

Abstract
Act1 is an essential adaptor molecule in IL-17-mediated signaling and is recruited to the IL-17 receptor upon IL-17 stimulation. Here, we report that Act1 is a client protein of the molecular chaperone, Hsp90. The Act1 variant (D10N) linked to psoriasis susceptibility is defective in its interaction with Hsp90, resulting in a global loss of Act1 function. Act1-/- mice modeled the mechanistic link between Act1 loss of function and psoriasis susceptibility. Although Act1 is necessary for IL-17-mediated inflammation, Act1-/- mice exhibited a hyper TH17 response and developed spontaneous IL-22-dependent skin inflammation. In the absence of IL-17-signaling, IL-22 is the main contributor to skin inflammation, providing a molecular mechanism for the association of Act1 (D10N) with psoriasis susceptibility.
INTRODUCTION
Psoriasis is a chronic inflammatory disease of the skin, which affects approximately 2% of the general population. Defining histological features of psoriatic skin lesions include epidermal hyperplasia, with aberrant differentiation and hyperproliferation of keratinocytes, as well as marked infiltration of leukocytes into the dermis1, 2. Although psoriasis was initially classified as a TH1-mediated disease, recent studies have highlighted a role for TH17 cells and their cytokine network in propagating and amplifying skin inflammation3. TH17 cells are a population of proinflammatory CD4+ effector T cells that are distinct from TH1 and TH2 cells due to their ability to produce IL-17A, IL-17F, IL-21 and IL-224-7, all of which are elevated in psoriatic lesions8-10.
While IL-17 is required for host defense against extracellular microorganisms11, 12, it is also a critical mediator of the pro-inflammatory processes in various autoimmune and inflammatory disorders. The IL-17-signaling cascade requires a key signaling molecule, Act1 (Traf3ip2/CIKS), to propagate downstream signaling events13-15. Upon IL-17 stimulation, Act1 is recruited to the IL-17R through a SEFIR-SEFIR-dependent interaction, exerting K63-linked polyubiquitination of TRAF6 followed by the activation of TAK1 and the IKK complex, resulting in NF-κB activation16, 17. The absence of Act1 leads to resistance to IL-17A-mediated inflammation in murine models of experimental autoimmune encephalomyelitis (EAE) and asthma14, 18, 19. Although Act1 is necessary for IL-17-mediated inflammatory responses, Act1-/- mice developed spontaneous autoimmune diseases, including skin inflammation20, which was also seen in mice with a spontaneous point mutation that introduced an early stop codon in the Act1 gene21. Notably, three independent genome-wide association studies (GWAS) recently linked a genetic variance of Act1—Act1 (D10N)—to psoriasis susceptibility22-24. However, important questions regarding the impact of this variant on the function of Act1 and how this variant might predispose patients to psoriasis remain unanswered.
One important goal of this study was to investigate the effects of the D10N mutation on Act1 function. One possibility is that Act1 (D10N) might somehow enhance IL-17 signaling and IL-17-dependent effector function in skin inflammation. However, we found the contrary: Act1 (D10N) was unable to interact with any of the known signaling components in the IL-17 pathway (including IL-17R, TRAFs and IKKi25, 26) and resulted in abolished IL-17-dependent gene expression. Here we showed that the N-terminus of Act1 contains a highly conserved region necessary for Act1’s interaction with the molecular chaperone, Hsp90. Hsp90 is one of the most abundant cellular chaperone proteins and plays a crucial role in regulating a wide array of proteins essential for cell homeostasis27, 28. Mutations that affect the N-terminus of Act1, like D10N, led to the loss of regulation by Hsp90 and consequently, a dead Act1 protein that is unable to propagate IL-17-mediated responses. Using our Act1-/- mice as a model for this dead mutation, we found that the spontaneous skin inflammation was the result of the hyper TH17 response in Act1-/- mice, suggesting immune dysregulation in the absence of a functional Act1. In the absence of IL-17-signaling, IL-22 appeared to be the main contributor of skin inflammation, as neutralization of IL-22 resulted in alleviation of the skin phenotype in the Act1-/- mice. Furthermore, we demonstrated that the hyper TH17 response is T cell-intrinsic, since T-cell-specific Act1-/- mice also developed a hyper TH17 response. Consistent with this, Act1-/- T cells transduced with Act1 (D10N) exhibited a hyper TH17 response compared to Act1-/- T cells that were transduced with Act1 (WT), indicating that the nonfunctional Act1 (D10N) is associated with the dysregulation of T cell function.
RESULTS
Act1 is a client protein of Hsp90
To search for novel Act1-interacting proteins, lysates from HeLa cells and MEFs (mouse embryonic fibroblasts) treated with IL-17 were immunoprecipitated for Act1 followed by Mass Spectrometry (Mass Spec) analysis. One protein in particular, heat-shock protein 90 (Hsp90), was repeatedly detected by Mass Spec as an IL-17-induced, Act1-interacting protein. We confirmed the interaction by co-immunoprecipitation experiments and found that Act1 indeed interacted with Hsp90 and IL-17 stimulation further promoted the interaction (Fig. 1a).
Figure 1. Act1 is a client protein of HSP90.
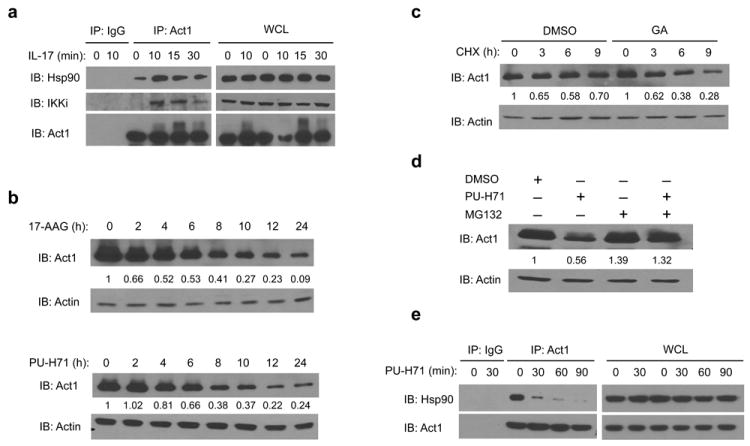
(a) Lysates from MEFs treated with IL-17 (50ng/ml) for the indicated times were immunoprecipitated with anti-Act1 or IgG control, followed by immunoblot analysis for Hsp90, IKKi and Act1. Immunoprecipitated (IP) products are shown on left, whole cell lysates (WCL) are shown on right. (b) MEFs were treated with Hsp90 inhibitors 17-AAG (1μM) or PU-H71 (1μM) for the indicated times. Actin is used as loading control. The relative amount of total Act1 to actin was quantified by densitometry. The Act1 to actin ratio in untreated MEFs was defined as 1. (c) DMSO (control) or GA pretreated MEFs were incubated with 10 μg/ml cycloheximide (CHX) for the indicated times. The relative amount of total Act1 to actin was quantified by densitometry. The Act1 to actin ratio prior to CHX treatment was defined as 1. (d) MEFs were treated with DMSO, PU-H71 (0.5μM) alone, MG-132 (1μM) alone, or PU-H71 (0.5μM) plus MG-132 (1μM) for 12 hours. The relative amount of total Act1 to actin was quantified by densitometry. The Act1 to actin ratio in DMSO-treated MEFs was defined as 1. (e) Lysates from MEFs treated with PU-H71 (1μM) for the indicated times were immunoprecipitated with anti-Act1 or IgG control. The data are representative of three independent experiments.
Hsp90 is an ATPase that facilitates the folding and assembly of its “client proteins”29-34. Loss of Hsp90 chaperone activity results in misfolding of client proteins, leading to their degradation29-31. As such, Hsp90 inhibitors were used in this study to determine Act1’s potential as an Hsp90 client protein. Hsp90 inhibitors such as geldanamycin (GA), a naturally Hsp90 inhibitor, binds to the ATP binding pocket at the N-terminus of Hsp90 to inhibit ATP binding and ATP-dependent Hsp90 chaperone function31. 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG), a synthetic derivative of GA, and PU-H71, a purine scaffold Hsp90 inhibitor, inhibit Hsp90 through similar mechanisms as GA30, 34.
Using Hsp90 inhibitors (17-AAG, and PU-H71), we found a time-dependent reduction in Act1 protein amounts in MEFs and Hela cells (Fig. 1b and data not shown). Using cycloheximide (CHX) to block new protein translation, we found that GA accelerated Act1 protein decay suggesting that Act1 protein stability is dependent on Hsp90 (Fig. 1c). In addition, use of the proteasome inhibitor, MG132, blocked PU-H71–induced decay of Act1 suggesting that the degradation of Act1 upon Hsp90 inhibition occurs through the proteasome (Fig. 1d). Consistent with reports suggesting that Hsp90 inhibitors lead to the dissociation of Hsp90 with its client proteins34, we found that after treatment with PU-H71, Act1 dissociates with Hsp90 in a short time period (Fig. 1e). Taken together, these results demonstrate that Act1 is a client protein of Hsp90.
Hsp90 activity is required for IL-17- signaling
Because Act1 is essential in IL-17-dependent signaling in autoimmune and inflammatory diseases14, 15, 18, 19, we sought to determine whether Hsp90 inhibition could interfere with IL-17 signaling and IL-17-dependent gene expression. MEFs were pretreated with Hsp90 inhibitors for 1 hour to disrupt the interaction of Act1 with Hsp90 (with indiscernible Act1 degradation), followed by IL-17 stimulation. In the presence of the inhibitors, IL-17-dependent phosphorylation of IκBα, Erk and Jnk were nearly abolished (Fig. 2a-b). Upon IL-17 stimulation, Act1 modifications (such as phosphorylation) help to govern specificity for downstream events35, 36. IL-17-induced modification of Act1 was greatly attenuated by Hsp90 inhibitors, indicating that Act1 is no longer recognized as a substrate of upstream kinases like IKKi. (Fig. 2a-b, arrow). Consistent with IL-17-induced signaling, IL-17 (IL-17A and IL-17F)-induced gene expression was greatly reduced after treatment with PU-H71 (Fig. 2c-d). To understand how the Hsp90 inhibitors blocked IL-17 signaling, we examined the impact of the inhibitors on IL-17-mediated interaction of Act1 with other signaling components in the IL-17 pathway. Pre-treatment with PU-H71 abolished the IL-17-dependent interaction of Act1 with TRAF6, TRAF3, TRAF2 and IKKi, as it also disrupted the interaction between Act1 and Hsp90 (Fig. 2e). These results suggest that Hsp90 activity plays an essential role in the integrity of Act1 function. Inhibition of Hsp90 chaperone function disrupts the interaction of Act1 with Hsp90 and with other IL-17 signaling molecules, resulting in the loss of IL-17-induced signaling and gene expression.
Figure 2. Hsp90 activity is required for IL-17-induced Act1-mediated signaling.
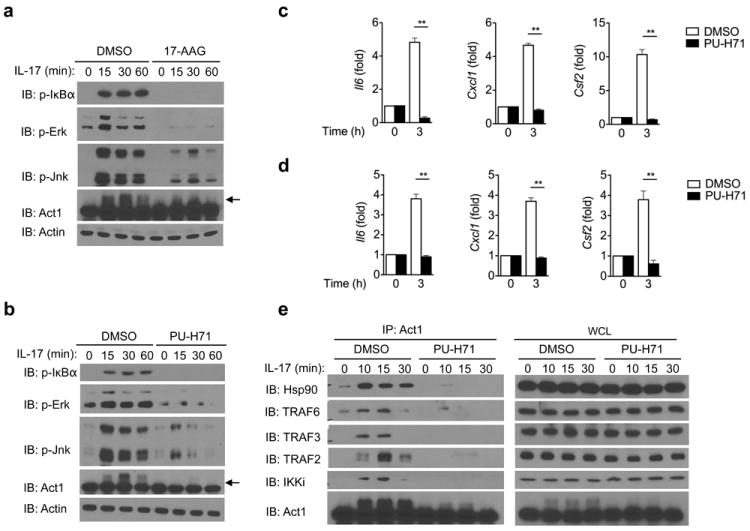
MEFs pretreated with 17-AAG (a) or PU-H71 (b) for 1 hour were stimulated with IL-17 (50ng/ml) for the indicated times, followed by immunoblot analysis for phosphorylated (p-) IκBα, p-Erk, p-Jnk, and Act1. Arrow indicates Act1 modification. (c) MEFs pretreated with PU-H71 for 1 hour were stimulated with IL-17 (50ng/ml) for the indicated times, followed by quantitative RT-PCR analysis for Il6, Cxcl1 and Csf2 expression. Results are presented as fold induction relative to time 0. (d) MEFs pretreated with PU-H71 for 1 hour were stimulated with IL-17F (50ng/ml) for the indicated times, followed by RT-PCR analysis of Il6, Cxcl1 and Csf2 expression. Results are presented as fold induction relative to time 0. (e) MEFs pretreated with PU-H71 for 1 hour were stimulated with IL-17 (50ng/ml) for the indicated times. Lysates were then immunoprecipitated with anti-Act1, followed by immunoblot analysis for Hsp90, TRAF6, TRAF3, TRAF2, IKKi and Act1. Immunoprecipitated (IP) products are shown on left, whole cell lysates (WCL) are shown on right. *p < 0.05, **p < 0.01 (Student’s t-test). The data are representative of three independent experiments.
The main function of IL-17-induced Act1-mediated signaling is to coordinate local tissue inflammation via the up-regulation of pro-inflammatory and neutrophil-mobilizing cytokines and chemokines. We next examined the impact of Hsp90 inhibition on IL-17-induced pulmonary inflammation in vivo. Wild-type mice were pretreated with DMSO or PU-H71 for two days prior to intranasal injection with recombinant IL-17. Twenty-four hours after IL-17 challenge, the mice were analyzed for Bronchoalveolar lavage (BAL) cells and lung inflammation. Infiltrating cells, like neutrophils, were significantly reduced in IL-17-challenged PU-H71-pretreated mice compared to IL-17-challenged DMSO-pretreated mice (Supplementary Fig. 1a). The decreased inflammatory phenotype correlated with decreased concentrations of CXCL1 (a potent neutrophil-recruiting chemokine) in the BAL and decreased Cxcl1 expression in the lungs (Supplementary Fig. 1b-c). Furthermore, IL-17-induced expression of Csf2 and Il6 were also abolished in the lungs of IL-17-challenged, PU-H71-pretreated mice compared to IL-17-challenged, DMSO-pretreated mice (Supplementary Fig. 1b). These results indicate the importance of Hsp90 function in IL-17-induced pulmonary inflammation.
Act1 N-terminus interacts with Hsp90
To determine the region(s) that is required for Act1 to interact with Hsp90, wild-type (WT) and truncation mutants of Act1 were transfected into HEK293 cells, followed by immunoprecipitation and immunoblot analysis. The deletion analysis showed that the deletion of amino acids 1-50 of Act1 resulted in a loss of interaction with Hsp90 (Fig. 3a). These results indicate that the N-terminus of Act1 is required for the interaction with Hsp90 (Fig. 3b). To further determine the minimum region that is sufficient for interaction with Hsp90, we transfected HEK293 cells with constructs coding for partial regions of Act1 (Supplementary Fig. 2). Our data indicates that the region between 1-100 amino acids of Act1 is sufficient for the interaction with Hsp90 (Supplementary Fig. 2). These findings suggest that the N-terminus of Act1 is necessary and sufficient for the interaction with Hsp90.
Figure 3. Loss of interaction of psoriasis-associated variant, Act1 (D10N), with Hsp90.
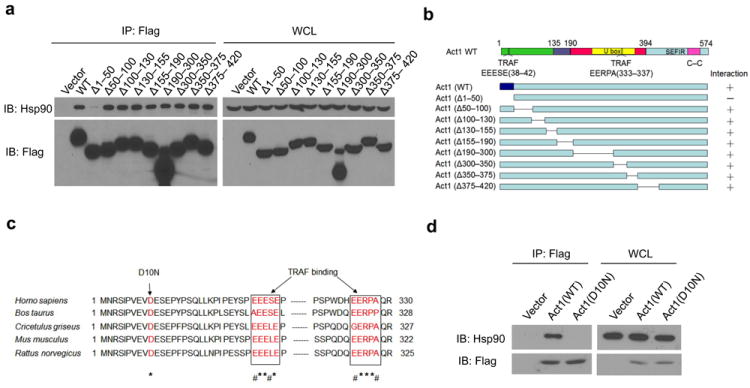
(a) HEK293 cells were transiently transfected with vector, Flag-tagged human wild-type (WT) Act1, or Flag-tagged deletion mutants of human Act1: Act1Δ1-50, Act1Δ50-100, Act1Δ100-130, Act1Δ130-155, Act1Δ155-190, Act1Δ190-300, Act1Δ300-350, Act1Δ350-375, or Act1Δ375-420. Lysates were immunoprecipitated with anti-Flag, followed by immunoblot analysis for Hsp90 and Flag. (b) Schematics of Act1-deletion mutants. Right column indicates interaction with Hsp90. (c) Alignment of Act1 sequences from Homo sapiens (human), Bos taurus (cow), Cricetulus griseus (bacteria), Mus musculus (mouse), and Rattus norvegious (rat). Asterisks (*) denote identical amino acids and the number sign (#) denotes conserved substitutions. (d) HEK293 cells were transfected with vector, Flag-tagged human Act1 (WT) and Act1 (D10N). Cell lysates were immunoprecipitated with anti-Flag, followed by immunoblot analysis for Hsp90 and Flag. Immunoprecipitated (IP) products are shown on left, whole cell lysates (WCL) are shown on right. The data are representative of three independent experiments (a, d).
Act1 (D10N) fails to interact with Hsp90
The N-terminus of Act1 consists of a highly conserved motif (Fig. 3c). Recent GWAS identified a psoriasis susceptibility locus at Act1 (Traf3ip2), including a coding variant of Act1 altering Aspartic acid at position 10 to Asparagine (D10N, rs33980500) as the most significant associated SNP (p=1.13×10-20, odds ratio=1.95)24 (Fig. 3c). Since this Act1 variant (D10N) is located within the N-terminus of Act1, we tested the impact of the point mutation on the interaction of Act1 with Hsp90. WT and Act1 (D10N) were transfected into HEK293 cells, followed by immunoprecipitation and immunoblot analysis (Fig. 3d). Indeed, Act1 (D10N) was unable to interact with Hsp90, suggesting that the Aspartic acid at position 10 plays an essential role in the interaction of Act1 with Hsp90.
It has been suggested that Act1 (D10N) is mainly defective in its binding with TRAF624. We thus explored whether the interaction of Act1 with Hsp90 is TRAF6-dependent. We have previously identified two putative TRAF binding sites in Act1 (residues 38-42 [TB1] and 333-337 [TB2])17 (Fig. 4a). Mutation of both TB1 and TB2 (TB12) sites resulted in a complete loss of interaction between Act1 and TRAF2 and TRAF6 but has no effect on the interaction of Act1 with Hsp90 (Fig. 4b). Furthermore, when WT and Traf6-/- MEFs were treated with IL-17, followed by co-immunoprecipitation and immunoblot analysis, we found that Act1 from both WT and Traf6-/- MEFs were able to interact with Hsp90, indicating that the IL-17-induced Hsp90 interaction is TRAF6-independent (Fig. 4c).
Figure 4. IL-17-induced Act1-Hsp90 interaction is TRAF6 independent.
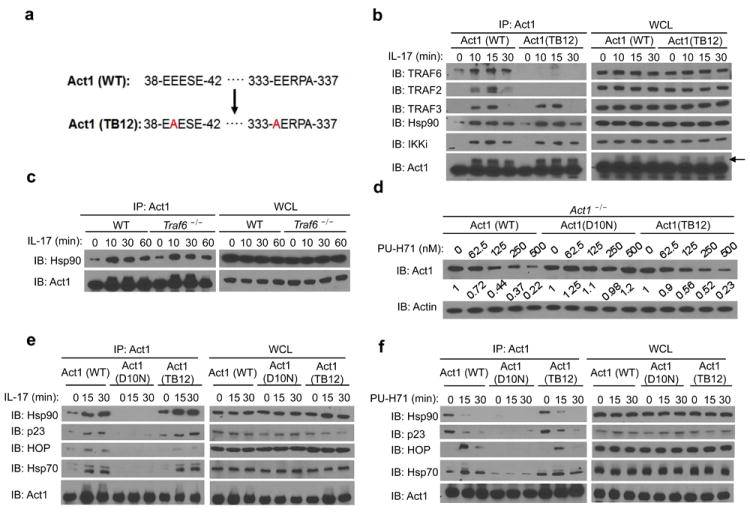
(a) Schematics of TRAF-binding site mutant (TB12) of Act1. (b) Act1-/- MEFs transduced with Act1 (WT) or Act1 (TB12) were treated with IL-17 (50ng/ml) for the indicated times. Cell lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis for TRAF6, TRAF2, TRAF3, Hsp90, IKKi and Act1. (c) Lysates from WT or Traf6-/- MEFs treated with IL-17 (50ng/ml) for the indicated times were immunoprecipitated with anti-Act1, followed by immunoblot analysis for Hsp90 and Act1. (d) Act1-/- MEFs transduced with Act1 (WT), Act1 (D10N), or Act1 (TB12) were left untreated (0) or treated with varying concentrations of PU-H71 for 24 hours. The relative amount of total Act1 to actin was quantified by densitometry. The Act1 to actin ratio in untreated MEFs was defined as 1. (e) Act1-/- MEFs transduced as in (d) were treated for the indicated times with IL-17 (50ng/ml). Cell lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis for Hsp90, p23, HOP, Hsp70 and Act1. (f) Act1-/- MEFs transduced as in (d) were treated for the indicated times with PU-H71 (1μM). Cell lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis for Hsp90, p23, HOP, Hsp70 and Act1. Immunoprecipitated (IP) products are shown on left, whole cell lysates (WCL) are shown on right. Data are representative of three independent experiments.
It is important to point out that Hsp90 functions as part of a chaperone machinery to guide client protein maturation or degradation in two main forms of Hsp90 complexes. While an Hsp90-cdc37-p23 complex stabilizes the client protein, an Hsp90-Hsp70-HOP complex targets it for proteasomal degradation34. Since the progression of the Hsp90 complex towards the stabilizing form requires the binding and hydrolysis of ATP, Hsp90 inhibitors such as PU-H71, tend to drive the complex towards the proteasome-targeting form, leading to increased proteasomal degradation of the client29. Interestingly, heat-shock protein 70 (Hsp70) was also repeatedly detected by Mass Spectrometry as an IL-17-induced, Act1-interacting protein, suggesting that Act1 may interact with the whole chaperone machinery in addition to Hsp90. Therefore, we decided to examine the interaction of Act1 (WT), Act1 (D10N), and Act1 (TB12) with co-chaperone proteins. While IL-17 induced the interaction of Act1 (WT) and Act1 (TB12) with Hsp70, HOP and p23, there was no detectable interaction of Act1 (D10N) with these co-chaperone proteins in response to IL-17 stimulation (Fig. 4e). Consistent with these observations, PU-H71 treatment led to the degradation of Act1 (WT) and Act1 (TB12), but Act1 (D10N) remained unaffected by the treatment (Fig. 4d). We then examined the impact of PU-H71 on the interaction of Act1 with co-chaperone proteins. As expected, PU-H71 enhanced the interaction of Act1 (WT) and Act1 (TB12) with Hsp70 and HOP, but decreased the interaction with Hsp90 and p23 (Fig. 4f). However, Act1 (D10N) still failed to show interaction with the chaperone proteins in the presence of PU-H71 (Fig. 4f). These observations indicate that the D10N mutation fails to interact with Hsp90 co-chaperone proteins, which renders this Act1 variant resistant to Hsp90-dependent regulation.
Act1 (D10N) is a loss of function variant
Because the D10N mutation resulted in the loss of interaction with Hsp90, we examined the impact of the mutation on IL-17 signaling. We reconstituted Act1-/- MEFs with Act1 (WT), Act1 (D10N), or vector DNA (Fig. 5a). Re-expression of (WT) but not Act1 (D10N) in Act1-/- MEFs promoted IL-17-induced phosphorylation of Jnk and Erk as well as activation of NF-κB (shown by phosphorylation of IκBα, NF-κB DNA binding, and NF-κB-dependent luciferase activity, Fig. 5b-d). While IL-17 induced the interaction of Act1 (WT) with IL-17R, TRAFs (2, 3 and 6) and IKKi, there was no detectable interaction of Act1 (D10N) with these proteins in response to IL-17 stimulation (Fig. 5e), suggesting the inability of Act1 (D10N) to interact with all of its known interacting proteins in the IL-17-signaling cascade. Interestingly, the Act1 (D10N) mutation also abolished the interaction of Act1 with CD40 and BAFFR (Supplementary Fig. 3 and data not shown). Previously, a report suggested that the N-terminus of Act1 contains a novel TRAF6-binding motif (PVEVDE at amino acids 6-1137). Mutation at this putative TRAF6 binding site (PVEVDE was mutated to PVAVAA, Act1 T6BM) abolished the interaction of Act1 (T6BM) with TRAF6. We found that this mutation exhibited the same mechanism of loss of function as that of D10N (Supplementary Fig. 4): impaired interaction with Hsp90 and with the signaling components in the IL-17 pathway.
Figure 5. Act1 (D10N) is a loss of function variant.
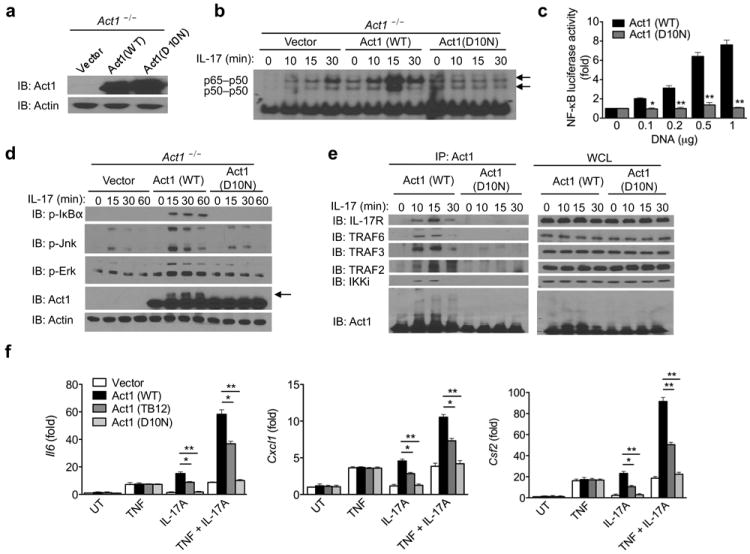
(a) Act1-/- MEFs transduced with vector, Act1 (WT), or Act1 (D10N) were subjected to immunoblot analysis for Act1 expression. (b) Act1-/- MEFs transduced as in (a) were treated with IL-17 (50ng/ml) for the indicated times. Cell lysates were subjected to gel-shift assay for NF-κB activation. (c) HEK293 cells transfected with E-selectin–luciferase reporter (100ng) and the indicated amounts of human Act1 or human Act1 (D10N) DNA, followed by luciferase assay analysis of NF-κB activity. (d) Act1-/- MEFs transduced as in (a) were treated with IL-17 (50ng/ml) for the indicated times, followed by immunoblot analysis. Arrow indicates Act1-modification. (e) Act1-/- MEFs transduced as in (a) were treated with IL-17 (50ng/ml) for the indicated times. Cell lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis for IL-17R, TRAF6, TRAF3, TRAF2, IKKi and Act1. Immunoprecipitated (IP) products are shown on left, whole cell lysates (WCL) are shown on right. (f) Act1-/- MEFs transduced with vector, Act1 (WT), Act1 (TB12), or Act1 (D10N) were left untreated or treated for 3 hours with TNF (10ng/ml), IL-17A (50ng/ml), IL-17F (50ng/ml), or TNF in combination with IL-17A, followed by RT-PCR analysis for Il6, Cxcl1 and Csf2 expression. The data are shown as fold induction over untreated (UT). *p < 0.05, **p < 0.01 and ***p < 0.005 (Student’s t-test). Data are representative of three independent experiments.
Act1 (D10N) is clearly distinct from Act1 (TB12) since Act1 (TB12), but not Act1 (D10N), still retained the interaction with Hsp90, TRAF3 and IKKi (Fig. 4b and 5e). Moreover, in contrast to Act1 (D10N), IL-17 stimulation was able to induce modification on Act1 (TB12) at similar amounts to that of Act1 (WT) (Fig. 4b, arrow). Although Act1 (D10N) protein was expressed at a comparable level to Act1 (WT), IL-17 failed to induce modifications on Act1 (D10N) (Fig. 5d, arrow), an observation seen earlier with Hsp90 inhibition (Fig. 2a-b). Consistent with these observations, IL-17 (A and F) and IL-17 plus TNF induced expression of Il6, Cxcl1 and Csf2 were restored in Act1-/- MEFs reconstituted with Act1 (WT), but not in Act1-/- MEFs reconstituted with Act1 (D10N) (Fig. 5f). IL-17-induced gene expression was only partially lost in the TB12 mutant cells (Fig. 5f). These results suggest that while TB1 and TB2 are specific for TRAF6 binding and TRAF6-dependent IL-17 signaling, the D10N mutation probably perturbed the function of the N-terminal region of Act1 critical for the binding with Hsp90, resulting in the loss of a broader function of Act1 than TRAF6-binding. Furthermore, IL-17-mediated mRNA stability could not be restored in MEFs reconstituted with Act1 (D10N), whereas it was restored in MEFs reconstituted with Act1 (WT) (Supplementary Fig. 5). Collectively, these observations implicate that the D10N mutation might have caused a major conformational change in Act1 due to the absence of appropriate folding by Hsp90, resulting in the inability of Act1 (D10N) to respond to and propagate IL-17-signaling events.
IL-22-mediated skin inflammation in Act1-/- mice
Our results indicate that the loss of function of Act1 (D10N) is due to the impaired regulation of Act1 by Hsp90. Given that IL-17A is strongly linked to the pathogenesis of psoriasis, it is puzzling that a loss-of-function Act1 variant is associated with psoriasis susceptibility. Although Act1-/- mice are resistant to IL-17-dependent inflammation, such as EAE, we have previously reported that Act1-/- mice develop spontaneous autoimmune phenotypes, including skin inflammation20, characterized by epidermal hyperplasia and cell infiltration. Immunostaining showed that there were substantially more CD3+, CD4+, CD11b+ and Gr-1+ infiltrating cells in the Act1-/- skin tissue than there were in the controls, indicating the participation of T cells, macrophages and neutrophils in the skin inflammation (Fig. 6a). Furthermore, more IL-17+ and IL-22+ CD4+ T cells were detected in the spleen and lymph nodes of Act1-/- mice compared to controls (Fig. 6b and Supplementary Fig. 6), and TH17 cytokine transcripts (Il17a, Il17f, and Il22) were increased in the skin of Act1-/- mice (Fig. 6d). Cytokine production by skin infiltrates isolated from Act1-/- mice also indicated elevated IL-17A and IL-22 production compared to skin infiltrates isolated from the control mice, whereas IL-4, IL-6, TNF, and IL-21 were undetectable (Figure 6c). Surprisingly, IL-21 transcripts were undetectable in the lymph nodes or the skin of Act1-/- mice. These results suggest that Act1 deficiency led to a hyper TH17 response with a bias towards IL-17-IL-22 expression evident in the periphery (spleen and LNs) as well as in the skin.
Figure 6. IL-22 neutralization attenuates skin inflammation in Act1-/- mice.
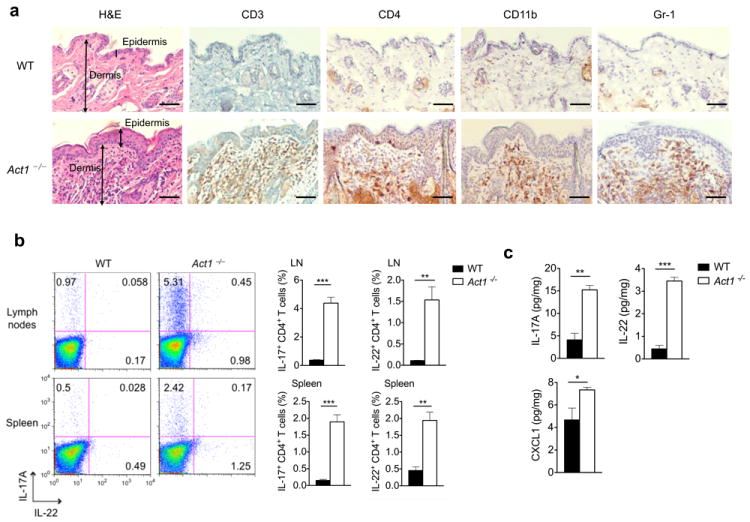
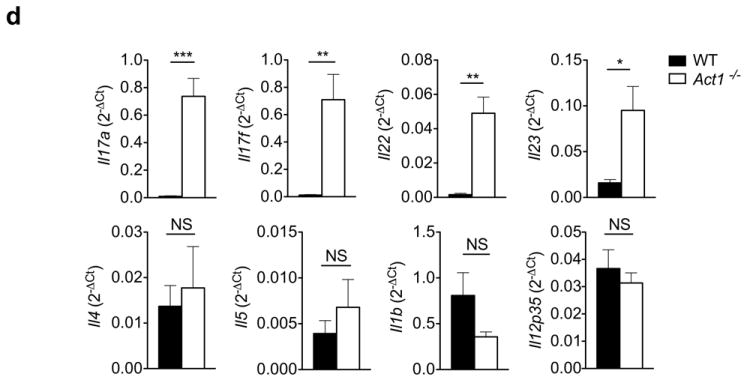
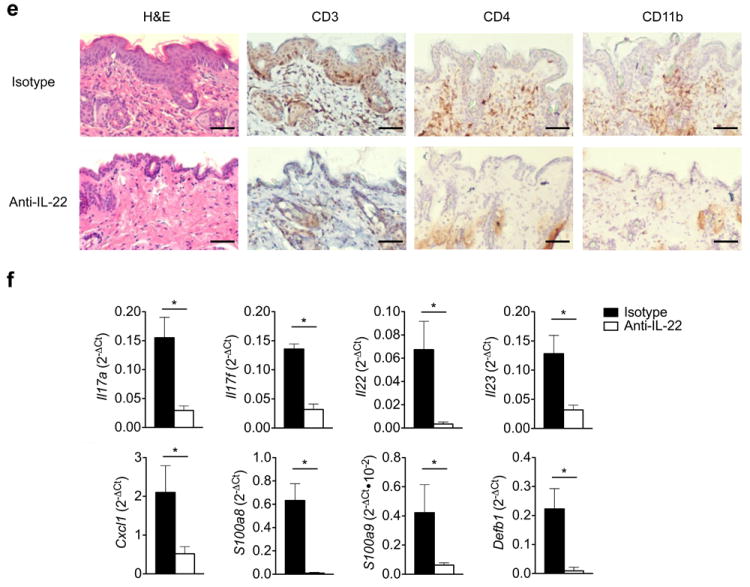
(a) Skin sections from 6 weeks old Act1 (WT) or Act1-/- mice were stained with hematoxylin and eosin (H&E) or with antibodies against cell surface markers for T cells (CD3, CD4), macrophages (CD11b), or neutrophils (Gr-1). CD3+, CD11b+, Gr-1+ and CD4+ cells are stained brown. Scale bar indicates 50μm. (b) Cells isolated from the spleen and lymph nodes (cervical, axillary, and inguinal) of 6 weeks old WT or Act1-/- mice were stimulated with PMA (20ng/ml) plus ionomycin (500ng/ml) for 5 hours followed by intracellular staining for IL-17A and IL-22. Flow plots are gated on CD4+ T cells. Right graphs indicate the percentage of IL-17+ and IL-22+ CD4+ T cells in the spleen and lymph nodes. (c) Cytokine production from skin infiltrates detected by ELISA. Skin infiltrates were isolated from the skin as described and cultured with anti-CD3/anti-CD28 for 12 hours. Cytokine production was normalized to skin tissue weight. (d) RT-PCR analysis of cytokine transcripts in the skin of 6 weeks old WT or Act1-/- mice. Data are graphed as mean 2-ΔCt ± SEM, where ΔCt=Cttarget – Ctactin. (e) Act1-/- mice were treated with 500μg of anti-IL-22 (I.P. injection) every other day for 3 weeks starting at 21 days of age. Skin sections from anti-IL-22 or isotype-treated mice were stained with hematoxylin and eosin or with anti-CD3, anti-CD4, or anti-CD11b. Scale bar indicates 50μm. (f) RT-PCR analysis of cytokine transcripts in the skin of anti-IL-22 or isotype treated mice. Data are graphed as mean 2-ΔCt ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005, NS (not significant) (Student’s t-test). Data are representative of three independent experiments with 3-5 mice per group per experiment.
Given that IL-17A/F signaling is abrogated in Act1-/- mice, and IL-21 is undetectable, we hypothesized that IL-22 is the major contributor to skin inflammation. To test this hypothesis, we neutralized Act1-/- mice with anti-IL-22 neutralizing antibody followed by histopathology analysis. Treatment with anti-IL-22 neutralizing antibody reduced epidermal hyperplasia and inflammatory cell infiltration in the skin of Act1-/- mice (Fig. 6e). Consistent with histopathology analysis, anti-IL-22 neutralizing antibody substantially reduced the expression of Il17a, Il17f, Il22 and Il23 in the skin of Act1-/- mice (Fig.6f). Since immune cells do not express the IL-22 receptor, IL-22 likely contributes to the inflammatory disease process through keratinocytes by inducing the expression of anti-microbial peptides and cytokines that amplify and maintain the pro-inflammatory environment necessary to sustain TH17 cells. Consistent with the literature38, 39, IL-22 was able to induce the expression of Il8, Defb1, S100a8, S100a9 and in human keratinocytes (Supplementary Fig. 7). We indeed observed that neutralizing IL-22 substantially reduced the expression of Cxcl1, S100a8, S100a9 and Defb1 in the skin of Act1-/- mice (Fig. 6f). Taken together, these results suggest that the TH17-derived cytokine, IL-22, plays a critical role in the skin inflammation in Act1-/- mice.
Like the Act1-/- mice, where the IL-17 signaling is abrogated, the Il17rc-/- mice also exhibited a hyper TH17 response. Increased TH17 cells were detected in the spleen and lymph nodes of Il17rc-/- mice compared to controls (Supplementary Fig. 8a and data not shown). Previous EAE experiments have shown that T cells from Il17rc-/- mice had higher antigen-specific TH17 cell response upon MOG33-55 immunization than wild-type controls40. Il17rc-/- mice also developed skin inflammation depicted by epidermal hyperplasia and T cell infiltration (Supplementary Fig. 8b). Consistent with the histopathology analysis, we observed increased expression of Il17a, Il17f, Il22, S100a8, S100a9 and Defb1 in the skin of Il17rc-/- mice (Supplementary Fig. 8c). We also detected elevated TH17 cytokines, IL-17A and IL-22, produced by skin infiltrating cells from Il17rc-/- mice compared to littermate controls (Supplementary Fig. 8d). These results suggest that the impairment of IL-17-induced Act1-mediated signaling resulted in increased TH17 cells and IL-22-dependent skin inflammation in Act1-/- mice. Indeed, neutralization of IL-22 in Il17rc-/- mice alleviated the inflammatory skin phenotype (Supplementary Fig. 8e-f).
Act1 D10N T cells exhibit hyper TH17 response
To eliminate the hyper TH17 response in Act1-/- mice, we generated Act1-/- Il23r-/- mice because it is well known that IL-23 plays a critical role in TH17 cell maintenance and expansion4, 6. TH17 cells and cytokine transcripts (including IL-22) were indeed reduced in the lymph nodes and the skin of Act1-/- Il23r-/- mice compared to Act1-/- mice (Fig. 7a and Supplementary Fig. 9a). Histopathology analysis showed that epidermal hyperplasia and T cell infiltration were also decreased in the skin of Act1-/- Il23r-/- mice, supporting the critical role of TH17 cells in the skin inflammation of Act1-/- mice (Fig. 7b). Consistent with histopathology analysis, we observed decreased expression of TH17 cytokines (Il17a, Il17f and Il22), S100a8, S100a9 and Defb1 in the skin of Act1-/- Il23r-/- mice compared to Act1-/- mice (Fig. 7c). A previous study reported a spontaneous point mutation that introduced a premature stop codon in Act1 led to spontaneous skin inflammation with elevated TH17 cytokine transcripts in the mutant mice21. Like this study, we have previously observed hyper IgE production in our Act1-/- mice attributed to the loss of negative regulation in B cells20. We now found that this hyper IgE can also be reversed in the Act1-/- Il23r-/- mice (Supplementary Fig. 9b), suggesting a link between the hyper TH17 response and the hyper IgE production in these mice.
Figure 7. Skin inflammation is attenuated in Act1-/- Il23r-/-mice.
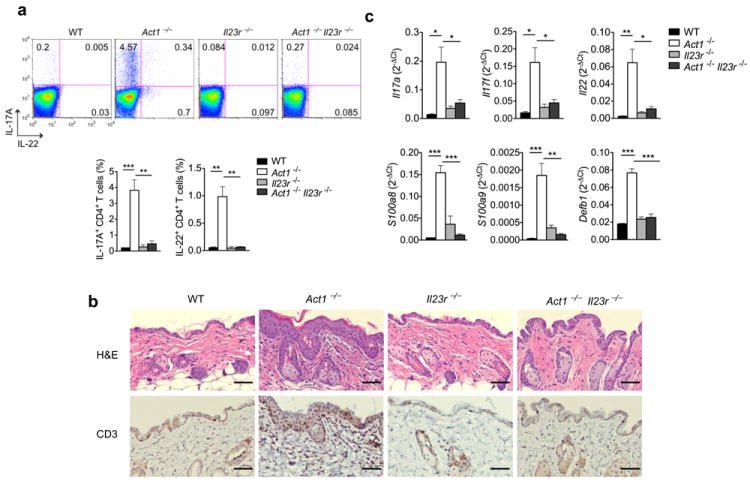
(a) Cells isolated from the lymph nodes of 6 weeks old WT, Act1-/-, Il23r-/-, or Act1-/- Il23r-/- mice were stimulated with PMA (20ng/ml) plus ionomycin (500ng/ml) for 5 hours followed by intracellular staining for IL-17A and IL-22. Right graph indicates the percentage of IL-17+ and IL-22+ CD4+ T cells in the lymph nodes. Flow plots are gated on CD4+ T cells. (b) Skin sections from 6 weeks old WT, Act1-/-, Il23r-/-, or Act1-/- Il23r-/- mice were stained with hematoxylin and eosin or with anti-CD3. Scale bar indicates 50μm. (c) RT-PCR analysis of cytokine transcripts in the skin of 6 weeks old WT, Act1-/-, Il23r-/-, or Act1-/- Il23r-/- mice. Data are graphed as mean 2-ΔCt ± SEM. *p < 0.05, **p < 0.01 (Student’s t-test). Data are representative of two independent experiments with 4 mice per group per experiment.
Next, we sought to determine the cell type that is responsible for the hyper TH17 response. Using T cell-specific Act1-deficient mice (Lck-Cre+ Act1fl/-), we examined the T cell contribution to this phenotype. We also observed skin inflammation along with CD4+, CD8+, and CD11b+ infiltrates in the skin of Lck-Cre+ Act1fl/- mice whereas it was not observed in littermate controls (Fig. 8a). Furthermore, skin infiltrates isolated from Lck-Cre+Act1fl/- mice produced more IL-17A and IL-22 than skin infiltrates from control mice (Fig. 8b). When naïve T cells were polarized to TH17 cells ex vivo, Lck-Cre+ Act1fl/- T cells showed more IL-17+ CD4+ T cells than controls, implicating the critical role of Act1 in modulating TH17 polarization (Fig. 8c). Consistent with this, we detected more Il17a and Il22 expression in the Lck-Cre+ Act1fl/- TH17 cells and more IL-17 and IL-22 in the culture supernatant of Lck-Cre+ Act1fl/- TH17 cells than the controls (Fig. 8d). To determine whether the D10N mutation in T cells was sufficient for the hyper TH17 response, we transduced Act1-/- T cells with retrovirus carrying vector, Act1 (WT), or Act1 (D10N) and polarized the T cells under TH17 skewing conditions. GFP+ cells were sorted for RT-PCR analysis of Il17a and Il22 expression. The Act1-/- T cells transduced with Act1 (D10N) exhibit a hyper TH17 response compared to Act1-/- T cells that were transduced with Act1 (WT) (Fig. 8e). Furthermore, when we injected Act1-/- T cells transduced with retrovirus carrying vector, Act1 (WT), or Act1 (D10N) into RAG1-/- mice and examined the lymph nodes and spleens two weeks later, we observed that Act1 (D10N) conferred a hyper TH17 response (Fig. 8f-g). Taken together, these results suggest that the nonfunctional Act1 (D10N) is associated with the dysregulation of T cell function.
Figure 8. Skin inflammation is observed in T cell-specific Act1-/- mice.
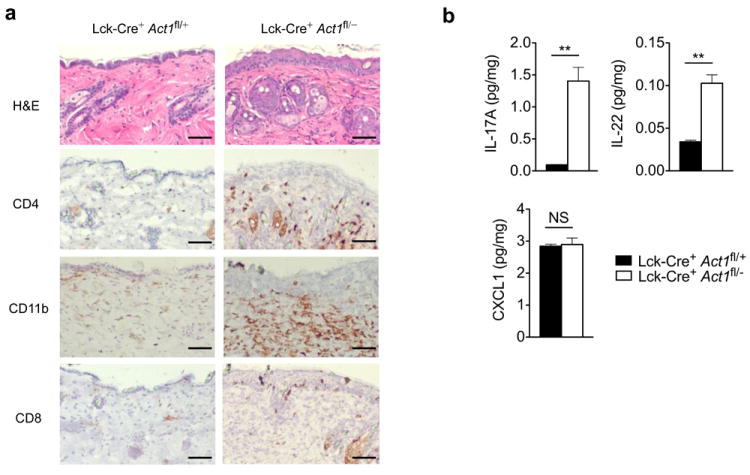
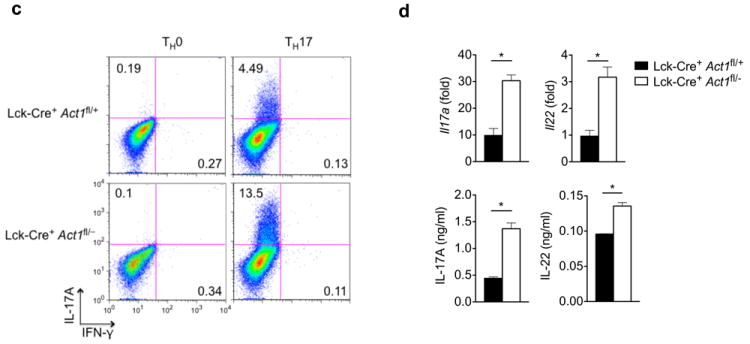
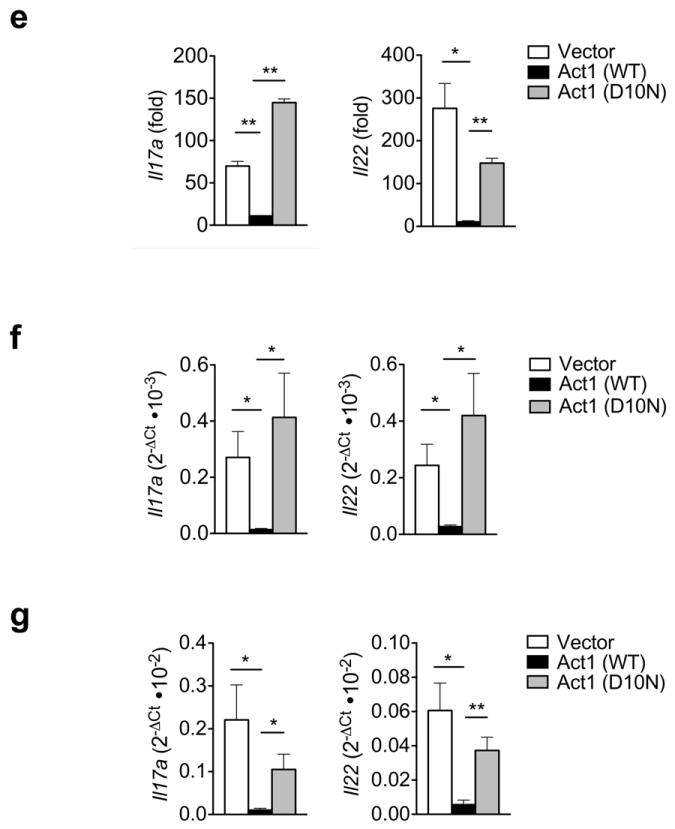
(a) Skin sections from Lck-Cre+ Act1fl/+ and Lck-Cre+ Act1fl/- mice were stained with H&E or antibodies against cell surface markers. CD4+, CD11b+ and CD8+ cells are stained brown. Scale bar indicates 50μm. (b) Cytokine production by skin infiltrates from Lck-Cre+ Act1fl/+ and Lck-Cre+ Act1fl/- mice. Cytokine production was normalized to tissue weight. (c) Naïve T cells were isolated from Lck-Cre+ Act1fl/- or Lck-Cre+ Act1fl/- mice and cultured on plate-bound anti-CD3/anti-CD28 under TH0 or TH17 skewing conditions. Following 3 days of culture, cells were restimulated with PMA (20ng/ml) and ionomycin (500ng/ml) for 5 hours followed by intracellular staining for IL-17A. (d) Ex vivo polarized TH17 cells were subjected to RT-PCR analysis for Il17a and Il22 expression after 3 days of culture (top). Culture supernatants were subjected to ELISA for IL-17A and IL-22 production (bottom). (e) Act1-/- T cells were transduced with retrovirus carrying vector, WT, or Act1 (D10N) and polarized to TH17 cells with anti-CD3/anti-CD28 in the presence of TGF-β and IL-6. GFP+ cells were sorted for RT-PCR analysis for Il17a and Il22 expression. The data are shown as fold induction of polarized TH17 cells over non-polarized T cells. (f-g) Act1-/- T cells were transduced with retrovirus carrying vector, WT, or Act1 (D10N). GFP+ cells were sorted and injected intravenously into RAG1-/- mice at 5×106 cells per mouse. Two weeks post adoptive transfer, lymph nodes (f) and spleen (g) were isolated from RAG1-/- mice, followed by RT-PCR analysis for Il17a and Il22 expression. *p < 0.05, **p < 0.01, ***p < 0.005, NS (not significant) (Student’s t-test). Data are representative of three (a-c) or two (d-g) independent experiments.
DISCUSSION
In this study, we found that although Act1 is necessary for IL-17-mediated inflammatory responses, Act1-/- mice exhibited a hyper TH17 response and developed spontaneous skin inflammation, which was attenuated by IL-22 neutralization. These results demonstrate that, in the absence of IL-17-Act1 signaling, the consequent hyper TH17 response is IL-22 biased, which acts as the main contributor of skin inflammation. Considering the critical role of IL-22 in human psoriasis, this mouse model provides a plausible interpretation for how the Act1 variant (D10N) is linked to psoriasis susceptibility. We found that Act1 is a client protein of the molecular chaperone, Hsp90. The Act1 variant (D10N) is defective in its interaction with Hsp90, resulting in a complete loss of function of Act1. Importantly, Act1-/- T cells restored with the D10N variant, but not the wild-type Act1, showed a hyper TH17 phenotype, demonstrating that the nonfunctional Act1 is associated with a dysregulation of T cell function, a risk factor for autoimmune diseases including psoriasis1, 2, 41.
The Act1 D10N mutation is within the highly conserved motif at the N-terminus of Act1, which contains an Hsp90-binding region that when altered, as in Act1 (D10N) and Act1 (T6BM), leads to a broad loss of function of Act1 and consequently, defective IL-17A-signaling. The loss of Act1 modifications during Hsp90 inhibitor use indicates that the chaperone activity of Hsp90—and thus, the proper folding of Act1—is critical for Act1 to be recruited to the receptor complex and to be recognized as a substrate of upstream kinases during IL-17 signaling. Mutations that affect Act1’s ability to bind to Hsp90—like Act1 (D10N) and Act1 (T6BM)—most likely resulted in major structural and conformational changes in Act1 that abolished IL-17-mediated signaling. Interestingly, we also observed that the interaction between Act1 and Hsp90 could be enhanced by IL-17A stimulation, which suggests that in addition to its chaperone function, Hsp90 may act as a scaffolding protein in the IL-17A signaling pathway. Further evaluation would be necessary to decipher the possible role of Hsp90 in the signaling cascade.
Because IL-17A is strongly linked to the pathogenesis of psoriasis, it appears contradictory that the loss of function of Act1 (D10N) and consequent impaired IL-17 signaling are linked to increased susceptibility for psoriasis. Since the D10N mutation renders Act1 nonfunctional, we attempted to use the Act1-/- mice to model the in vivo impact of this dead mutation. Interestingly, although Act1-/- mice are defective in IL-17-mediated signaling, IL-22 functions as the major player in the pathogenesis of skin inflammation in Act1-/- mice. In the normal state, IL-22 acts as a skin repair agent to induce keratinocyte proliferation and maintain barrier integrity. In the pathogenic state, elevated and dysregulated IL-22 production leads to keratinocyte hyperproliferation and aberrant differentiation8. In murine models of psoriasis, including data shown in this report, skin inflammation can be attenuated by IL-22 neutralization42-44. Elevated IL-22 protein and gene transcripts are also found in the serum and skin lesions of patients with psoriasis9. It is possible that in patients with psoriasis who carry the Act1 variant(s), the pathogenesis of the disease is mediated by IL-22 compensating for the loss of IL-17 signaling. Thus, these findings may provide an individualized therapeutic approach for patients carrying the Act1 variant. Currently, no animal model has been able to mimic all of the characteristics of human psoriasis. However, given the genetic association of Act1 (D10N) to psoriasis susceptibility in humans, our findings of complete loss of function of Act1 (D10N) and the skin inflammation in Act1-/- mice underscore a possible mechanistic link between the skin inflammation in Act1-/- mice and the pathogenesis of psoriasis in patients with the Act1 variant.
Intriguingly, in an IL-23-induced skin inflammation model of psoriasis, IL-23 injection led to more IL-22 gene transcripts in the skin of the Il17a-/- mice than in control mice45. Taken together, these findings suggest that the loss of IL-17 signaling may result in a hyper TH17 response, implicating a negative regulatory role of IL-17 signaling in TH17 cell polarization in vivo. Future studies are required to understand the detailed molecular mechanism for how IL-17 signaling may modulate the TH17 cell response. It is important to point out that even though there is an increase in the TH17 cell population, Il17rc-/- and Act1-/- mice remain resistant to IL-17-dependent pathogenesis like experimental autoimmune encephalomyelitis (EAE)14, 18, 40. These published data and the results presented in this study collectively suggest that TH17-mediated diseases are cytokine-specific. While IL-17 plays a dominant role in TH17-mediated EAE, IL-22 is responsible for the TH17-mediated skin inflammation in Act1-/- and Il17rc-/- mice.
We demonstrated that the hyper TH17 response in Act1-/- mice is T cell-intrinsic since skin inflammation and hyper TH17 response were also observed in Lck-Cre+ Act1fl/- mice. Furthermore, Act1-/- T cells reconstituted with Act1 (D10N) exhibited a hyper TH17 response while cells reconstituted with wild-type Act1 did not, indicating that the Act1 D10N mutation and its loss of function was sufficient to cause the hyper TH17 phenotype. Our data from the Il17rc-/- mice indicate a possible negative feedback of IL-17 signaling on TH17 cells. However, since Act1 also functions under signaling mediated by the other IL-17 family members, it would thus be important to determine whether T cell-specific Il17rc-/- mice also exhibit a hyper TH17 response in the future.
The increase in TH17 cells, cytokine transcripts, and skin inflammation in Act1-/- mice was reversed in Act1-/- Il23r-/- mice, further supporting the critical role of TH17 cells in the skin inflammation of Act1-/- mice. We also found that the hyper IgE level in Act1-/- mice can be reversed in Act1-/- Il23r-/- mice, suggesting a causal link between the hyper TH17 response and the hyper IgE production in these mice. Although serum IgE concentrations are not among the common diagnostic tests for psoriasis, nor have serum IgE concentrations been well-documented in patients with psoriasis, elevated IgE have been reported in a cohort of patients with psoriatic erythroderma (PE), a severe type of psoriasis46. Serum IgE concentrations were elevated in 81.3% of the PE group, compared to 6.3% of the control group. In addition, case studies involving psoriasiform lesions and elevated IgE concentrations have been similarly documented47, 48. It would thus be interesting to study the IgE concentrationss in patients carrying the Act1 (D10N) variant to determine whether this mutation defines a specific cohort of patients with psoriasis.
In addition to Act1’s role as a component in the IL-17R-signaling cascade in fibroblasts, endothelial cells, epithelial cells, astrocytes, and macrophages, Act1 plays a negative role in the CD40–CD40L and BAFF–BAFFR signaling pathways in B cells to control B cell maturation and survival, respectively20. The loss of Act1 thus results in increased numbers of B cells, which culminates in splenomegaly, lymphadenopathy, hypergammaglobulinemia, and autoantibody production. The Act1 (D10N) mutation also abolished the interaction of Act1 with CD40 and BAFFR. Therefore, it is possible that the loss of negative regulation of B cell function in Act1-/- mice might also contribute to the pathogenesis of the skin inflammation in mice and possibly in patients with the D10N mutation. However, we found that Act1-/- μ-/- mice still developed antigen-specific hyper TH17 cell response upon MOG33-55 immunization and had increased TH17 cells in their spleen and lymph nodes compared to μ-/- mice, suggesting that the hyper TH17 response in the Act1-/- mice is most likely independent of B cells (unpublished data, X.L). Nevertheless, it would be interesting to determine whether patients with psoriasis who carry the Act1 variant have other autoimmune co-morbidities.
MATERIAL and METHODS
Mice
Act1-/- mice were generated as described previously20. Il23r-/- mice were generated as described49 and bred to Act1-/- mice to generate Act1-/- Il23r-/- mice. Lck-Cre+ Act1fl/- mice were generated as previously described50. Littermate controls were used as WT mice for all in vivo experiments in this manuscript except in Supplemental Fig. 1, where WT mice were purchased from Taconic Farms, Inc. RAG1-/- mice were purchased from Jackson Laboratories. The Cleveland Clinic Institutional Animal Care and Use Committee reviewed and approved all animal experiments.
Cell culture and reagents
Primary MEFs were isolated from WT and Act1-/- embryos at embryonic day 14.5. HEK293 cells, and WT and Act1-/- MEFs were maintained in DMEM supplemented with 10% (volume/volume) FBS (Hyclone), penicillin G (100mg/ml) and streptomycin (100mg/ml). Recombinant IL-17A, IL-17F, and TNF were purchased from R&D Systems; geldanamycin GA (G3381), 17-AAG (A8476) and PU-H71 (P0030) were from Sigma; anti-Flag (M2; F3165) was from Sigma; antibody to phosphorylated Jnk (92516), phosphorylated IκBα (2859), IKKi (3416) was from Cell Signaling; antibody to Hsp90 (ADI-SPA-835), HOP (ADI-SRA-1500), p23 (ADI-SPA-670) were from Enzo Life Sciences; antibody to phosphorylated Erk (sc-7383), anti-IkB (sc-371), anti-TRAF3 (sc-1828), anti-TRAF6 (sc-7221), anti-TRAF2 (sc-876), human anti-Act1 (sc-13112), anti-IL17RA (sc-30175), anti-Hsp70 (SC-33575), anti-actin (sc-1615) were from Santa Cruz Biotechnology.
Immunoprecipitations and luciferase assays
Cells were lysed in lysis buffer [0.5% Triton X-100, 20 mM Hepes (pH 7.4), 150mM NaCl, 12.5mM β-glycerophosphate, 1.5mM MgCl2, 10mM NaF, 2mM dithiothreitol, 1mM sodium orthovanadate, 2mM EGTA, 20mM aprotinin, 1mM phenylmethylsulfonyl fluoride]. Cell extracts were incubated with 1 μg of the appropriate antibodies overnight at 4°C with 20 μl of protein A Sepharose beads. After incubation, beads were washed four times with lysis buffer, resolved by SDS-PAGE, and analyzed by Western blotting. NFκB luciferase reporter assays were performed as previously described17.
Retroviral production
For infection of MEFs, viral supernatants were collected 36 hours after transfection of Phoenix cells with 5 μg Act1, Act1 (D10N) and Act1 (T6BM) cloned into pMx-IP. For T cell infection, viral supernatant were collected 36 hours after transfection of Phoenix cells with 4 μg Flag-tagged Act1 and Act1 (D10N) cloned into pMSCV-IRES-GFP.
Quantitative Real-time PCR
Total RNA was extracted from MEF cells or skin with TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Skin tissues were homogenized using an OMNI TH tissue homogenizer (Omni International) prior to RNA extraction. The cDNA was synthesized with random hexamers (Applied Biosystems) using M-MLV reverse transcriptase (Promega). All skin gene-expression results are expressed as 2-ΔCt where ΔCt=Cttarget − Ctactin.
Gel-shift assay
The probe for gel-shift assays was bought from SANTA CRUZ (sc-2505). Complementary oligonucleotides were end-labeled with [γ-32P] ATP using polynucleotide kinase (Roche). Approximately 20,000 c.p.m. of probe were used per assay. Cytoplasmic extracts were prepared as described14. Binding reactions were incubated for 20 min at 25 °C in a total volume of 20 ml containing 20 mM HEPES buffer, 10 mM KCl, pH 7.0, 0.1% (volume/volume) Nonidet P-40, 0.5 mM dithiothreitol, 0.25 mM phenylmethanesulfonyl fluoride and 10% (volume/ volume) glycerol.
Histological analysis
Tissues were fixed with 10% formalin and processed into paraffin tissue blocks using routine methods by AML Laboratories (Baltimore, Maryland) or embedded in OCT and sectioned or serially sectioned to obtain consecutive levels. Paraffin embedded sections were stained with H&E and anti-CD3 (Abcam). Frozen sections were stained with anti-CD4 (BD Pharmigen), anti-CD11b (BD Pharmigen), and anti-Gr1 (BD Pharmigen). Immunohistochemistry staining for CD3 were done on paraffin-embedded sections that had been antigen-retrieved with sodium citrate. All images were captured using a DP71 digital camera (Olympus) attached to an Olympus BX41 microscope.
Intracellular staining for IL-17+ and IL-22+ cells
Single cell suspensions were obtained from the lymph nodes or the spleen of mice and cultured for 5 hours with PMA (20ng/ml) plus ionomycin (500ng/ml). GolgiStop (BD Biosciences) was added during the last two hours of incubation. Following stimulation, cells were washed and fixed with 2% PFA followed by cell permeabilization (Permeabilization Buffer, eBiosciences). Cells were stained with anti-CD3 PerCP (BD Biosciences), anti-CD4 FITC (BD Biosciences), anti-IL-17A APC (BioLegend), and anti-IL-22 PE (eBiosciences) and analyzed by FACSCalibur.
IL-22 neutralization in vivo
Anti-IL-22 neutralization antibodies (clone 8E11) were in-house generated by Genetech. Starting at 21 days of age, mice were injected intraperitoneally with 500ug of anti-IL-22 antibodies in a volume of 200uL every other day until 6 weeks of age.
Isolation of skin infiltrates and cytokine detection
Skin tissue removed from animals were weighed and incubated in 2U/ml of dispase overnight at 4°C. The epidermis was subsequently peeled from the dermis and incubated in complete RPMI medium. The dermis was minced and incubated in complete RPMI media. Following incubation at 37°C overnight, cells were collected by centrifugation and cultured on anti-CD3 (3μg/ml) and anti-CD28 (3μg/ml)-coated plates. Following 12 hours of culture, the supernatant was collected for ELISA to determine cytokine production. Cytokine concentrations were standardized to tissue weight. IL-17A ELISA kit was purchased from BioLegend. All other ELISA kits were purchased from R&D.
T cell polarization and retroviral transduction
Naïve CD4+ T cells were isolated from the spleen and lymph nodes of mice using Miltenyi CD4+CD62L+ isolation kit and cultured for 3 days on plate bound anti-CD3 (3μg/ml) and anti-CD28 (3μg/ml) under TH0 (anti-IL-4 and anti-IFN-γ) or TH17-skewing (IL-6, TGF-β, anti-IL-4, and anti-IFN-γ) conditions. Anti-IL-4 (1μg/ml), anti-IFN-γ (2μg/ml), IL-6 (20ng/ml), and TGF-β (0.1ng/ml). For retroviral transduction, naïve CD4+ T cells were isolated as above and activated for 24hrs on plate-bound anti-CD3 (1μg/ml) and anti-CD28 (2μg/ml). Following activation, cells were collected, resuspended in retroviral supernatant with 8μg/ml polybrene, and spun at 2500rpm for 90mins at 20°C. Fresh media containing IL-6 (20ng/ml) and TGF-β (0.1ng/ml) (TH17-skewing condition) or IL-2 (20ng/ml) were added to the cells and incubated at 37°C. Cells were collected at 48hrs after transduction and sorted for GFP+ cells. For adoptive transfer of transduced T cell into RAG1-/- mice, T cells were transduced as above with IL-2. 48hrs after transduction, cells were sorted for GFP and injected into mice at 5×106 cells per mouse.
Intranasal instillation of IL-17
Mice were anesthetized with isoflurane. Carrier-free recombinant mouse IL-17 (R&D Systems) resuspended in sterile saline (0.9%) was instilled into the nasal opening in an aliquot of 50μl (1μg) per mouse.
BAL fluid and tissue collection
HL-1 medium (0.7 ml; BioWhittaker) was used to obtain BAL fluid through the trachea with a blunt needle and 1-ml syringe. Cytospin slide preparations where obtained with Shandon CytoSpin III Cytocentrifuge (Shandon–Thermo Scientific). Lungs were collected and then immediately ‘snap-frozen’ in liquid nitrogen. Total RNA was obtained with TRIzol (Invitrogen) and an OMNI TH tissue homogenizer (Omni International).
Statistical analysis
The significance of differences between two groups was determined by Student’s t-test (Two-tailed). Unless otherwise specified, all results are shown as mean and the standard error of the mean (mean±SEM). A p value <0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank AML Laboratories and the LRI Histology Core for processing tissue samples for histology; Jin Ma and Wen Qian at the LRI for technical support; Nina Volokh and Danielle Kish at the LRI for critical discussions. This work was supported by NIH grants (1R01NS071996 and 1P01 HL103453) and the Sandler Award for Asthma Research to X.L.; L.W. was supported in part by NIH T32 GM007250 to the Case MSTP and NIH T32 AI 89474-1 to the Case Immunology Training Program.
Footnotes
AUTHOR CONTRIBUTIONS
C.W. and L.W. performed the experiments and analyzed the data; K.B., J.A.Z, B.N.M., T.H., Z.K., and H.X. contributed to the experiments; C.L. provided constructs; J.A.C., A.D., and J.G. are part of the collaborative team at BMS that analyzed the mass spec data; S.H. and K.D.B. contributed retroviral stock; J.S.K. provided clinical expertise; V.K.K., and O.W. provided reagents; L.W., C.W., and X.L. wrote the manuscript; L.W., S.M., J.A.Z., K.D.B., and X.L. edited the paper.
References
- 1.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 2.Nickoloff BJ, Nestle FO. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest. 2004;113:1664–1675. doi: 10.1172/JCI22147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan JR, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 5.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 6.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouyang W. Distinct roles of IL-22 in human psoriasis and inflammatory bowel disease. Cytokine Growth Factor Rev. 2010;21:435–441. doi: 10.1016/j.cytogfr.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Res PC, et al. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS One. 2010;5:e14108. doi: 10.1371/journal.pone.0014108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caruso R, et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat Med. 2009;15:1013–1015. doi: 10.1038/nm.1995. [DOI] [PubMed] [Google Scholar]
- 11.Cho JS, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conti HR, et al. TH17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 14.Qian Y, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 15.Sonder SU, et al. IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J Biol Chem. 2011;286:12881–12890. doi: 10.1074/jbc.M110.199547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000;191:1233–1240. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C, et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. 2009;2:ra63. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang Z, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swaidani S, et al. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. J Immunol. 2009;182:1631–1640. doi: 10.4049/jimmunol.182.3.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian Y, et al. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity. 2004;21:575–587. doi: 10.1016/j.immuni.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Matsushima Y, et al. An atopic dermatitis-like skin disease with hyper-IgE-emia develops in mice carrying a spontaneous recessive point mutation in the Traf3ip2 (Act1/CIKS) gene. J Immunol. 2010;185:2340–2349. doi: 10.4049/jimmunol.0900694. [DOI] [PubMed] [Google Scholar]
- 22.Strange A, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellinghaus E, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huffmeier U, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun D, et al. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF) Nat Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu S, et al. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J Exp Med. 2010;207:2647–2662. doi: 10.1084/jem.20100703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 28.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerchietti LC, et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat Med. 2009;15:1369–1376. doi: 10.1038/nm.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marubayashi S, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–3593. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orthwein A, et al. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weigert O, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012;209:259–273. doi: 10.1084/jem.20111694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz R, et al. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J Exp Med. 2012;209:275–289. doi: 10.1084/jem.20111117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waza M, et al. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- 35.Bulek K, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho AW, et al. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol. 2010;185:1063–1070. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryzhakov G, Blazek K, Udalova IA. Evolution of vertebrate immunity: sequence and functional analysis of the SEFIR domain family member Act1. J Mol Evol. 2011;72:521–530. doi: 10.1007/s00239-011-9450-7. [DOI] [PubMed] [Google Scholar]
- 38.Liang SC, et al. IL-22 induces an acute-phase response. J Immunol. 2010;185:5531–5538. doi: 10.4049/jimmunol.0904091. [DOI] [PubMed] [Google Scholar]
- 39.Guilloteau K, et al. Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1{alpha}, and TNF-{alpha} Recapitulates Some Features of Psoriasis. J Immunol. 2010 doi: 10.4049/jimmunol.0902464. [DOI] [PubMed] [Google Scholar]
- 40.Hu Y, et al. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4307–4316. doi: 10.4049/jimmunol.0903614. [DOI] [PubMed] [Google Scholar]
- 41.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol. 2009;5:325–331. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 42.Zheng Y, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 43.Ma HL, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Belle AB, et al. IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol. 2012;188:462–469. doi: 10.4049/jimmunol.1102224. [DOI] [PubMed] [Google Scholar]
- 45.Rizzo HL, et al. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol. 2011;186:1495–1502. doi: 10.4049/jimmunol.1001001. [DOI] [PubMed] [Google Scholar]
- 46.Li LF, Sujan SA, Yang H, Wang WH. Serum immunoglobulins in psoriatic erythroderma. Clin Exp Dermatol. 2005;30:125–127. doi: 10.1111/j.1365-2230.2004.01717.x. [DOI] [PubMed] [Google Scholar]
- 47.Saraceno R, Scotto G, Chiricozzi A, Chimenti S. Urticaria associated with hyper-IgE in a patient with psoriasis undergoing treatment with efalizumab. Acta Derm Venereol. 2009;89:412–413. doi: 10.2340/00015555-0613. [DOI] [PubMed] [Google Scholar]
- 48.Li XL, et al. Unusual psoriasiform lesions in a patient with Hyper-IgE syndrome. J Eur Acad Dermatol Venereol. 2007;21:424–426. doi: 10.1111/j.1468-3083.2006.01918.x. [DOI] [PubMed] [Google Scholar]
- 49.Awasthi A, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swaidani S, et al. T cell-derived Act1 is necessary for IL-25-mediated Th2 responses and allergic airway inflammation. J Immunol. 2011;187:3155–3164. doi: 10.4049/jimmunol.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.