

Abstract
Cardiac myosin binding protein-C (cMyBP-C), a sarcomeric protein with 11 domains, C0-C10, binds to the myosin rod via its C-terminus, while its N-terminus binds regions of the myosin head and actin. These N-terminal interactions can be attenuated by phosphorylation of serines in the C1-C2 motif linker. Within the sarcomere, cMyBP-C exists in a range of phosphorylation states, which may affect its ability to regulate actomyosin motion generation. To examine the functional importance of partial phosphorylation, we bacterially expressed N-terminal fragments of cMyBP-C (domains C0-C3) with 3 of its phosphorylatable serines (S273, S282, and S302) mutated in combinations to either aspartic acids or alanines, mimicking phosphorylation and dephosphorylation respectively. The effect of these C0-C3 constructs on actomyosin motility were characterized in both the unloaded in vitro motility assay and in the load-clamped laser trap assay where force:velocity (F:V) relations were obtained. In the motility assay, phosphomimetic replacement (i.e. aspartic acid) reduced the slowing of actin velocity observed in the presence of C0-C3 in proportion to the total number phosphomimetic replacements. Under load, C0-C3 depressed the F:V relationship without any effect on maximal force. Phosphomimetic replacement reversed the depression of F:V by C0-C3 in a graded manner with respect to the total number of replacements. Interestingly, the effect of C0-C3 on F:V was well fitted by a model that assumed C0-C3 acts as an effective viscous load against which myosin must operate. This study suggests that increasing phosphorylation of cMyBP-C incrementally reduces its modulation of actomyosin motion generation providing a tunable mechanism to regulate cardiac function.
Keywords: Force-Velocity, viscosity, motility assay, laser trap, protein kinase A, contractility
Introduction
Striated muscle's ability to generate motion under externally applied loads was first described by A.V. Hill as a hyperbolic relationship between force and velocity, with slower velocities associated with greater loads (Hill 1938). Unloaded shortening velocity differs dramatically among the various muscle types (e.g. smooth, cardiac, and skeletal). It was in 1967 that Michael Bárány elegantly demonstrated that variation in shortening velocities were directly correlated to the underlying hydrolysis rate of MgATP (Bárány 1967); a process involving the cyclic interaction between myosin and actin. In addition, both Michael and Kate Bárány contributed significantly to our understanding of how phosphorylation of muscle contractile and regulatory proteins provided a means of modulating muscle's force and motion generation (for review see Bárány and Bárány 1980). Cardiac myosin binding protein-C (cMyBP-C) is one such regulatory protein that modulates actomyosin function in the heart in a phosphorylation-dependent manner, which is the focus of the work presented below.
Mutations in cMyBP-C are a leading cause of hypertrophic cardiomyopathy (HCM), emphasizing its importance to normal cardiac function (Harris et al. 2011). In the sarcomere, cMyBP-C is localized in 7-9 stripes across the A-band (Craig and Offer 1976), due to its attachment to the thick filament via its C-terminus (Moos et al. 1975). The N-terminus of cMyBP-C can bind actin, myosin S2, regulatory light chain, and possibly the myosin head; presumably these interactions govern cMyBP-C's capacity to modulate actomyosin motion generation (Moos et al. 1978; Gruen and Gautel 1999; Ratti et al. 2011; Witt et al. 2001). Specifically, cMyBP-C slows myosin's translocation of actin filaments in the in vitro motility assay (Razumova et al. 2006; Saber et al. 2008; Shchepkin et al. 2010) and its elimination in fibers either chemically or genetically leads to faster unloaded shortening velocities and kinetics of tension recovery in response to a length change (Hofmann et al. 1991a; Stelzer et al. 2006a). This inhibitory effect could result from cMyBP-C's N-terminus binding myosin and restricting the motion of the myosin head or altering the kinetics of myosin's motion generation (Hofmann et al. 1991b; Calaghan et al. 2000). Alternatively, cMyBP-C's N-terminus could bind actin and affect thin filament regulation (Razumova et al. 2008), compete for myosin binding sites (Saber et al. 2008), or act as a tether to provide a viscous load that myosin must work against (Palmer et al. 2004; Weith et al. 2012).
Phosphorylation of cMyBP-C in response to β-adrenergic stimulation is a major determinant of enhanced contractility (Barefield and Sadayappan 2010). There are 4 phosphorylatable serines (in mice: S273, S282, S302, and S307), which are located in a cardiac specific insert within the highly conserved motif linker between domains C1 and C2 (Gautel et al. 1995; Jia et al. 2010). These sites are phosphorylated hierarchically, with S282 phosphorylated first, which is permissive for S302 and possibly S273 phosphorylation (Gautel et al. 1995; Sadayappan et al. 2011). Phosphorylation may regulate cMyBP-C's effects on actomyosin contractility. For example, in cardiac fibers, force at sub-maximal calcium levels is increased with cMyBP-C phosphorylation (McClellan et al. 2001) as are rates of force development and thus crossbridge kinetics following a stretch (Stelzer et al. 2006b; Tong et al. 2007). These changes in actomyosin force generation may result from a reduction in cMyBP-C's ability to bind the myosin S2 and/or actin due to phosphorylation (Gruen et al. 1999; Shaffer et al. 2009; Weith et al. 2012), though its effect on actin binding is still debated (Rybakova et al. 2011). cMyBP-C is substantially phosphorylated in the hearts of humans and mice (van Dijk et al. 2009; Sadayappan et al. 2005; Sadayappan et al. 2006; Jacques et al. 2008; Copeland et al. 2010), which may be important in maintaining contractile reserve (Nagayama et al. 2007), whereas cMyBP-C dephosphorylation is characteristic of heart failure (Sadayappan et al. 2005; Jacques et al. 2008; Copeland et al. 2010). However, data are lacking that characterize how the overall level of cMyBP-C phosphorylation influences cMyBP-C function, let alone the contribution of the individual phosphorylation sites. Is the effect of phosphorylation on cMyBP-C function graded with increasing levels of overall phosphorylation or is phosphorylation of a specific site or sites sufficient to give the same effect as the fully phosphorylated motif?
To address this question, we characterized the inhibitory effect of bacterially expressed N-terminal cMyBP-C fragments (C0-C3) on myosin's translocation of actin filaments in the motility assay both in the absence and presence of an applied load (Debold et al. 2005). We have shown previously that this short N-terminal fragment recapitulates the inhibition of actin filament velocity as observed for whole cMyBP-C (Weith et al.2012). In the present study, 8 mutant C0-C3 constructs were generated with combinations of either aspartic acids or alanines substituted for S273, S282 and S302 to mimic phosphorylation and dephosphorylation, respectively (see Fig.1). The results demonstrated that C0-C3 slows actomyosin motion generation in the absence of load and depresses the force:velocity (F:V) relationship for a small ensemble of myosin molecules without any effect on myosin's maximum force generation. This depression (i.e. inhibition) was modulated by C0-C3 phosphomimetic replacement in a graded manner proportional to the total number of phosphomimetics at S273, S282, and S302 without any apparent site-specific dependence. The depressed F:V in the presence of C0-C3 was modeled assuming that C0-C3 acted as a viscous load to alter actomyosin kinetics.
Fig. 1.
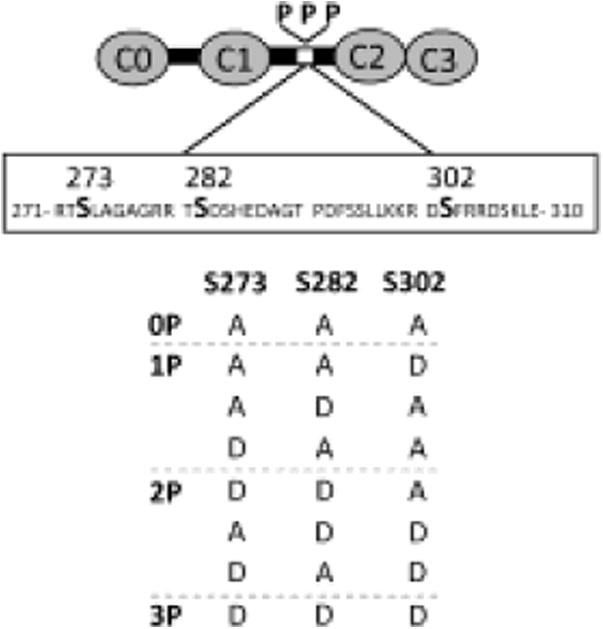
Schematic of bacterially-expressed C0-C3 fragments. Three serines within the cMyBP-C motif (S273, S282, S302) were replaced with a combination of either alanines (A) to mimic dephosphorylation or aspartic acids (D) to mimic phosphorylation, generating 8 constructs with each D as a phosphomimetic (P).
Materials and Methods
Proteins
Myosin and actin were prepared from chicken pectoralis (Margossian and Lowey 1982; Pardee and Spudich 1982). Actin filaments were fluorescently labeled with equimolar tetramethyl-rhodamine-phalloidin (TRITC) (Warshaw et al. 1990). C0-C3 fragments were bacterially expressed from mouse cardiac cDNA using a pET expression system (Novagen, Madison, WI) (Sadayappan et al. 2005). Eight different phosphomimetic C0-C3 fragments were expressed, with either aspartic acids (D) to mimic phosphorylation or alanines (A) to mimic dephosphorylation at S273, S282 or S302 (Figure 1). The various mutant constructs will be referred to as C0-C3XXX, where X is either a D or A replacement and XXX refers to residue order 273, 282, and 302. As a control, C0-C3 was fully phosphorylated (i.e. 96%) at all 4 serines following protein kinase A (PKA) treatment as described previously (Weith et al. 2012). A C0-C3 construct was also expressed containing a biotin-tag on its C-terminus that was used to quantify the abundance of C0-C3 on the motility surface.
In Vitro Motility Assay
The in vitro motility assay has been described previously (Palmiter et al. 2000). In short, 100 μg/ml of myosin was added to a nitrocellulose-coated flowcell, which was then blocked with bovine serum albumin (BSA). Next, unlabeled actin was incubated in the flowcell for 1 minute and then washed with 1 mM ATP actin buffer (AB) (25 mM KCl, 1 mM EGTA, 10 mM DTT, 25 mM imidazole, 4 mM MgCl2, adjusted to pH 7.4; containing an oxygen scavenging system (0.1 μg/mL glucose oxidase, 0.018 μg/mL catalase, 2.3 μg/mL glucose) to eliminate myosin that irreversibly binds actin in an ATP-insensitive manner (Palmiter et al. 2000). TRITC-actin was incubated in the flow cell for 1 minute and then the flow cell was washed three times with AB. Motility buffer (AB containing 1 mM ATP, 0.5% methyl cellulous and 1 μM of the desired C0-C3 fragment) was added to the flow cell and actin motility was observed at 30° C after 3 minutes of incubation. A Nikon Eclipse Ti-U epiflourescent microscope and a Mega Z camera (Stanford Photonics) utilizing Piper software were used to acquire the images at 10 frames/s, the image stacks were down-sampled to 2 frames/s, and Dia Track 3.03 software for Windows (Semasopht) was employed for data analysis.
Load-Clamp Laser Trap Assay
The load-clamp laser trap assay and its use to determine the F:V for a small ensemble of myosin has been described in detail (Debold et al. 2005; Debold et al. 2007). The assay is a modification of the traditional three bead laser trap assay (Guilford et al. 1997) where the addition of a feedback system allows constant loads to be applied to an actin filament being translocated by myosin. Since the laser trap acts like a Hookean spring, maintaining a constant distance between the trap center and a bead attached to one end of an actin filament, results in a constant force being applied. The position of the bead relative to the trap center was monitored on a quadrant photodiode detector and this signal was used in a custom software program to drive an acousto-optic deflector (NEOS Technologies) so that desired loads could be applied. A series of loads were applied, increasing from 1-8 pN and then descending back to 1 pN with the duration of each load varying between 100-180 ms. Velocity at each applied load was determined from the linear regression through the displacement-time data for each load (Fig. 2).
Fig. 2.
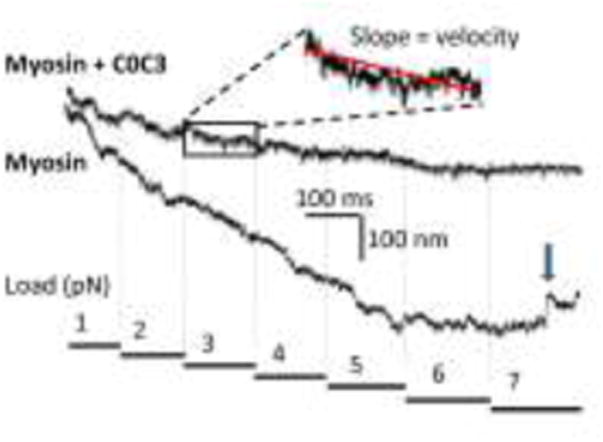
Example force-clamp laser trap assay data used to generate F:V. A series of constant loads (figure bottom) are applied to the actin filament against which the myosin must operate. Shown are two raw data traces, one for myosin alone and one for myosin in the presence of C0-C3. The traces show actin filament displacement versus time. To analyze such data, a section of the trace corresponding to a single load is fit with a linear regression, the slope of which is the velocity at that load. Note the slower velocities in the presence of C0-C3. The arrow in the myosin trace shows a slip (rapid backward movement of the actin).
Experiments took place in a 20 μl nitrocellulose-coated flowcell. Myosin (30μg/ml) was first adhered to the coverslip containing 3μm diameter beads that served as pedestals. The coverslip surface was then blocked with 0.5 mg/ml BSA for 12 minutes. Trapping buffer (AB with100 μM ATP) containing TRITC-biotin-labeled actin and 1.4 μm neutravadin-coated polystyrene beads was added to the flow cell. The actin was effectively biotinylated by incubating 1 μM actin with 0.5 μM TRITC-phalloidin and 0.5 μM biotin-phalloidin (Invitrogen, Carlsbad, CA). The neutravidin beads were prepared by incubating the beads in 20 μL AB containing 0.2 mg of neutravidin (overnight, 22 °C) followed by 4 washes in AB by reconstitution and pelleting by centrifugation to remove excess neutravidin. To manipulate a single actin filament, the neutravidin-coated beads were captured in two separate laser traps and attached at each end of an actin filament. Once an actin filament was captured, it was lowered onto the surface pedestal and as soon as the myosin engaged the actin filament, the force feedback was initiated. Control data were initially obtained in the absence of C0-C3 and then using the same flowcell, trapping buffer containing 0.2 μM C0-C3XXX was introduced and allowed to incubate for 8 minutes. The flowcell was washed three times to remove any unbound C0-C3, then trapping buffer containing TRITC-biotin-labeled actin and 1.4 μm neutravadin coated polystyrene beads was added to the flowcell and the F:V experiments were repeated. This 0.2 μM C0-C3 concentration was chosen through an iterative process so that substantial reductions in velocities at low loads nearly matched the slowing of velocity observed under unloaded conditions in the motility assay. A minimum of 500 loads over many pedestals were acquired for each condition.
Estimating C0-C3 Motility Surface Binding
The abundance of C0-C3 on the surface of nitrocellulose-coated flowcells was determined by labeling biotinylated C0-C3 with streptavadin-coated Qdots (Em:565 nm; Invitrogen) and then quantifying the density of surface-adhered Qdot-labeled C0-C3. For Qdot labeling, a 1 μM aliquot of Qdots was mixed with a 1 μM aliquot of the C0-C3 diluted in AB (1 hour, 22°C). Next, a solution was prepared that contained a 1:8000 dilution of Qdot-labeled and unlabeled C0-C3, respectively by mixing a 0.5 nM aliquot of Qdot-labeled C0-C3 with an equal aliquot of 4 μM unlabeled C0-C3. This sample mixture was serially diluted in motility buffer to give final total concentrations of C0-C3 between 0.125- 4.0 μM. The surface of the nitrocellulous-coated flowcells was prepared to match that of the motility assay experiments with an initial 1 mg/mL BSA incubation for 1 minute followed by two, 20 μl infusions of the C0-C3 mixtures. After 3 minutes, the fluorescent image of the surface with its bound Qdot-labeled and unlabeled C0-C3 (Fig. 3 A) was recorded for 100 frames at 10 frames/s in 6 separate visual fields. To determine the number of Qdots per visual field, the intensity threshold command (ImageJ, NIH) was used to make each image binary, such that Qdots were white and the background black. Then the number of Qdots was counted using the ImageJ Analyze Particles command. The number of Qdots per μm2 was multiplied by 8000 to account for the Qdot-labeled C0-C3 dilution factor (see above). The average number of Qdots per μm2 ± SD were reported from the 6 movies for each C0-C3 concentration. The data were well fitted (R2 = 0.99) with a one site binding model (Fig 3B) described by: y = Bmax(x)/(Kd +x), where y is the number of C0-C3 molecules per μm2, x is the concentration of C0-C3, and Bmax and Kd are constants of the fit. The surface of the motility chamber was nearly saturated at 4 μM C0-C3, indicating that the C0-C3 is binding to the BSA-coated surface of the flowcell rather than to itself. Extrapolation from the curve indicates that 2,100 C0-C3 molecules per μm2 are adhered to the motility surface during F:V experiments when 0.2 μM C0C3 is used.
Fig. 3.
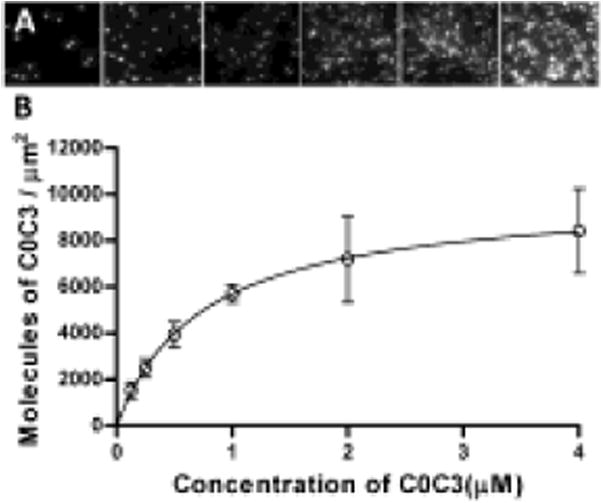
Quantification of abundance of C0-C3 on the motility surface. a) Qdot-labeled C0-C3 molecules were directly observed on the surface of nitrocellulose-coated flow cells at multiple concentrations (left to right: 0.015, 0.03, 0.0625, 0.125, 0.25, 0.50 nM; 77.6 μm2 of visual area shown for each) while in the presence of 8,000 fold excess of unlabeled C0-C3. b) The number of C0-C3 molecules per μm2 (y) with respect to C0-C3 concentration (x) was fitted with a one site binding model described by: y = Bmax(x)/(Kd +x), where Bmax = 9955 ± 95.30 (SE) C0-C3 molecules per μm2 and Kd = 0.748 ± 0.020 (SE) μM. y was obtained by multiplying the number of observed Qdot-labeled C0-C3 by 8,000 to account for the dilution with unlabeled C0-C3.
Data Analysis and Statistics
The F:V data obtained for myosin alone in the absence of C0-C3 were fitted by the classical Hill F:V equation: (F+a)(V+b) = (Fmax +a)b, where Fmax is the maximum isometric force, and a and b are constants of the fit (Hill 1938). We previously proposed that cMyBP-C may act as a viscous load to slow actin filament velocity in the motility assay (Weith et al. 2012). Therefore, we fitted the F:V data collected in the presence of the various C0-C3 fragments with a modified Hill F:V equation in which the force term, F, was replaced by F= Fapplied + Fviscous. Fapplied is the externally applied feedback force while C0-C3's effective viscous force is Fviscous = νV, where ν the viscosity coefficient. Because the data are paired (i.e. each flowcell had both a myosin control and myosin in the presence of C0-C3), values for the constants Fmax, a, and b from the myosin control F: V fit were then used in the modified Hill F:V fit to the paired C0-C3 data so that ν was the only fit parameter.
Data are presented as mean ± standard error, and all experiments were performed a minimum of 3 times. Statistical comparisons were done using the ANOVA test with a Bonferroni correction for multiple comparisons. P < 0.05 was considered significant.
Results
Actin Filament Velocity Slowing by C0-C3 Phosphomimetic Constructs
We recently reported that C0-C3 slowed actin filament velocity in the motility assay in a concentration-dependent manner and that 96% phosphorylation at all four serines with PKA treatment reduced C0-C3's inhibitory effect (Weith et al. 2012). With the use of the C0-C3 phosphomimetic mutants in the motility assay, we could then establish the relative importance of individual phosphorylation sites in modulating C0-C3's inhibitory capacity versus the effect of changing the overall phosphorylation level. We used 1 μM of the various C0-C3 phosphomimetic constructs as our standard of comparison since this concentration of PKA-treated C0-C3 previously showed a nearly complete reversal in the inhibition of actin filament velocity (Weith et al. 2012). Unphosphorylated C0-C3 and its mutant counterpart, C0-C3AAA, significantly (p < 0.01) decreased actin sliding velocity by 43% to 2.9±0.1 μm/s compared to 5.1 ± 0.3 μm/s for myosin alone in the absence of C0-C3 (Fig. 4). As a group, the single-site phosphomimetics (C0-C3DAA, C0-C3ADA, C0-C3AAD) were less inhibitory, reducing actin filament velocity 31% to 3.5 ± 0.2 μm/s (p < 0.05). The double-site phosphomimetics (C0-C3ADD, C0-C3DAD, C0-C3DDA) were even less inhibitory, giving a 16% reduction in velocity of 4.3 ± 0.2 μm/s (p < 0.01). Finally, the triple-site phosphomimetic, C0-C3DDD, had the same minimal effect on velocity (4.6 ± 0.2 μm/s) as the PKA-treated C0-C3 (4.7 ± 0.2 μm/s). The single- and double-site phosphomimetics were grouped for comparison purposes since there was no significant difference in the inhibitory capacity of fragments with the same number of phosphomimetic replacements regardless of the aspartic acid(s) position.
Fig. 4.
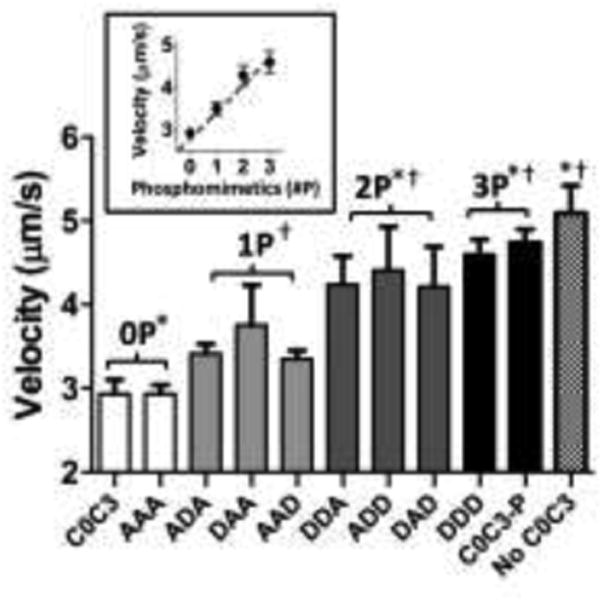
Actin filament velocities in the presence of C0-C3 phosphomimetic fragments. Data are mean ± standard error (n = 4). † represents significant difference from unphosphorylated C0-C3 (0P) (p<0.05). * represents significant difference from single-site phosphomimetics (1P) (p<0.05). Inset shows velocities averaged across levels of phosphomimetic replacement. There were no significant differences between C0-C3 phosphomimetics at the same level of phosphorylation. C0-C3-P is PKA-treated C0-C3.
F:V Relations are Depressed by C0-C3 Phosphomimetic Constructs
The F:V curve for a small ensemble of skeletal muscle myosin molecules (∼10 heads; Harris et al. 1994), experiencing 1-8 pN of applied load, was well fitted by the classic Hill equation (See Methods) (Fig. 5 (all black F:V), Table 1), with a curvature parameter defined by a/Fmax of 0.3, similar to that observed in whole skeletal muscle (Woledge et al. 1985). The maximum force estimated from the F:V fit was 11±1 pN, giving an estimate of 1.1 pN per myosin head. When unphosphorylated C0-C3 was added to the flowcell (Fig. 5 A), there was an overall depression of the F:V with the velocity at 1 pN load reduced by 53%, similar to that observed in the motility assay under unloaded conditions (Fig. 4). As with the motility assay experiments, a similar depression in the F:V was observed for the C0-C3AAA , once again confirming that this construct serves as a model for unphosphorylated C0-C3. All three single-site phosphomimetic constructs had lesser impacts on F:V (Fig 5B), which was even more pronounced in the double-site phosphomimetic constructs (Figure 5C). Neither the triple-site phosphomimetic C0-C3DDD nor the PKA-treated C0-C3 altered the F:V and thus were no different than the F:V for myosin alone (Figure 5D).
Fig. 5.
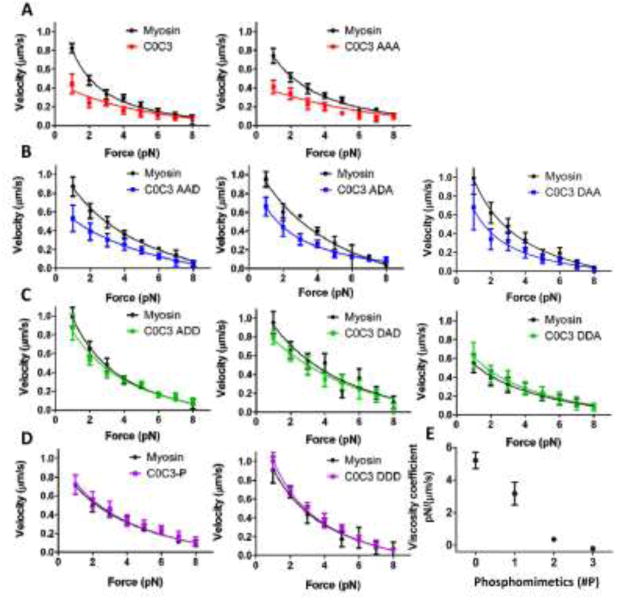
F:V for C0-C3 phosphomimetics in the load-clamped laser trap assay. Panels (a-d) show paired data where myosin F:V data were obtained first as control and then C0-C3 fragments were introduced into the same flowcell and the F:V determined again. Myosin curves were fit to the Hill F:V, while all C0-C3 fragment curves were fit to a modified F:V with an added viscous force term (see Methods). Each curve contains a minimum of 500 individual loads. Values are mean ± standard error. a) F:V of unphosphorylated C0-C3 constructs. b) Single-site phosphomimetic C0-C3 constructs. c) Double-site phosphomimetic C0-C3 constructs. d) PKA-treated and triple-site phosphomimetic C0-C3 construct. Parameters of the fits are listed in Table 1. e) Viscosity coefficients determined from modified F:V fit to the phosphomimetic C0-C3 data. Viscosity coefficients were averaged by number of phosphomimetic replacements. Error bars represent standard deviation. For each construct, individual viscosity coefficients are listed in Table 1.
Table 1.
F:V parameters. F:V data for myosin alone and in the presence of the various C0-C3 phosphomimetic constructs were fitted to a modified Hill F:V relationship with the addition of a viscous force term (see Methods). The actual curve fits are shown in Fig. 5. The fit parameters are provided as the mean ± standard error of the fit.
| Construct | Viscosity coefficient | a | b | Fmax |
|---|---|---|---|---|
| pN/(μm/s) | (pN) | (μm/s) | (pN) | |
| C0C3 | 4.7 ± 1.0 | 1.0 ± 0.4 | 0.1 ± 0.1 | 15 ± 4 |
| AAA | 5.7 ± 1.3 | 4.5 ± 1.0 | 0.2 ± 0.1 | 15 ± 4 |
| AAD | 4.1 ± 0.5 | 2.4 ± 0.8 | 0.5 ± 0.1 | 10 ± 1 |
| ADA | 2.7 ± 0.5 | 4.7 ± 2.5 | 0.4 ± 0.2 | 9 ± 1 |
| DAA | 2.7 ± 0.8 | 2.0 ± 0.6 | 0.4 ± 0.1 | 9 ± 1 |
| ADD | 0.5 ± 0.2 | 2.0 ± 0.3 | 0.3 ± 0.04 | 10 ± 1 |
| DAD | 0.8 ± 0.3 | 4.5 ± 3.9 | 0.5 ± 0.4 | 12 ± 4 |
| DDA | −0.2 ± 0.1 | 6.6 ± 2.4 | 0.5 ± 0.2 | 11 ± 1 |
| DDD | −0.2 ± 0.1 | 2.7 ± 1.0 | 0.4 ± 0.1 | 9 ± 1 |
| C0C3-P | −0.2 ± 0.2 | 4.2 ± 1.0 | 0.4 ± 0.1 | 11 ± 1 |
| Myosin | na | 3.0 ± 0.8 | 0.3 ± 0.1 | 11 ± 1 |
Our previous work suggested that C0-C3 may act as a viscous load on myosin (Weith et al. 2012). Thus, assuming this model, we fitted the F:V data for all C0-C3 constructs with a modified Hill equation (average R2=0.94) that included a viscous load, Fviscous (see Methods). By fitting the F:V with this model, we could estimate C0-C3's effective viscosity coefficient, ν, which served as a means for comparing the inhibitory effects of the various C0-C3 constructs on F:V. ν varied in a graded manner as a function of the number of phosphomimetic replacements (Fig. 5E, Table 1). The highest ν were associated with C0-C3 (4.7±1.0 pN·s/μm) and C0-C3AAA (5.7±1.3 pN·s/μm) with the lowest, effectively zero, for the PKA-treated C0-C3 (−0.2±0.2 pN·s/μm) and its triple-site phosphomimetic model, C0-C3DDD (−0.2±0.1 pN·s/μm), suggesting that fully phosphorylated C0-C3 has no appreciable effect on the F:V relationship. The Fmax being similar for all the constructs (Table 1) shows that C0-C3, regardless of the number of phosphomimetic replacements, has no effect on myosin's maximum force generation. Although the a and b Hill equation fit parameters may differ for the various phosphomimetic constructs, we do not draw any conclusions from these differences as investigators in the past have not been able to relate these parameters to any meaningful physiological property of the underlying contractile system (Woledge et al. 1985). However, a more recent model proposes that strained myosin attached to a moving actin filament exists in a mechanical feedback loop that depends on the rate of actin filament sliding and that the model provides an analytical basis for the Hill F:V parameters (Yadid et al. 2011).
C0-C3 Prevents Sudden Displacements of the Actin Filament Under Load
We have reported previously (Debold et al. 2005) in similar F:V experiments that as myosin moves an actin filament against a constant load, forward motion is occasionally interrupted by a rapid backward movement of the actin filament (i.e. >20 nm within 3 ms), termed a “slip” (see inset in Figure 6, and arrow in Figure 2). In the present study similar 37±3 nm slips were observed with myosin alone with the frequency of slips increasing with applied load (Fig. 6). However, in the presence of unphosphorylated C0-C3, the frequency of slips at nearly every load was dramatically reduced (Fig. 6). Phosphomimetic replacement appears to mitigate the retarding effect of unphosphorylated C0-C3 on slip frequency. This effect is once again proportional to the number of replaced serines with the slip frequency in the presence of the triple-site phosphomimetic nearly equal to myosin alone. The slip distance was unaffected by any of the phosphomimetic constructs (33-40 nm) compared to myosin alone.
Fig. 6.

Backward slips of the actin filament in the load-clamped laser trap assay. Inset in graph is an example of a slip, a sudden, rapid and backward movement of the actin filament. Slip frequency in the absence of C0-C3 fragments (Myosin) and in the presence of C0-C3 with various levels of phosphomimetic replacements (P). Data are mean ± standard error for slip frequencies for all constructs within a group (i.e., 1P, 2P, 3P).
Discussion
The physiological range of cMyBP-C motif phosphorylation varies substantially in vivo, being highly phosphorylated in normal states(van Dijk et al. 2009; Sadayappan et al. 2005; Sadayappan et al. 2006; Jacques et al. 2008; Copeland et al. 2010), increased in response to β-adrenergic stimulation (Gruen et al. 1999, Nagayama et al. 2007), and dephosphorylated in disease states(Sadayappan et al. 2005; Jacques et al. 2008; Copeland et al. 2010) . Although in vitro work highlights the importance of phosphorylation in regulating cMyBP-C's function, most studies used fully phosphorylated cMyBP-C or did not quantify the extent of cMyBP-C phosphorylation (Weith et al. 2012; Shaffer et al. 2009; Rybakova et al. 2011). Therefore, we used expressed, N-terminal C0-C3 fragments with either aspartic acids and alanines replacing for serines S273, S282, and S302 to determine how the extent of phosphorylation (i.e., effectively single-, double- or triple-site phosphorylation) affects cMyBP-C's modulation of force and motion generation by myosin in the motility and laser trap assays. Our work demonstrates that phosphomimetic replacement (i.e. aspartic acid) reduces the slowing of actin velocity observed in the presence of C0-C3 in proportion to the total number phosphomimetic replacements.
Partial phosphorylation lessens cMyBP-C's effect on actomyosin motion generation in a graded manner
The ability of cMyBP-C to reduce actomyosin shortening velocity and power generation in muscle fibers was inferred from studies involving chemical extraction or genetic elimination of MyBP-C (Hofmann et al. 1991a; Korte et al. 2003). Similarly, motif phosphorylation in response to PKA treatment in fibers resulted in increased actomyosin kinetics as evidenced by greater power generation and faster rates of tension redevelopment following a stretch (Tong et al. 2008; Sadayappan et al. 2009). Therefore, cMyBP-C phosphorylation effectively reduces cMyBP-C's role in modulating actomyosin function. The data presented here support this view as unphosphorylated C0-C3 depressed unloaded actin filament velocity (Fig. 4) and actomyosin F:V relationships (Fig. 5). In addition, cMyBP-C's N-terminus is largely responsible for this effect since C0-C3 recapitulates the inhibitory effects of whole cMyBP-C (Weith et al. 2012). However, with each additional aspartic acid replacement of serines 273, 282, and 302, regardless of position, the influence of cMyBP-C on unloaded actin filament velocity and F:V was incrementally decreased (Figs. 4, 5). We did not uncover any hierarchical dependence on the site of phosphomimetic replacement, at least within the resolution of our assays. The motif contains a fourth phosphorylatable serine (S307) (Jia et al. 2010), which is much less characterized than S273, S282, or S302. In our assays, the triple-site phosphomimetic (C0-C3DDD) was as capable of limiting C0-C3's effect on actomyosin function as PKA-treated C0-C3 (Fig. 4, 5), which has all 4 serines phosphorylated (Weith et al. 2012). S307 does not appear to impart any greater effect once the other three serines have been effectively phosphorylated, agreeing with fiber studies (Sadayappan et al. 2009).
How motif phosphorylation modulates cMyBP-C's capacity to regulate actomyosin function is a matter of speculation. Presumably, motif phosphorylation alters cMyBP-C's interaction with actin and/or myosin. Phosphorylation may simply change cMyBP-C's surface charge and thus alter electrostatic interactions with actin and myosin. Alternatively, cMyBP-C's N-terminal domain may adopt a more stable conformation, once one or more serines have been phosphorylated. This stabilized conformation may lower the affinity of cMyBP-C's binding interface for actin (Weith et al. 2012; Schaffer et al. 2009) and/or myosin (Gruen et al. 1999). A recent NMR-structural study of the motif suggests that phosphorylation of S273 extends an α-helix within the motif, which could be part of this conformational change (Howarth et al. 2012). In addition, using site-directed mutagenesis, Gautel and coworkers showed that S282 is phosphorylated before S273 or S302 (Gautel et al. 1995), suggesting that S282 phosphorylation may be required to induce a conformational change in the motif to allow kinases access to other phosphorylatable serines (Gautel et al. 1995; Sadayappan et al. 2011). This hierarchy in the order of phosphorylation does not appear to translate into a hierarchical mechanical effect on actomyosin function as measured in this study.
C0-C3 behaves as a viscous load
C0-C3's depression of unloaded velocity and F:V (Fig. 4, 5) must be caused by either C0-C3 binding to myosin and/or actin, since there are no other proteins in our study. However, without direct readouts of such binding it is impossible to distinguish between these two possibilities. For instance, cMyBP-C binding to myosin's S2 segment (Moos et al. 1978, Gruen and Gautel 1999, Harris et al. 2004), regulatory light chain (Ratti et al. 2011), or head (Witt et al. 2001) could alter myosin's kinetics directly or given that cMyBP-C's C-terminus binds simultaneously to the myosin rod, cMyBP-C could then act as a tether or strut to limit myosin's force and motion generating conformational changes. Alternately, cMyBP-C's N-terminal binding to actin could create a load to slow myosin's motion generation. Although the significance and specificity of this binding is still debated (Rybakova et al. 2011), several studies do support stereospecific actin binding in vitro (Weith et al. 2012; Mun et al. 2011; Lu et al. 2011) and the potential for this to occur in vivo (Luther et al. 2011). Based on our previous work where C0-C3 binds actin reversibly in the laser trap, and tethers actin filaments to the motility surface, we proposed that cMyBP-C might act as a viscous load (Weith et al. 2012). Therefore, we fit the F:V data in the presence of the various C0-C3 constructs by a modified Hill equation with the addition of a viscous load term (see Methods). It is important to note that cMyBP-C's inhibitory effect through myosin binding could also be described in general terms as a viscous load. Therefore, regardless of its site of action, the viscous load F:V model proposed here offers a means of quantifying cMyBP-C's mechanical impact on actomyosin function. However, a caveat to these studies is that although our in vitro model system allows us to control the protein stoichiometry, it does not retain the sarcomeric spatial relationships that exist between cMyBP-C and both actin and myosin. More importantly, cMyBP-C's location within the sarcomere is restricted to the C-zone, which raises the question does cMyBP-C only exert its mechanical effects on the actomyosin crossbridges with which it interacts, which cannot be addressed by the present studies. A potential intermediate step that could bridge the motility assay and the muscle fiber is the use of native myosin thick filaments.
If C0-C3 acts as a viscous element, then the force it exerts will be directly proportional to velocity of the actin filament. Therefore, C0-C3's viscous load should be most pronounced in the motility assay under unloaded conditions (Fig. 4) and in F:V at the faster velocities where the external loads are lowest, which in fact was the case (Fig. 5). Based on the viscosity coefficients, ν, from the F:V fits in the presence of C0-C3 and C0-C3AAA (Fig. 5A, Table 1), the slower velocities at 1 pN compared to myosin alone would be due to a viscous load equivalent to 2.0-2.5 pN. Given the C0-C3 surface density determined from the Qdot-labeled C0-C3 surface binding assay (see Methods; Fig. 3), and the geometric constraints that limit the amount of C0-C3 in contact with the actin filament in the laser trap (Walcott et al. 2009), we estimate that ∼10 C0-C3 are in contact with the filament in the assay. Therefore, each C0-C3 molecule imparts ∼0.20-0.25 pN of viscous load, similar to the 0.12 pN viscous load that the actin-binding protein, α-actinin, exerts on actin filaments in the motility assay (Greenberg and Moore 2010). Interestingly, at zero velocity where the viscous load no longer exists, the estimated maximal force from F:V in the presence of any C0-C3 construct was similar to that for myosin alone (Table 1), agreeing with previously published data suggesting that cMyBP-C has no effect on maximal force generation in fibers (Korte et al. 2003; Sadayappan et al. 2009).
The reduction in the frequency of slips (Fig. 6) at any applied load in the presence of C0-C3 also supports the fragment acting as a viscous element. Slips, or reversals of forward motion during a loaded movement by a small ensemble of myosin molecules in the laser trap, have been described previously (Debold et al. 2005). Since myosin binding to actin is a stochastic process, the probability exists that at any given time there will be too few myosin attached to the actin filament to support the load resulting in detachment and the actin filament being pulled back rapidly to the laser trap center. This slip continues until one or more myosin molecules rebind and resume moving the actin filament forward. Unphosphorylated C0-C3 greatly reduced the number of slips compared to myosin alone, suggesting that C0-C3 tethers the actin filament to the surface, effectively reducing the number of slips.
Partial phosphorylation of cMyBP-C in the heart
The physiological effects of several of the phosphomimetic constructs used in this study have been examined within the context of transgenic mice expressing mutant cMyBP-C. In the absence cMyBP-C, the null mouse exhibits a hypertrophic heart (Sadayappan et al. 2005; Harris et al. 2002). Expression of cMyBP-CDDD in the null background (AllP+ mouse; Sadayappan et al. 2006) was able to prevent the hypertrophic phenotype and protected the heart from ischemic-reperfusion injury, whereas cMyBP-CAAA expression did not rescue the normal phenotype (AllP-mouse; Sadayappan et al. 2005). In addition, the cMyBP-CAAA mouse myocardium had compromised contractile reserve in response to increased heart rate or adrenergic stimulation (Gruen et al. 1999, Nagayama et al. 2007). These data coupled with human studies showing that normal cMyBP-C phosphorylation is substantial (van Dijk et al. 2009; Sadayappan et al. 2005; Sadayappan et al. 2006; Jacques et al. 2008; Copeland et al. 2010) and that dephosphorylation is a characteristic of heart failure and ischemia (Sadayappan et al. 2005; Jacques et al. 2008; Copeland et al. 2010), emphasize the importance of cMyBP-C and its phosphorylation on normal cardiac structure and function. In fact, in transgenic mice expressing cMyBP-CADA, cMyBP-CDAD,or cMyBP-CSAS (a construct in which only S282 was mutated to alanine), these expressed mutants failed to completely rescue the cMyBP-C null hypertrophic phenotype (Sadayappan et al. 2011). The cMyBP-CDAD mice had worse fibrosis and more hypertrophy than the cMyBP-CADA, suggesting that irreversible hyperphosphorylation (i.e. aspartic acid replacement), at least at S273 and S302, may be more detrimental than constitutive dephosphorylation (i.e. alanine replacement) at those sites.
Partial phosphorylation of cMyBP-C is the norm in human hearts with 90% of cMyBP-C having one or more serines phosphorylated (Copeland et al 2010). Given our results that incremental increases in phosphorylation lead to a gradual decrease in cMyBP-C's inhibition of actomyosin F:V, then as long as cMyBP-C is partially phosphorylated in vivo, the heart can call upon cMyBP-C phosphorylation to tap into its contractile reserve in response to increased cardiovascular demand and adrenergic stimulation; a perfect example of how phosphorylation of a regulatory protein can have dramatic effects on muscle contractility as described by Michael and Kate Barany, pioneers in the field of post-translational modification of muscle proteins.
Acknowledgments
We thank G. Kennedy, from the Instrumentation and Modeling Facility, for imaging expertise. National Institutes of Health funds supported AW (HL007944); MP (HL07647); JG, JR, and DW (HL059408). The Fondation Leducq supported JR.
References
- Bárány M. ATPase activity of myosin correlated with speed of muscle shortening. J Gen Physiol. 1967;50(1):197–218. doi: 10.1085/jgp.50.6.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bárány M, Bárány K. Phosphorylation of the myofibrillar proteins. Annu Rev Physiol. 1980;42:275–92. doi: 10.1146/annurev.ph.42.030180.001423. [DOI] [PubMed] [Google Scholar]
- Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2011;48:866–75. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calaghan SC, Trinick J, Knight PJ, White E. A role for C-protein in the regulation of contraction and intracellular Ca2+ in intact rat ventricular myocytes. J Physiol. 2000;528 Pt 1:151–6. doi: 10.1111/j.1469-7793.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J, Marston SB. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J Mol Cell Cardiol. 2010;49:1003–11. doi: 10.1016/j.yjmcc.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Craig R, Offer G. The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B Biol Sci. 1976;192:451–61. doi: 10.1098/rspb.1976.0023. [DOI] [PubMed] [Google Scholar]
- Debold EP, Patlak JB, Warshaw DM. Slip sliding away: load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophys J. 2005;89:L34–6. doi: 10.1529/biophysj.105.072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, Seidman C, Warshaw DM. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284–91. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–60. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MJ, Moore JR. The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton. 2010;67:273–85. doi: 10.1002/cm.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen M, Gautel M. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–49. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- Gruen M, Prinz H, Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999;453:254–9. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- Guilford WH, Dupuis DE, Kennedy G, Wu J, Patlak JB, Warshaw DM. Smooth muscle and skeletal muscle myosins produce similar unitary forces and displacements in the laser trap. Biophys J. 1997;72:1006–21. doi: 10.1016/S0006-3495(97)78753-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DE, Work SS, Wright RK, Alpert NR, Warshaw DM. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15:11–9. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–64. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SP, Rostkova E, Gautel M, Moss RL. Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ Res. 2004;95:930–6. doi: 10.1161/01.RES.0000147312.02673.56. [DOI] [PubMed] [Google Scholar]
- Hill AV. The heat of shortening and the dynamic constants of muscle. Proc R Soc London B Biol Sci. 1938;126:136–195. [Google Scholar]
- Hofmann PA, Greaser ML, Moss RL. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol. 1991a;439:701–15. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann PA, Hartzell HC, Moss RL. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J Gen Physiol. 1991b;97:1141–63. doi: 10.1085/jgp.97.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth JW, Ramisetti S, Nolan K, Sadayappan S, Rosevear PR. Structural Insight into Unique Cardiac Myosin-binding Protein-C Motif: A Partially Folded Domain. J Biol Chem. 2012;287:8254–62. doi: 10.1074/jbc.M111.309591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, Tsang VT, Marston SB. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol. 2008;45:209–16. doi: 10.1016/j.yjmcc.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Jia W, Shaffer JF, Harris SP, Leary JA. Identification of novel protein kinase A phosphorylation sites in the M-domain of human and murine cardiac myosin binding protein-C using mass spectrometry analysis. J Proteome Res. 2010;9:1843–53. doi: 10.1021/pr901006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res. 2003;93:752–8. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- Lu Y, Kwan AH, Trewhella J, Jeffries CM. The C0C1 fragment of human cardiac myosin binding protein C has common binding determinants for both actin and myosin. J Mol Biol. 2011;413:908–13. doi: 10.1016/j.jmb.2011.09.026. [DOI] [PubMed] [Google Scholar]
- Luther PK, Winkler H, Taylor K, Zoghbi ME, Craig R, Padron R, Squire JM, Liu J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc Natl Acad Sci U S A. 2011;108:11423–8. doi: 10.1073/pnas.1103216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982;85 Pt B:55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- McClellan G, Kulikovskaya I, Winegrad S. Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys J. 2001;81:1083–92. doi: 10.1016/S0006-3495(01)75765-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos C, Mason CM, Besterman JM, Feng IN, Dubin JH. The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J Mol Biol. 1978;124:571–86. doi: 10.1016/0022-2836(78)90172-9. [DOI] [PubMed] [Google Scholar]
- Moos C, Offer G, Starr R, Bennett P. Interaction of C-protein with myosin, myosin rod and light meromyosin. J Mol Biol. 1975;97:1–9. doi: 10.1016/s0022-2836(75)80017-9. [DOI] [PubMed] [Google Scholar]
- Mun JY, Gulick J, Robbins J, Woodhead J, Lehman W, Craig R. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C) J Mol Biol. 2011;410:214–25. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, Kass DA. Control of in vivo left ventricular contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 2007;116:2399–408. doi: 10.1161/CIRCULATIONAHA.107.706523. [DOI] [PubMed] [Google Scholar]
- Palmer BM, Noguchi T, Wang Y, Heim JR, Alpert NR, Burgon PG, Seidman CE, Seidman JG, Maughan DW, LeWinter MM. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ Res. 2004;94:1615–22. doi: 10.1161/01.RES.0000132744.08754.f2. [DOI] [PubMed] [Google Scholar]
- Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell Motil. 2000;21:609–20. doi: 10.1023/a:1005678905119. [DOI] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Cell Biol. 1982;24:271–89. doi: 10.1016/s0091-679x(08)60661-5. [DOI] [PubMed] [Google Scholar]
- Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286:12650–8. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razumova MV, Bezold KL, Tu AY, Regnier M, Harris SP. Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. J Gen Physiol. 2008;132:575–85. doi: 10.1085/jgp.200810013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, Harris SP. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J Biol Chem. 2006;281:35846–54. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- Rybakova IN, Greaser ML, Moss RL. Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J Biol Chem. 2011;286:2008–16. doi: 10.1074/jbc.M110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber W, Begin KJ, Warshaw DM, VanBuren P. Cardiac myosin binding protein-C modulates actomyosin binding and kinetics in the in vitro motility assay. J Mol Cell Cardiol. 2008;44:1053–61. doi: 10.1016/j.yjmcc.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Robbins J. Cardiac myosin binding protein-C phosphorylation in a {beta}-myosin heavy chain background. Circulation. 2009;119:1253–62. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ Res. 2011;109:141–50. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156–63. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–23. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer JF, Kensler RW, Harris SP. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem. 2009;284:12318–27. doi: 10.1074/jbc.M808850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchepkin DV, Kopylova GV, Nikitina LV, Katsnelson LB, Bershitsky SY. Effects of cardiac myosin binding protein-C on the regulation of interaction of cardiac myosin with thin filament in an in vitro motility assay. Biochem Biophys Res Commun. 2010;401:159–63. doi: 10.1016/j.bbrc.2010.09.040. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ Res. 2006a;98:1212–8. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006b;99:884–90. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–82. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–83. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- Walcott S, Fagnant PM, Trybus KM, Warshaw DM. Smooth muscle heavy meromyosin phosphorylated on one of its two heads supports force and motion. J Biol Chem. 2009;284:18244–51. doi: 10.1074/jbc.M109.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaw DM, Desrosiers JM, Work SS, Trybus KM. Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J Cell Biol. 1990;111:453–63. doi: 10.1083/jcb.111.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weith A, Sadayappan S, Gulick J, Previs MJ, Vanburen P, Robbins J, Warshaw DM. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J Mol Cell Cardiol. 2012;52:219–27. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt CC, Gerull B, Davies MJ, Centner t, Linke WA, Thierfelder L. Hypercontractile properties of cardiac muscle fibers in a knock-in mouse model of cardiac myosin-binding protein-C. J Biol Chem. 2001;276:5353–9. doi: 10.1074/jbc.M008691200. [DOI] [PubMed] [Google Scholar]
- Woledge RC, Curtin NA, Homsher E. Energetic aspects of muscle contraction. Monogr Physiol Soc. 1985;41:1–357. [PubMed] [Google Scholar]
- Yadid M, Sela G, Amiad Pavlov D, Landesberg A. Adaptive control of cardiac contraction to changes in loading: from theory of sarcomere dynamics to whole-heart function. Pflugers Arch. 2011;462:49–60. doi: 10.1007/s00424-011-0966-x. [DOI] [PubMed] [Google Scholar]