and protein expression. Methylation of the GPX1 promoter was identified in approximately 20% of primary breast cancers and a highly significant correlation between TFAP2C and GPX1 expression was confirmed when considering only those tumors with an unmethylated promoter, whereas the related factor, TFAP2A, failed to demonstrate a correlation. The results demonstrate that TFAP2C regulates the expression of GPX1, which influences the redox state and sensitivity to oxidative stress induced by peroxides. Given the established role of GPX1 in breast cancer, the results provide an important mechanism for TFAP2C to further influence oncogenesis and progression of breast carcinoma cells.
INTRODUCTION
Breast cancer is the most common cancer and the second leading cause of cancer death in women in the United States, with an annual incidence of 207,000 breast cancer cases in 2010 (1). Developing novel treatment strategies and targeted therapy for cancer has relied on characterizing tumors for expression of genes that influence sensitivity to specific drugs or treatments. The different clinical phenotypes of breast cancer have defined patterns of gene expression, which are predictive of recurrence and overall survival (2). Luminal A breast cancers have the best prognosis and are characterized by expression of estrogen receptor-alpha (ERα) and non-amplified expression of ErbB2/Her2. Luminal B breast cancers, which demonstrate a greater tendency to recur, also express ERα and commonly have amplified expression of ErbB2/Her2. The HER2 phenotype has low or absent expression of the steroid receptors, ERα and progesterone receptor (PgR), and amplified expression of ErbB2/Her2. The basal or “triple-negative” phenotype carries the worst prognosis and is characterized by a relatively undifferentiated cell lacking expression of the steroid receptors and ErbB2/Her2. The characteristic patterns of gene expression, in particular ERα, PgR and ErbB2/Her2, have important implications for outcome in breast cancer and further predict sensitivity to certain therapies, such as anti-estrogens and trastuzumab (3). However, we are only now beginning to define the transcriptional mechanisms that establish the patterns of expression characteristic of the various clinical phenotypes of breast cancer.
TFAP2C is a member of the retinoic acid-inducible, developmentally regulated family of AP-2 transcription factors that include five members—TFAP2A (AP-2α), TFAP2B (AP-2β), TFAP2C (AP-2γ), TFAP2D (AP-2δ) and TFAP2E (AP-2ε) (4–8). AP-2 factors bind to GC-rich regulatory elements in the promoters of target genes through a helix-loop-helix motif in the DNA binding domain, which, except for TFAP2D, is highly homologous in all the AP-2 family members (9). Interestingly, AP-2 factors have been implicated in the regulation of ERα and Her2 expression in breast cancer. TFAP2C regulates expression of ERα and several pathways of hormone response (10, 11) and recent findings demonstrated significant overlap in the promoters targeted by ERα and TFAP2C (12). In addition, TFAP2A and TFAP2C were able to induce expression of the cloned ERBB2 gene promoter (5, 13–15). Similarly in MDA-MB-453 cells, TFAP2C was found to target the ERBB2 promoter and knock down of TFAP2C significantly reduced ErbB2/Her2 expression (16). In BT474 breast carcinoma cells, both TFAP2A and TFAP2C were shown to participate in the regulation of ErbB2/Her2 expression (17). Furthermore, a correlation has been reported between AP-2 expression and the expression of ErbB2/Her2 in primary breast cancers (17–19). Hence, there is mounting evidence that AP-2 factors, and in particular TFAP2C, plays a central role in the regulation of genes known to have important prognostic significance in breast cancer.
By using a combination of gene expression arrays and chromatin immunoprecipitation with direct sequencing (ChIP-seq), we previously defined a set of target genes directly regulated by TFAP2C in MCF-7 breast carcinoma cells (20). Many of the TFAP2C target genes identified were known to be expressed as part of the luminal A breast cancer gene expression signature and/or part of the estrogen-regulated pathways. Notably, we found no evidence for TFAP2C binding to the ERBB2 gene in MCF-7 cells despite other studies demonstrating that the ERBB2 gene is regulated by TFAP2C in cells that overexpress ErbB2/Her2. This finding led to the hypothesis that TFAP2C can be directed to a different set of genes in breast cancers of different phenotypes. This hypothesis was further supported by studies of the ERα gene promoter showing that epigenetic chromatin modifications, including CpG methylation and histone deacetylation, was associated with chromatin inaccessibility, which blocked TFAP2C from binding to the AP-2 regulatory region in ERα-negative cells (21). Since TFAP2C is known to regulate the expression of many genes that comprise the luminal phenotype expression signature, we further hypothesized that genes differentially targeted by TFAP2C comparing breast cancer cell lines having different phenotypic expression profiles would likely identify regulatory regions of genes with a clinically significant impact. The goal of this study was to compare ChIP-seq data for TFAP2C in a panel of breast cancer cell lines representing the luminal A, luminal B and HER2 breast cancer phenotypes. We sought to identify genes with clear differences in the ability of TFAP2C to bind to the regulatory region comparing the different cell lines and to determine the molecular basis for differential gene targeting. In this way, we hoped to more fully understand the role of TFAP2C in regulating the expression of genes that impact the clinically significant breast cancer phenotypes.
RESULTS
GPX1 Promoter is Targeted by TFAP2C in Breast Cancer Cell Lines
Previous studies identified an important role for TFAP2C in establishing patterns of gene expression that are highly predictive of the clinical phenotype in breast cancer. There was also the suggestion that important differences may exist in the genes targeted by TFAP2C comparing breast cancer cells from different phenotypes. We hypothesized that comparing different breast cancer cell lines for TFAP2C target genes could identify genes regulated by TFAP2C that play a critical role in determining clinically-relevant phenotypic differences. We chose to compare four breast cancer cell lines representing the luminal and HER2 cancer phenotypes (22). MCF-7 is an accepted model for the luminal A breast cancer phenotype, expressing ERα, non-amplified expression of HER2, TFAP2C and relatively low expression of TFAP2A (see Figure 1a). BT-474 expresses ERα and HER2 and is a model for the luminal B phenotype. The relatively high level of TFAP2C expression in BT-474 is consistent with the overexpression of TFAP2C noted in more aggressive ERα-positive cancers. Based on the expression pattern for ERα and HER2, MDA-MB-453 and SKBR-3 would be classified as the HER2 breast cancer phenotype; however, the expression pattern in these cell lines suggest a luminal origin. Since all four cell lines express endogenous TFAP2C, we performed ChIP-seq using anti-TFAP2C antibody without needing to overexpress the factor. Examples of the ChIP-seq data from the four breast cancer cell lines are shown in Figure 1B, which illustrates the high quality and reproducibility of the data. The data for the genomic regions from two genes—KRT8 and GPX1—are presented demonstrating the cell type-specificity for TFAP2C binding at these two loci. In the first panel, a region of approximately 15 kbp is shown from the cytokeratin 8 (KRT8) gene and in this diagram, transcription for KRT8 reads right to left. The TFAP2C binding pattern is similar to what is found in most target genes—the pattern is identical for all four cell lines, there is a series of peaks clustered in and around the transcriptional start site and an additional peak within 1 kbp of the last exon. KRT8 is one of the core genes representing the luminal cytokeratin cluster signature. We analyzed the ChIP-seq data to identify promoters that demonstrated differential targeting of TFAP2C comparing the different breast cancer phenotypes. One gene identified in this analysis was glutathione peroxidase (GPX1), which encodes a selenoenzyme in the glutathione peroxidase family that detoxifies peroxide and is a key antioxidant in humans, protecting cells from reactive oxygen species (ROS). As seen in Figure 1B, TFAP2C binds strongly to a region just upstream of the transcriptional start site (TSS) for the GPX1 gene in BT-474 and MDA-MB-453 cell lines but there is no binding to this genomic region in MCF-7 or SKBR-3 cell lines. Interestingly, the expression pattern of GPX1 is highly variable in primary breast cancer tumors, as noted in Figure 1C. These data indicate that GPX1 is differentially targeted by TFAP2C comparing the four cell lines, thus further experiments were designed to demonstrate the functional role of TFAP2C in regulation of GPX1 and the molecular basis for differential gene targeting.
Figure 1. Expression of TFAP2C and ChIP-seq in Breast Cancer Lines.
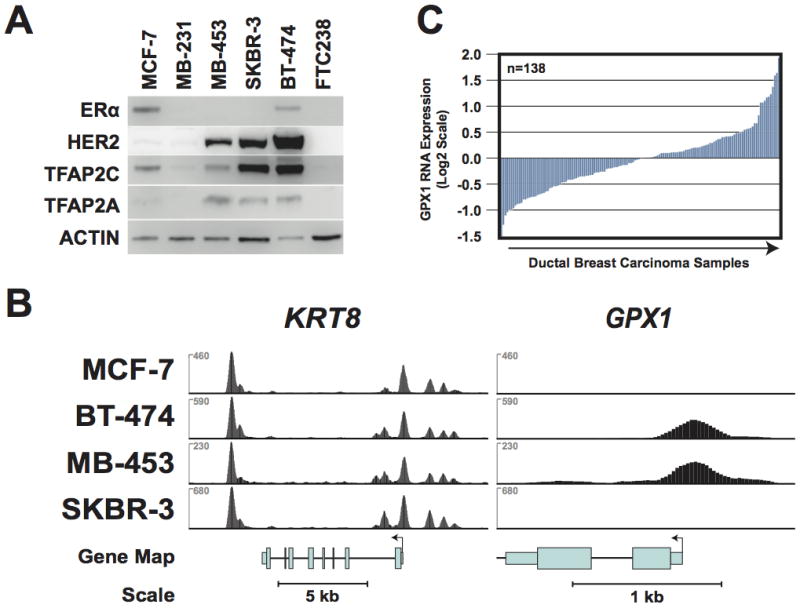
A. Western blots of cell lines shown demonstrate expression pattern for ERα, HER2, TFAP2C and TFAP2A. B. ChIP-seq data for TFAP2C genomic binding in breast cancer cell lines with the genome browser graphical output for the KRT8 and GPX1 genes. The y-axis represents normalized coverage (reads per million mapped) for all four cell lines with KRT8 and GPX1 presented to the same scale. C. Oncomine data of expression (log2 scale) of GPX1 in primary breast cancers.
TFAP2C Regulates Expression of GPX1
We evaluated GPX1 RNA and protein expression in the four breast carcinoma lines. As shown in Figure 2A, GPX1 was expressed in BT-474 and MDA-MB-453 cell lines but the gene was essentially not expressed in MCF-7 or SKBR-3. TFAP2C and TFAP2A were knocked down using siRNAs that we have previously shown to specifically knockdown each family member (11). Knockdown of TFAP2C in BT-474 or MDA-MB-453 cells reduced GPX1 expression to 50–80%, whereas knockdown of TFAP2A had no significant effect on GPX1 expression (Figure 2B). Knockdown of either TFAP2C or TFAP2A had no effect on GPX1 expression in either MCF-7 or SKBR-3 (data not shown). The span of the TFAP2C peak from ChIP-seq shown in Figure 1B is approximately 200 bp. Using gel-shift analyses we refined the location of TFAP2C binding in the GPX1 promoter and a representative experiment is shown in Figure 2C,D. A 60 bp region of DNA from the GPX1 promoter centered at the ChIP-seq binding peak is shown in Figure 2C. This 60 bp sequence was used as a double-stranded DNA probe, which was labeled and used in gel-shift with TnT-synthesized TFAP2C. Competitors 5, 6 and 7 were 20 bp oligos from the left, middle or right-hand region spanning the 60 bp probe. As seen in Figure 2D, oligo 7 effectively competed for TFAP2C binding and the supershift with anti-TFAP2C antibody confirmed the identity of the TFAP2C complex. The sequence contained in oligo 7, 5′-CCCGGAGGG, matches the consensus site we reported for in vitro and genomic binding of TFAP2C (20), and the location of the site corresponds to the center of the peak from the ChIP-seq results. A second site matching the known AP-2 consensus sequence (GCCTGAGGG) was noted at the 3′ edge of oligo 5; however, since AP-2 binds poorly to oligos when the site is on the end, a modified oligo called 5* was created that was shifted by four bp to place the potential AP-2 site into the middle of the 20 bp oligo. As seen in Figure 2D, the 5* oligo competitor also competed for TFAP2C binding to the full-length probe. These data demonstrated that GPX1 is a TFAP2C target gene and there are two AP-2 binding sites that are targeted by TFAP2C in the GPX1 5′ regulatory region. In addition, the location of TFAP2C binding determined by ChIP-seq correctly predicted differential expression in these luminal breast cancer cell lines.
Figure 2. GPX1 Expression and TFAP2C regulation.

A. GPX1 expression by RT-PCR (top panel) and Western blot (bottom panels) in four breast cancer cell lines. B. GPX1 expression in BT-474 and MDA-MB-453 after transfection with siRNAs: Mock (mock transfected), non-targeting (NT), TFAP2A (A) or TFAP2C (C) or both siRNAs (A+C). C. The 60 bp region of the GPX1 gene promoter with consensus sequences of potential AP-2 binding sites (underlined) and relative location of additional oligonucleotide competitors used in gel-shift assay. D. Gel shift with radiolabeled probe shown in C with oligo competitors. The locations of free probe, TFAP2C complex and complex supershift are noted.
GPX1 Expression and Sensitivity to Oxidative Stressors
The differential expression of GPX1 suggests that TFAP2C has the potential of influencing the oxidative state and sensitivity to peroxide of breast cancer cells. The selenium-dependent glutathione peroxidase activity was measured in the four breast cancer cell lines. As seen in Figure 3A, relatively high levels of selenium-dependent GPX activity was found in BT-474 and MDA-MB-453 compared to minimal activity for MCF-7 and SKBR-3. These data demonstrate that GPX enzymatic activity correlates with the expression of GPX1 in the four cell lines. The functional effects of TFAP2C regulation of GPX1 was evaluated by comparing sensitivity of BT-474 to tert-butyl hydroperoxide with and without TFAP2C knock down. As seen in Figure 3B, knock down of TFAP2C increased sensitivity to tert-butyl hydroperoxide over a concentration range of 20 to 60 μM compared to NT siRNA. To further prove that these differences were due to GPX1, overexpression of exogenous GPX1 was induced in MCF-7 cells by an adenoviral vector expressing the GPX1 gene (Figure 3C). Expression of GPX1 was confirmed by Western blot and resulted in an increase in selenium-dependent GPX activity. Interestingly, overexpression of TFAP2C failed to alter GPX1 expression or induce selenium-dependent GPX activity in GPX1 negative MCF-7 cells. Next, we examined whether expression of GPX1 in MCF-7 cells altered the sensitivity to tert-butyl hydroperoxide (Figure 3D). MCF-7 cells induced with either Ad-empty, Ad-TFAP2C or mock infected had equal sensitivity to tert-butyl hydroperoxide with a LD50 of approximately 40 μM. However, with overexpression of GPX1, MCF-7 cells exhibited resistance to tert-butyl hydroperoxide with an LD50 that exceeded 105 μM. The data indicate an important role for TFAP2C in regulating the oxidative state and sensitivity to peroxides, particularly lipid peroxides, of breast cancer cells through the regulation of GPX1.
Figure 3. GPX Activity and Sensitivity to tert-butyl hydroperoxide.

A. Selenium-dependent GPX activity in four breast cancer cell lines demonstrated significantly greater activity in BT-474 and MDA-MB-453 compared to MCF-7 and SKBR-3. B. Sensitivity to tert-butyl hydroperoxide was measured at various concentrations comparing BT-474 transfected with siRNA to TFAP2C (siTFAP2C) or a non-targeting siRNA (siNT) and demonstrates increased drug sensitivity with knock down of TFAP2C over a concentration range of 20 to 60 μM. C. Selenium-dependent GPX activity was induced in MCF-7 cells by overexpression of GPX1 by infection with Ad-GPX1 (G) compared to Ad-empty (E) or Ad-TFAP2C (C), p<0.05. Western blots below graph confirm expression of proteins after viral infection. D. Sensitivity to tert-butyl hydroperoxide was measured at various concentrations comparing MCF-7 cells infected with adenoviruses indicated demonstrating that overexpression of GPX1 significantly increased resistance to tert-butyl hydroperoxide at concentrations of 40 μM and above.
TFAP2C Binding to GPX1 Promoter Regulated by CpG Methylation
The next critical issue to address was the molecular basis for the lack of TFAP2C binding to the regulatory region of GPX1 in MCF-7 and SKBR-3 breast cancer cell lines. The promoter region including the AP-2 regulatory region is GC rich and contains a number of potential DNA methylation sites. In addition, a recent study identified gene silencing of GPX1 in gastric cancers due to CpG methylation (23). To examine CpG methylation, we used bisulfite sequencing of the GPX1 promoter from the four cell lines and found evidence for extensive methylation of the CpGs in MCF-7 and SKBR-3 and little or no methylation of this region in BT-474 and MDA-MB-453 cells (Figure 4A,B). These findings suggest that the lack of TFAP2C binding in the GPX1 promoter identified in MCF-7 and SKBR-3 may be functionally related to DNA methylation in and around the AP-2 regulatory region. To further determine whether CpG methylation regulates TAP2C expression of GPX1, the four breast cancer cells were treated with 5-aza-2-deoxycytidine (5-aza dC) and expression of GPX1 mRNA and protein was examined. Treatment with 5-aza dC activated GPX1 expression (both GPX1 RNA and protein) in MCF-7 and SKBR-3 but had no effect on expression in BT-474 or MDA-MB-453 cell lines (Figure 4C). Furthermore, treating MCF-7 cells with 5-aza-dC induced binding of TFAP2C to the GPX1 promoter, whereas, no binding was detected in mock-treated cells (Figure 4D). These data support a model in which the promoter regions of certain genes, in this case GPX1, are epigenetically silenced through chromatin modifications that includes DNA methylation and are insensitive to reactivation by overexpression of a transcriptional regulatory factor alone. In addition, the data demonstrate that CpG methylation is functionally linked to gene silencing since GPX1 expression was activated by 5-aza-dC drug treatment.
Figure 4. Methylation of GPX1 promoter.

A. Results of bisulphite sequencing of CpG island of GPX1 promoter in cell lines shown with CpGs denoted by a circle. B. Example of sequencing reads from the GPX1 promoter CpG island from template prepared from MCF-7 and BT-474 cells demonstrating the presence of C preceding a G in the site indicated by arrows showed that these cytosines were methylated in MCF-7 cells. Unmethylated cytosines (C) were converted to uracils (T) in BT-474 cells. C. Expression of GPX1 by RT-PCR (top panel) and Western blot (bottom panels) after treatment with 5-aza dC for cell lines shown. D. ChIP analysis performed with anti-TFAP2C antibody compared to IgG at GPX1 promoter demonstrated significant increase in binding of TFAP2C after treatment with 5-aza-dC, whereas, no evidence for binding of TFAP2C in absence of drug (Mock). Data normalized to ChIP with IgG.
TFAP2C Regulates GPX1 Expression in Primary Breast Cancers
The results from cell lines indicate an important role for TFAP2C in regulating GPX1 expression in breast cancer. To determine if the cell line model mirrors regulation in primary cancers, a panel of primary breast cancer samples was analyzed for GPX1 expression and methylation of the GPX1 promoter (see Figure 5). DNA was extracted from archival breast cancer samples and subjected to analysis for CpG methylation at the GPX1 promoter using bisulfite DNA sequencing. Representative data for CpG methylation is shown in Figure 5A. Significant methylation in the region of the AP-2 regulatory regions was identified in approximately 20% of breast cancer samples. Expression of GPX1, TFAP2A and TFAP2C mRNA levels were determined using quantitative RT-PCR analysis. As shown in Figure 5B, no significant association was seen comparing GPX1 and TFAP2A expression. However, a highly significant correlation was demonstrated between GPX1 and TFAP2C expression, when considering samples with an unmethylated GPX1 promoter. Of note, one sample with high TFAP2C expression but low GPX1 expression was noted to have a methylated GPX1 promoter (tumor 3). The correlation between expression of GPX1 and TFAP2C had an R2 value of 0.91 (p<0.0001), which indicates that TFAP2C is a key factor in regulating the expression of GPX1 in breast cancers that have an unmethylated GPX1 promoter. However, with methylation of the GPX1 promoter, GPX1 expression is insensitive to TFAP2C transcriptional activity.
Figure 5. Methylation of GPX1 gene and expression of GPX1, TFAP2A and TFAP2C in primary breast cancer samples.

A. Examples of results from bisulfite sequencing demonstrating the methylation patterns among primary breast cancer samples showing high degree of methylated (1–4) and unmethylated (5–8) chromatin. B/C. Semi-quantitative RT-PCR used to determine a correlation between GPX1 expression and expression of TFAP2A (B) and TFAP2C (C) in panel of primary breast cancer samples (n=20). Results demonstrated strong association between expression of TFAP2C and GPX1 in unmethylated samples with R2= 0.908, p<0.0001. No significant correlation was found comparing GPX1 expression with TFAP2A expression with R2=0.022, p=0.58.
DISCUSSION
There is growing evidence that oxidative stress plays a critical role in oncogenesis and cancer progression (24, 25). Elevated levels of oxidative stress result in accumulation of reactive oxygen species (ROS) including superoxide, hydrogen peroxide and hydroxyl radicals. Elevated levels of ROS identified in cancer cells may result from several mechanisms including increased rates of metabolism, mitochondrial dysfunction or alteration in other enzymes in the ROS metabolic pathways. Since oxygen radicals can damage DNA, elevated ROS may play an important role in oncogenesis by contributing to genetic instability leading to transformation and cancer progression. Alternatively, increased ROS can induce a proliferative response through activation of several proliferative mechanisms including activation of the mitogen-activated protein kinases (MAPKs)/ERK1/2, increased signaling via phosphoinositide-3-kinase (PI3K)/Akt or activation of the NF-κB pathway. Alternatively, elevated levels of ROS can alter apoptotic signaling through the activation of c-Jun N-terminal kinases (JNKs), p53 or the forkhead transcription factors such as FOXO3a. Elevated levels of ROS have further been shown to be involved in metastasis by altering cell motility, cell adhesion and angiogenesis. On the other hand, elevated levels of ROS in cancer cells can also increase their susceptible to chemotherapeutic agents that are capable of increasing intracellular levels of ROS and peroxide (26, 27). Targeting the ROS system for cancer therapy is particularly attractive since novel chemotherapeutic agents have recently been developed that increase ROS and induce apoptosis in cancer cells with minimal effects on normal cells (28). Hence, mechanisms that modulate ROS in cancer cells influence several processes in oncogenesis and potentially offer a number of pathways for the development of novel treatment strategies.
GPX1 is an important antioxidant selenoenzyme that catalyzes the detoxification of hydrogen peroxide, resulting in the oxidation of glutathione (GSH) to glutathione disulphide (GSSG) (25). Mice genetically engineered to have a GPX1-null phenotype demonstrate increased liver peroxides, increased rates of hydrogen peroxide production and display growth retardation (29). Several studies have implicated an important role of GPX1 in breast cancer oncogenesis and progression. Individuals homozygous for a leucine at codon 198 of the GPX1 protein had an odds ratio for breast cancer risk that was approximately doubled compared to individuals inheriting a GPX1 polymorphism with a proline at this position (30, 31). Additional studies suggested that the increased breast cancer risk associated with the Pro198Leu GPX1 polymorphism might be further influenced by racial effects, inheritance of certain MnSOD polymorphisms, and environmental factors (32–34). Loss of heterozygosity (LOH) for the GPX1 locus has been reported to occur in 36% of breast cancers and 42% of colon carcinomas, suggesting that elimination of GPX1 activity has oncogenic effects (30, 35). Finally, the expression of GPX1 in breast cancer was reported to be inversely associated with expression of ERα and was also predictive of cancer survival (36, 37).
Particularly germane to breast cancer treatment, GPX1 activity has been directly implicated in sensitivity to chemotherapy. Overexpression of GPX1 in T47D breast carcinoma cells significantly increased their resistance to the anthracycline, doxorubicin (38). Drugs such as docosahexaenoic acid (DHA) have been shown to enhance the effectiveness of anthracyclines by blocking GPX1 activity (39). In a cardiomyocyte model, GPX1 deficiency resulted in increased susceptibility to ROS-mediated doxorubicin-induced cardiotoxicity compared to cardiomyocytes with wild-type GPX1 expression (40). Futhermore, β-phenylethyl isothiocyanate (PEITC) was described as a novel chemotherapeutic agent, which increases ROS-mediated cell killing through inactivation of GPX1, and highlights the important role of GPX1 in regulating ROS in transformed cells and the potential for targeting GPX1 to alter chemosensitivity of cancer cells (41). GPX1 may also influence the survival of metastatic lesions. Using an animal model of breast cancer, a recent report demonstrated that tumor entrained neutrophils (TENs) inhibit lung metastases through a peroxide-mediated cell killing mechanism (42). The expression of GPX1 in some breast tumors has the potential to protect metastatic cancer cells from TEN-mediated cell killing by detoxifying peroxide.
The current findings are significant in that they demonstrate an important role for TFAP2C in regulating ROS and sensitivity to peroxide in breast cancer through the control of GPX1 expression. The results from an examination of clinical samples indicate that GPX1 regulation in primary breast cancer is complex involving both trans-activation through an AP-2 regulatory region and epigenetic chromatin modification through CpG methylation. Furthermore, our data indicates a unique role for TFAP2C regulation of GPX1 compared to the related family member, TFAP2A. A recent study reported that interference with AP-2 expression in a murine breast cancer cell line increased apoptosis and augmented sensitivity to doxorubicin chemotherapy (43). Although GPX1 was not directly investigated in that study, the effects of TFAP2C in mediating ROS and sensitivity to anthracyclines through GPX1 could account for the effects of AP-2 in mediating anthracycline sensitivity. Additionally, AP-2 factors may alter ROS in cancer cells through the regulation of c-myc and MnSOD (44, 45). Previous studies demonstrated that p53 increases oxidative stress and apoptosis through the regulation of GPX1 expression (46) and the current findings add to the growing literature suggesting important co-regulatory mechanisms involving a cooperative association of p53 and AP-2 factors (47–49).
In addition to TFAP2C induction of the GPX1 promoter through a direct interaction with the regulatory site, activation of the GPX1 promoter is subject to epigenetic modifications. We have previously demonstrated that chromatin modifications involving DNA methylation and histone deacetylation can make the ERα promoter inaccessible to TFAP2C (21). The current data expand the role for epigenetic chromatin modification of TFAP2C target genes, characterized by CpG methylation in and around the AP-2 regulatory binding site, resulting in inaccessibility of the promoter to TFAP2C. CpG methylation of the AP-2 regulatory region was proven to be functionally linked to expression by showing that treatment with drugs that inhibit DNA methylation (5-aza-dC) can re-activate expression of GPX1. Studies examining methylation of the GPX1 promoter in gastric cancer estimated that 17% of tumors had significant promoter methylation (23); our data suggests that approximately 20% of breast cancers lack GPX1 expression due to promoter methylation. Of particular significance is that discordant expression of TFAP2C and GPX1 was identified in tumors with methylation of the GPX1 promoter. This finding highlights the concern that failure to consider epigenetic modifications or other potential complexities in gene regulation may frustrate the ability to define transcriptional mechanisms that establish clinically relevant patterns in gene regulation.
Several important conclusions can be drawn from the findings in the current study. The first is that the results highlight the important role of TFAP2C in regulating genes that establish the clinically relevant breast cancer phenotype. The approach of comparing cell lines for differences in genomic targeting of TFAP2C has been a fruitful means of identifying critical regulatory regions of genes that influence the clinical phenotype of breast cancers. All of the cell lines utilized in the current study are of luminal origin, hence, our data does not directly address whether TFAP2C-mediated regulation of GPX1 is specific to luminal breast cancer or whether other mechanisms of gene regulation play a role in GPX1 expression in the basal (triple-negative) phenotype. However, it has been shown that GPX1 expression is found as a characteristic of basal cancer lines (50). Additional studies examining the pattern of genomic binding of TFAP2C in various cell lines, particularly cell lines of different clinical phenotype, should be an ongoing means to define the regulatory regions of key breast cancer genes. Demonstrating that TFAP2C can regulate the oxidative state and sensitivity to peroxides in breast cancer cells through the control of GPX1 provides evidence that TFAP2C likely has substantial influence over several processes critical to the biology of breast cancer including oncogenesis, sensitivity to certain chemotherapeutic agents and progression of metastases. Further work to examine directly the role of TFAP2C in these various physiologic processes will help to elucidate the biologic role of TFAP2 in cancer development and progression.
MATERIAL AND METHODS
Chromatin Immunoprecipitation with Direct Sequencing (ChIP-Seq)
All manipulations were done as previously described (20). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (51) and are accessible through GEO Series accession number GSE36351 and are accessible for review through the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=tvmxvweeggmmafm&acc=GSE36351
Cell lines and tissue samples
Cell lines, MCF-7, BT-474, MDA-MB-453 and SKBR-3 were obtained from American Type Culture Collection. All cell lines were grown in appropriate medium at 37 °C in a humidified 5% CO2 incubator. Breast cancer tissue specimens were obtained from patient samples from surgical resection specimens under an IRB approved protocol.
RNA isolation, cDNA synthesis and Real-Time-PCR
Total RNA was isolated from cell lines and breast tissue samples using Rneasy Mini Kit (Qiagen). cDNA was synthesized from 1 μg of total RNA with d(N)6 primer using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression was monitored by quantitative real-time PCR. TaqMan primers and detection probes for genes were GPX1 Assay Hs00829989_gH and TFAP2C Assay Hs00231476_m1 (Apply Biosystems). Expression values were normalized to the mean of 18S rRNA 4319413E-0803037 (Applied Biosystems) as endogenous control.
DNA isolation and sodium bisulfite modification
Genomic DNA was isolated from cells and tissues using QIAamp DNA Mini kit (Qiagen). The bisulfite conversion and cleanup of DNA for methylation analysis was performed by EpiTect Bisulfite Kit (Qiagen). The EpiTect Whole Bisulfitome Kit (Qiagen) was used for amplify bisulfite converted DNA that was used for subsequent assays.
Western blots
Western blots were done using the following primary antibodies: TFAP2-α 3B5, TFAP2-γ 6E4/4, GPDH 6C5 (Santa Cruz Biotechnology) and GPX1 3120-1 (Epitomics).
Transient transfection
Transfections with siRNA for TFAP2A and TFAP2C were performed as described (11). Non-targeting siRNA was obtained from Dharmacon as Non-TARGET #2 siCONTROL (Cat# D-001208-14-20). Cells were harvested after 96h incubation.
5′-aza-deoxycytidine (5′-aza-dC) treatment
Cells (6 × 105 cells/100 mm dish) were seeded in 10% FCS in corresponding media. After 24 h the cells were treated with 2.5 μM 5′-aza-dC (Sigma-Aldrich). Cells were harvested after 120h incubation and used for both RNA extraction and Western Blot assays.
Bisulfite sequencing of genomic DNA
Bisulfite-converted genomic DNA was used as a template to amplify fragments of 220–351 bp with a high CpG content from around the TSS of GPX1. Primers designed using the MethPrimer software (http://www.urogene.org/methprimer/:), were GPX1_L1 AGGGAGTTTAGGTTTATAGGTTTTT GPX1_R1 TAACCTCCCCTTACAATACTTATTC; GPX1_L2 AGTATTGTAAGGGGAGGTTAGTAGG and GPX1_R2 AATTACCTAAACCAAACCAAACATAC. Amplified PCR fragments were sequenced directly.
Gel-Shift Assay
Gel Shift Assay Core System (Cat# E3050, Promega) was used, a 60bp probe (NCBI36/hg18 chr3:49,370,821-49,370,881) was labeled and TFAP2C protein was generated from pcDNA3.1-AP2C (52) using TNT T7 Quick Coupled Transcription/Translation System Cat#L1170 (Promega). Cold double stranded competitors were made as follows (possible binding sites are underlined): GPX1_5 CAGTTTTCCGGGCCTGAGGG, GPX1_6 TCCCGCCTCATCCGGCCCCC, GPX1_7 GCCCTACCCGGAGGGCTCGG, and GPX1_5* TTCCGGGCCTGAGGGTCCCG. 2 μg Abs AP-2γ (6E4/4) (Santa Cruz Biotechnology) were added per reaction as needed.
GPX Activity Assay
The activity of GPX1 was conducted using Glutathione Peroxidase Cellular Activity Assay Kit Cat# CPG1 (Sigma-Aldrich).
Tert-butyl-OOH sensitivity Assay
The tert-butyl hydroperoxide assay was performed as described with slight modifications (53). BT-474 were transfected in 6-well dishes with either NT or TFAP2C siRNA for 48 hours as described above. MCF-7 cells were seeded in 6-well dishes and transduced with 100 MOI of adenovirus harboring wild-type human TFAP2C, wild-type human GPX-1, or empty vector (all vectors obtained from the Gene Transfer Vector Core Facility, University of Iowa) and allowed to incubate for 24 hours. Treated cells were trypsinized and seeded into 96-well plates at approximately 104 cells/well. Following overnight incubation, cells were exposed to varying concentrations of tert-butyl hydroperoxide in phenol red- and serum-free DMEM (MCF-7) or Optimem (BT-474) for 24 hours. Media was removed and replaced with media containing 0.5 mg/ml MTT, and incubated for 3 hours at 37° C. This media was replaced with 50 ul of DMSO to solubilize the formazan crystals, and absorbance was read on a Tecan Infinite 200 plate reader at 570 nm with a reference of 630 nm.
Acknowledgments
We appreciate tumor samples provided by Dr. Ryan Askeland and Christine Hochstedler (Department of Pathology, University of Iowa) and statistical support from Dr. Junlin Liao (Department of Surgery, University of Iowa). This work was supported in part by the National Institutes of Health grants R01CA109294 (PI: R. J. Weigel) and R01CA115438 (PI: F. E. Domann). Anthony Cyr received salary support from NIH F30 AA019856 and Dr. Philip Spanheimer was supported by the NIH grant T32CA148062 (PI: R. J. Weigel). The work was also supported by a generous gift from the Kristen Olewine Milke Breast Cancer Research Fund.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buzdar AU. Role of biologic therapy and chemotherapy in hormone receptor-and HER2-positive breast cancer. Ann Oncol. 2009 Jun;20(6):993–9. doi: 10.1093/annonc/mdn739. [Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 4.Williams T, Admon A, Luscher B, Tjian R. Cloning and expression of AP-2, a cell-type-specific transcription factor that activates inducible enhancer elements. Genes Dev. 1988;2(12A):1557–69. doi: 10.1101/gad.2.12a.1557. [DOI] [PubMed] [Google Scholar]
- 5.Bosher JM, Totty NF, Hsuan JJ, Williams T, Hurst HC. A family of AP-2 proteins regulates c-erbB-2 expression in mammary carcinoma. Oncogene. 1996;13(8):1701–7. [PubMed] [Google Scholar]
- 6.Feng W, Williams T. Cloning and characterization of the mouse AP-2 epsilon gene: a novel family member expressed in the developing olfactory bulb. Mol Cell Neurosci. 2003;24(2):460–75. doi: 10.1016/s1044-7431(03)00209-4. [DOI] [PubMed] [Google Scholar]
- 7.Moser M, Imhof A, Pscherer A, Bauer R, Amselgruber W, Sinowatz F, et al. Cloning and characterization of a second AP-2 transcription factor: AP-2 beta. Development. 1995;121(9):2779–88. doi: 10.1242/dev.121.9.2779. [DOI] [PubMed] [Google Scholar]
- 8.Zhao F, Satoda M, Licht JD, Hayashizaki Y, Gelb BD. Cloning and characterization of a novel mouse AP-2 transcription factor, AP-2delta, with unique DNA binding and transactivation properties. J Biol Chem. 2001;276(44):40755–60. doi: 10.1074/jbc.M106284200. [DOI] [PubMed] [Google Scholar]
- 9.Eckert D, Buhl S, Weber S, Jager R, Schorle H. The AP-2 family of transcription factors. Genome Biol. 2005;6(13):246. doi: 10.1186/gb-2005-6-13-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McPherson LA, Baichwal VR, Weigel RJ. Identification of ERF-1 as a member of the AP2 transcription factor family. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4342–7. doi: 10.1073/pnas.94.9.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodfield GW, Horan AD, Chen Y, Weigel RJ. TFAP2C controls hormone response in breast cancer cells through multiple pathways of estrogen signaling. Cancer Res. 2007 Sep 15;67(18):8439–43. doi: 10.1158/0008-5472.CAN-07-2293. [DOI] [PubMed] [Google Scholar]
- 12.Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, et al. AP-2gamma regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. Embo J. 2011 Jul 6;30(13):2569–81. doi: 10.1038/emboj.2011.151. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Begon DY, Delacroix L, Vernimmen D, Jackers P, Winkler R. Yin Yang 1 cooperates with activator protein 2 to stimulate ERBB2 gene expression in mammary cancer cells. J Biol Chem. 2005 Jul 1;280(26):24428–34. doi: 10.1074/jbc.M503790200. [DOI] [PubMed] [Google Scholar]
- 14.Delacroix L, Begon D, Chatel G, Jackers P, Winkler R. Distal ERBB2 promoter fragment displays specific transcriptional and nuclear binding activities in ERBB2 overexpressing breast cancer cells. DNA Cell Biol. 2005 Sep;24(9):582–94. doi: 10.1089/dna.2005.24.582. [DOI] [PubMed] [Google Scholar]
- 15.Yang JW, Lee EY, Kang KW. ErbB2 overexpression in p53-inactivated mammary epithelial cells. FEBS Lett. 2006 Nov 27;580(27):6501–8. doi: 10.1016/j.febslet.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 16.Ailan H, Xiangwen X, Daolong R, Lu G, Xiaofeng D, Xi Q, et al. Identification of target genes of transcription factor activator protein 2 gamma in breast cancer cells. BMC Cancer. 2009;9:279. doi: 10.1186/1471-2407-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allouche A, Nolens G, Tancredi A, Delacroix L, Mardaga J, Fridman V, et al. The combined immunodetection of AP-2alpha and YY1 transcription factors is associated with ERBB2 gene overexpression in primary breast tumors. Breast Cancer Res. 2008;10(1):R9. doi: 10.1186/bcr1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner BC, Zhang J, Gumbs AA, Maher MG, Kaplan L, Carter D, et al. Expression of AP-2 transcription factors in human breast cancer correlates with the regulation of multiple growth factor signalling pathways. Cancer Res. 1998;58(23):5466–72. [PubMed] [Google Scholar]
- 19.Pellikainen J, Naukkarinen A, Ropponen K, Rummukainen J, Kataja V, Kellokoski J, et al. Expression of HER2 and its association with AP-2 in breast cancer. Eur J Cancer. 2004 Jul;40(10):1485–95. doi: 10.1016/j.ejca.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 20.Woodfield GW, Chen Y, Bair TB, Domann FE, Weigel RJ. Identification of primary gene targets of TFAP2C in hormone responsive breast carcinoma cells. Genes Chromosomes Cancer. 2010 Oct;49(10):948–62. doi: 10.1002/gcc.20807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodfield GW, Hitchler MJ, Chen Y, Domann FE, Weigel RJ. Interaction of TFAP2C with the estrogen receptor-alpha promoter is controlled by chromatin structure. Clin Cancer Res. 2009 Jun 1;15(11):3672–9. doi: 10.1158/1078-0432.CCR-08-2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4(7):e6146. doi: 10.1371/journal.pone.0006146. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jee CD, Kim MA, Jung EJ, Kim J, Kim WH. Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur J Cancer. 2009 May;45(7):1282–93. doi: 10.1016/j.ejca.2008.12.027. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 24.Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001;3(5):323–7. doi: 10.1186/bcr315. [Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010 May;44(5):479–96. doi: 10.3109/10715761003667554. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006 Sep;10(3):175–6. doi: 10.1016/j.ccr.2006.08.015. [Comment] [DOI] [PubMed] [Google Scholar]
- 27.Maiti AK. Genetic determinants of oxidative stress-mediated sensitization of drug-resistant cancer cells. Int J Cancer. 2012 Jan 1;130(1):1–9. doi: 10.1002/ijc.26306. [DOI] [PubMed] [Google Scholar]
- 28.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011 Jul 14;475(7355):231–4. doi: 10.1038/nature10167. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Esposito LA, Kokoszka JE, Waymire KG, Cottrell B, MacGregor GR, Wallace DC. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic Biol Med. 2000 Mar 1;28(5):754–66. doi: 10.1016/s0891-5849(00)00161-1. [In Vitro Research Support, U.S. Gov’t, P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu YJ, Diamond AM. Role of glutathione peroxidase 1 in breast cancer: loss of heterozygosity and allelic differences in the response to selenium. Cancer research. 2003 Jun 15;63(12):3347–51. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] [PubMed] [Google Scholar]
- 31.Ravn-Haren G, Olsen A, Tjonneland A, Dragsted LO, Nexo BA, Wallin H, et al. Associations between GPX1 Pro198Leu polymorphism, erythrocyte GPX activity, alcohol consumption and breast cancer risk in a prospective cohort study. Carcinogenesis. 2006 Apr;27(4):820–5. doi: 10.1093/carcin/bgi267. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 32.Cox DG, Tamimi RM, Hunter DJ. Gene x Gene interaction between MnSOD and GPX-1 and breast cancer risk: a nested case-control study. BMC Cancer. 2006;6:217. doi: 10.1186/1471-2407-6-217. [Research Support, N.I.H., Extramural] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhuo P, Goldberg M, Herman L, Lee BS, Wang H, Brown RL, et al. Molecular consequences of genetic variations in the glutathione peroxidase 1 selenoenzyme. Cancer research. 2009 Oct 15;69(20):8183–90. doi: 10.1158/0008-5472.CAN-09-1791. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J, Zhou GW, Wang N, Wang YJ. GPX1 Pro198Leu polymorphism and breast cancer risk: a meta-analysis. Breast cancer research and treatment. 2010 Nov;124(2):425–31. doi: 10.1007/s10549-010-0841-z. [Meta-Analysis] [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Benya RV, Carroll RE, Diamond AM. Allelic loss of the gene for the GPX1 selenium-containing protein is a common event in cancer. J Nutr. 2005 Dec;135(12 Suppl):3021S–4S. doi: 10.1093/jn/135.12.3021S. [Research Support, N.I.H., Extramural Review] [DOI] [PubMed] [Google Scholar]
- 36.Townsend AJ, Morrow CS, Sinha BK, Cowan KH. Selenium-dependent glutathione peroxidase expression is inversely related to estrogen receptor content of human breast cancer cells. Cancer Commun. 1991 Aug;3(8):265–70. doi: 10.3727/095535491820873119. [DOI] [PubMed] [Google Scholar]
- 37.Jenssen TK, Kuo WP, Stokke T, Hovig E. Associations between gene expressions in breast cancer and patient survival. Hum Genet. 2002 Oct;111(4–5):411–20. doi: 10.1007/s00439-002-0804-5. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 38.Gouaze V, Mirault ME, Carpentier S, Salvayre R, Levade T, Andrieu-Abadie N. Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol Pharmacol. 2001 Sep;60(3):488–96. [Research Support, Non-U.S. Gov’t] [PubMed] [Google Scholar]
- 39.Vibet S, Goupille C, Bougnoux P, Steghens JP, Gore J, Maheo K. Sensitization by docosahexaenoic acid (DHA) of breast cancer cells to anthracyclines through loss of glutathione peroxidase (GPx1) response. Free Radic Biol Med. 2008 Apr 1;44(7):1483–91. doi: 10.1016/j.freeradbiomed.2008.01.009. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 40.Gao J, Xiong Y, Ho YS, Liu X, Chua CC, Xu X, et al. Glutathione peroxidase 1-deficient mice are more susceptible to doxorubicin-induced cardiotoxicity. Biochim Biophys Acta. 2008 Oct;1783(10):2020–9. doi: 10.1016/j.bbamcr.2008.05.027. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006 Sep;10(3):241–52. doi: 10.1016/j.ccr.2006.08.009. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.] [DOI] [PubMed] [Google Scholar]
- 42.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011 Sep 13;20(3):300–14. doi: 10.1016/j.ccr.2011.08.012. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thewes V, Orso F, Jager R, Eckert D, Schafer S, Kirfel G, et al. Interference with activator protein-2 transcription factors leads to induction of apoptosis and an increase in chemo- and radiation-sensitivity in breast cancer cells. BMC Cancer. 2010;10:192. doi: 10.1186/1471-2407-10-192. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu L, Hitchler MJ, Sun W, Sarsour EH, Goswami PC, Domann FE. TFAP2A inhibits c-MYC induced oxidative stress and apoptosis in HaCaT human keratinocytes. J Oncol. 2009 doi: 10.1155/2009/780874. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu CH, Huang Y, Oberley LW, Domann FE. A family of AP-2 proteins down-regulate manganese superoxide dismutase expression. The Journal of biological chemistry. 2001 Apr 27;276(17):14407–13. doi: 10.1074/jbc.M009708200. [Research Support, U.S. Gov’t, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 46.Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer research. 2004 Apr 1;64(7):2350–6. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 47.McPherson LA, Loktev AV, Weigel RJ. Tumor Suppressor Activity of AP2alpha Mediated through a Direct Interaction with p53. J Biol Chem. 2002;277(47):45028–33. doi: 10.1074/jbc.M208924200. [DOI] [PubMed] [Google Scholar]
- 48.Scibetta AG, Wong PP, Chan KV, Canosa M, Hurst HC. Dual association by TFAP2A during activation of the p21cip/CDKN1A promoter. Cell Cycle. 2010 Nov 15;9(22):4525–32. doi: 10.4161/cc.9.22.13746. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hannay JA, Liu J, Zhu QS, Bolshakov SV, Li L, Pisters PW, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther. 2007 May;6(5):1650–60. doi: 10.1158/1535-7163.MCT-06-0636. [Research Support, N.I.H., Extramural] [DOI] [PubMed] [Google Scholar]
- 50.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast cancer research and treatment. 2004 Feb;83(3):249–89. doi: 10.1023/B:BREA.0000014042.54925.cc. [Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 51.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002 Jan 1;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McPherson LA, Weigel RJ. AP2alpha and AP2gamma: a comparison of binding site specificity and transactivation of the estrogen receptor promoter and single site promoter constructs. Nucleic Acids Res. 1999;27(20):4040–9. doi: 10.1093/nar/27.20.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johansson K, Jarvliden J, Gogvadze V, Morgenstern R. Multiple roles of microsomal glutathione transferase 1 in cellular protection: a mechanistic study. Free Radic Biol Med. 2010 Dec 1;49(11):1638–45. doi: 10.1016/j.freeradbiomed.2010.08.013. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]