

Summary
GRASP65 phosphorylation during mitosis and dephosphorylation after mitosis are required for Golgi disassembly and reassembly during the cell cycle. At least eight phosphorylation sites on GRASP65 have been identified, but whether they are modified in a coordinated fashion during mitosis is so far unknown. In this study, we raised phospho-specific antibodies that recognize phosphorylated T220/T224, S277 and S376 residues of GRASP65, respectively. Biochemical analysis showed that cdc2 phosphorylates all three sites, while plk1 enhances the phosphorylation. Microscopic studies using these antibodies for double and triple labeling demonstrate sequential phosphorylation and dephosphorylation during the cell cycle. S277 and S376 are phosphorylated from late G2 phase through metaphase until telophase when the new Golgi is reassembled. T220/224 is not modified until prophase, but is highly modified from prometaphase to anaphase. In metaphase, phospho-T220/224 signal localizes on both Golgi haze and mitotic Golgi clusters that represent dispersed Golgi vesicles and Golgi remnants, respectively, while phospho-S277 and S376 labeling is more concentrated on mitotic Golgi clusters. Expression of a phosphorylation-resistant GRASP65 mutant T220A/T224A inhibited mitotic Golgi fragmentation to a much larger extent than the expression of the S277A and S376A mutants. In cytokinesis, T220/224 dephosphorylation occurs prior to that of S277, but after S376. This study provides evidence that GRASP65 is sequentially phosphorylated and dephosphorylated during mitosis at different sites to orchestrate Golgi disassembly and reassembly during cell division, with phosphorylation of the T220/224 site being most critical in the process.
Key words: Golgi, Mitotic disassembly, GRASP65, Phosphorylation, Cell cycle
Introduction
In mammalian cells, the Golgi apparatus consists of over a hundred stacks of parallel-aligned membrane cisternae, which are further laterally linked to form a Golgi ribbon (Ladinsky et al., 1999). This complex structure undergoes dramatic morphological changes during the cell cycle. In early mitosis, the Golgi is disassembled into vesicles that can be observed as Golgi haze, and into tubulovesicular structures that are seen as mitotic Golgi clusters observed by fluorescence microscopy. In telophase, these fragments are evenly distributed into the two daughter cells where they are reassembled into a new Golgi apparatus each. The exact details of how the Golgi structure is maintained and how their assembly and disassembly is orchestrated are not well understood. One key element is a protein network called the “Golgi matrix” (Xiang and Wang, 2011), which is comprised of golgins, GRASP proteins and other Golgi structural proteins. The two GRASP proteins, GRASP65 (65 kDa) and GRASP55 (55 kDa), were first identified as Golgi stacking factors in an in vitro system that reconstitutes mitotic Golgi disassembly and post-mitotic Golgi reassembly (Barr et al., 1997; Shorter et al., 1999). Later research demonstrated that these proteins are involved in additional functions, including Golgi ribbon linking, cell cycle progression, trafficking, and unconventional secretion (D'Angelo et al., 2009; Duran et al., 2008; Feinstein and Linstedt, 2008; Gee et al., 2011; Kinseth et al., 2007; Kondylis et al., 2005; Kuo et al., 2000; Puthenveedu et al., 2006; Sütterlin et al., 2005).
At the molecular level, it has been shown that each GRASP protein forms trans-oligomers through the PDZ domains at each respective N-terminus. This oligomerization possibly mediates tethering of the cisternae into stacks as well as linking of Golgi stacks into a ribbon (Puthenveedu et al., 2006; Sengupta et al., 2009; Truschel et al., 2011; Wang et al., 2005; Wang et al., 2003; Xiang and Wang, 2010). Whether the oligomerization of the GRASP proteins is also helping to regulate protein trafficking and unconventional secretion remains unknown. Oligomerization of the GRASP proteins is believed to be regulated by phosphorylation (Vinke et al., 2011; Wang and Seemann, 2011). GRASP65 is known to be the major target of mitotic kinases on the Golgi membranes (Wang et al., 2003). The protein is phosphorylated by cdc2 (also called cdk1) and polo-like kinase 1 (plk1) during mitosis (Lin et al., 2000; Preisinger et al., 2005; Sütterlin et al., 2001; Tang et al., 2008). GRASP55, on the other hand, is phosphorylated by the MAP kinase ERK2 (Jesch et al., 2001; Xiang and Wang, 2010). Phosphorylation of GRASP65 and GRASP55 causes their de-oligomerization and thus disrupts Golgi cisternal stacking and Golgi ribbon linking (Feinstein and Linstedt, 2008; Vinke et al., 2011; Wang et al., 2003; Xiang and Wang, 2010). This subsequently allows Golgi membrane vesiculation that yields a large number of vesicles dispersed throughout the cell.
At the end of mitosis, GRASP65 is dephosphorylated by the protein phosphatase PP2A, which leads to the reformation of GRASP65 trans-oligomers (Tang et al., 2008). Golgi fragments then fuse in a coordinated fashion to form new cisternae, which are subsequently tethered into stacks and ultimately the Golgi ribbon (Vinke et al., 2011; Wang, 2008; Wang and Seemann, 2011). It has been shown that the expression of a phospho-deficient GRASP65 mutant (mG), in which 5 serine residues (S216, S217, S277, S367 and S376) and 2 threonine moieties (T220 and T224) were mutated to alanines, inhibited Golgi fragmentation during mitosis in the cell (Tang et al., 2010b). This strongly suggests that GRASP65 phosphorylation plays an important role in regulating Golgi architecture throughout cell division. GRASP65 can also be phosphorylated on residue S277 in interphase cells, particularly upon growth factor stimulation, which mediates the remodeling of the Golgi to facilitate membrane trafficking in migrating cells (Bisel et al., 2008; Yoshimura et al., 2005). Many features of this Golgi remodeling process during interphase are similar to that seen during mitosis, although architectural changes happen to a much lesser degree. Finally, the residue S367 is also found to be phosphorylated under interphase conditions in an in vitro phosphorylation assay (Preisinger et al., 2005). It is not clear whether activation of this site is, like S277, involved in Golgi remodeling in interphase cells.
One of the previously used phospho-deficient mutant of GRASP65, mG, contains seven known/putative phosphorylation sites mutated to alanines (Tang et al., 2010b). Expression of this mutant inhibited Golgi fragmentation during mitosis, suggesting that the phosphorylation of at least one of these sites is necessary for Golgi disassembly during mitosis. Several lines of evidence suggest that each site may be phosphorylated in a distinct fashion to help carry out different functions. For example, distinct sets of phospho-peptides were generated when GRASP65 was treated with either cdc2 or plk1 followed by trypsinization and 2D gel analysis (Lin et al., 2000), indicating that cdc2 and plk1 modify different residues on GRASP65. In another in vitro phosphorylation assay using recombinant GRASP65, seven phosphorylation sites were identified by mass spectrometry. Six of the seven sites were also present in the mG mutant; an additional site, S400, was identified. In this analysis four sites were shown to be phosphorylated by cdc2. Modification of GRASP65 by cdc2 recruits plk1 binding and allows plk1 to phosphorylate the protein (Preisinger et al., 2005). Yet another phosphorylation site, S189, was identified as a plk1 target (Sengupta and Linstedt, 2010). Its activation disrupts GRASP65 oligomers (Truschel et al., 2012). Whether the different phosphorylation sites present in GRASP65 are modified simultaneously or sequentially at the onset of mitosis, and whether they are equally important for mitotic Golgi fragmentation, remains poorly understood.
In this study we utilized five GRASP65 phospho-specific antibodies that target three different phosphorylation sites. We show that the three sites are phosphorylated and dephosphorylated sequentially during the process of cell division. In early mitosis, S277 and S376 are phosphorylated in late G2 phase, while phosphorylation of T220/T224 by cdc2 begins in prophase. During mitosis, phosphorylation of T220/T224 is most critical for mitotic Golgi disassembly, as expression of the T220A/T224A mutant of GRASP65 inhibited mitotic Golgi fragmentation to a much greater extent than that expression of either S277A or S376A mutants. In late mitosis during cytokinesis, S376 is dephosphorylated, prior to T220/T224, while S277 remains phosphorylated even in late cytokinesis. Our study demonstrated that GRASP65 is sequentially phosphorylated and dephosphorylated at different sites during the cell cycle, which allows regulated mitotic Golgi disassembly and reassembly.
Results
Development and characterization of phospho-specific antibodies that recognize various GRASP65 phosphorylation sites in rodent cells
To better understand the role of GRASP65 phosphorylation in mitotic Golgi disassembly, we raised and subsequently characterized phospho-specific antibodies against different phosphorylation sites. We prepared phosphorylated GRASP65 by treatment of recombinant wild type (WT) His-tagged rat GRASP65 full-length protein with mitotic cytosol. In a first approach this phospho-protein was used to immunize mice. One resulting monoclonal mouse antibody, LX108, was found to recognize only phosphorylated GRASP65 but not its non-phosphorylated form. In a second approach we generated three single chain variable fragment monoclonal phospho-specific antibodies (scFv antibodies), R3G3, R3F2 and RB7 using an in vitro approach (Vielemeyer et al., 2009) (supplementary material Table S1). To determine the precise epitope of each phospho-specific antibody on GRASP65, purified recombinant WT GRASP65 and 10 different phosphorylation-resistant mutants (Fig. 1A) were incubated with either mitotic (Fig. 1B) or interphase (Fig. 1C) cytosol followed by Western blot analysis using these phospho-antibodies. If mutation of a particular site led to a weakening of the signal of a particular antibody, despite treatment with mitotic cytosol, it was assumed that a conformation created by phosphorylation of the non-mutated site was specifically recognized by the antibody. As shown in Fig. 1, LX108 recognized GRASP65 only after treatment with mitotic but not interphase cytosol. Among the mutants treated with mitotic cytosol, LX108 recognized phosphorylated mB, mC, mF and mJ mutants, but not mA, mD, mE, mG, mH and mI. Modification of S216/S217 and T220/T224 abolished the signal (mA, mD, mE and mG in Fig. 1B); while mutation of T220/T224 but not S216/S217 also abrogated it (mH and mI in Fig. 1B). The only common site that was altered in all mutant constructs affecting the immunoreactivity of LX108 was T220/T224, strongly suggesting that LX108 specifically recognizes this site after phosphorylation. Using the same approach, we showed that both R3F2 and RB7 scFv antibodies specifically recognize the mitotically phosphorylated S376 epitope (Vielemeyer et al., 2009) while the scFv antibody R3G3 recognizes phospho-S277, which is phosphorylated in both interphase and mitosis, with a weaker signal after interphase cytosol treatment (Fig. 1), consistent with the previous reports that S277 is phosphorylated during interphase (Bisel et al., 2008; Yoshimura et al., 2005).
Fig. 1. Determination of the phosphorylation sites on GRASP65 that the phospho-specific antibodies recognize.

(A) Schematic illustration of known and putative phosphorylation sites on GRASP65 and its mutants in which indicated phosphorylation sites were mutated to alanines. (B,C) Purified His-tagged full-length wild type GRASP65 or its phosphorylation deficient mutants were incubated with either buffer alone (lane 1 in each blot) or with cytosol from either mitotic cells (B), or interphase cells (C) as indicated. Equal amounts of protein were analyzed by Western blot. Five anti-GRASP65 antibodies (supplementary material Table S1) were used, 7E10 being a non-phospho specific anti-GRASP65 antibody. LX108 (a mouse monoclonal antibody) failed to recognize any constructs where T220/T224 is mutated or when proteins were incubated with interphase cytosol. R3G3 (scFv recombinant antibody) did not bind mutants of S277; R3F2 and RB7 (two different scFv antibodies) both fail to bind mutated S376. In each case, loss of signal for a given mutant indicated that at least one of the mutated phospho-residues must be phosphorylated in the wild type protein for antibody binding. Thus LX108 recognizes phosphorylated T220/T224, R3G3 specifically binds pS277, while R3F2 and RB7 bind pS376.
To determine whether these phospho-specific antibodies recognize GRASP65 in cells, we performed Western blot analysis of HeLa cells stably expressing either wild type (WT) rat GRASP65-GFP, mG-GFP, or simply GFP (enhanced green fluorescent protein). A monoclonal antibody, 7E10, which recognizes rat GRASP65 regardless of its phosphorylation state, was used as a control. During interphase, WT GRASP65-GFP (but not the mG mutant) was recognized by R3G3 but not by LX108 or R3F2 (Fig. 2A–D). During mitosis, WT GRASP65-GFP was recognized by all three phospho-antibodies (Fig. 2E). These results suggest that the phospho-antibodies are able to recognize phospho-rat-GRASP65 in vivo, and also indicate that those antibodies do not recognize the endogenous human GRASP65 in HeLa cells even after phosphorylation. This was confirmed by immunofluorescence microscopy. Rat GRASP65-GFP was recognized by LX108 in mitotic but not interphase cells (Fig. 3, A vs B), validating that LX108 is a phospho-specific antibody for rat GRASP65. LX108 also recognized endogenous GRASP65 in mitotic NRK (normal rat kidney) cells (Fig. 3C; supplementary material Fig. S1). Similar results were reported for R3G3, R3F2 and RB7 (Vielemeyer et al., 2009). Furthermore, all phospho-specific antibodies, LX108, R3G3, R3F2 (Fig. 3D–I) and RB7 (not shown) detected WT but not the mG mutant of rat GRASP65 in mitotic cells.
Fig. 2. LX108, R3G3 and R3F2 recognize mitotically phosphorylated GRASP65 in cell lysate by Western blot.

(A–D) HeLa cells (lane 1 in each blot), or HeLa cells stably expressing GFP, GFP-tagged rat wild type (WT) GRASP65, or the mG mutant with all 7 phosphorylation sites mutated to alanines (Fig. 1A) (lanes 2–4 in each blot) were analyzed by Western blot using the indicated antibodies. Only S277 (detected by R3G3) is phosphorylated in interphase cells but at a relatively lower level than in mitosis (C). (E) Non-synchronized interphase (Int) or nocodazole-blocked, synchronized mitotic (Mit) HeLa cells expressing WT or mG GRASP65-GFP were analyzed by Western blot, LX108 and R3F2 were able to detect the phospho-protein in WT transfected synchronized cells (lane 2 in each blot).
Fig. 3. The phospho-specific antibodies LX108, R3G3 and R3F2 recognize WT GRASP65 but not its phosphorylation deficient mG mutant in mitotic cells.
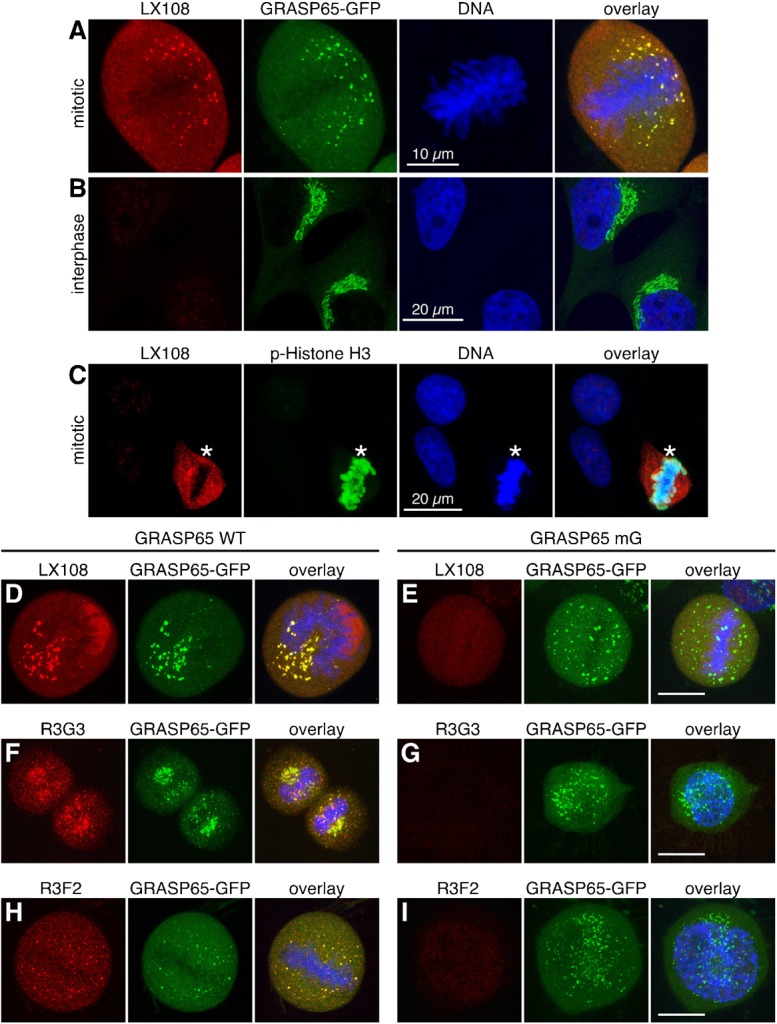
(A,B) Mitotic (prometaphase) (A) or interphase (B) HeLa cells stably expressing WT GRASP65-GFP were stained by LX108 (red), GFP (green), and DNA (blue). Note the LX108 signal in mitotic but not interphase cells. (C) Rat origin NRK cells were stained by LX108 (red), phospho-Histone H3 (p-Histone H3, green) and DNA (blue). Asterisks indicate mitotic cells. (D–I) Immunofluorescence images of mitotic HeLa cells expressing WT GRASP65-GFP (D,F,H) or its phosphorylation mutant mG (E,G,I) were stained with LX108 (T220/T224) (D,E), R3G3 (S277) (F,G), or R3F2 (S376) (H,I) antibodies. GFP epifluorescence is shown in green. Scale bar in all except B,C: 10 µm. Scale bar in B,C: 20 µm.
The T220/T224 site is phosphorylated by cdc2
Previous work has shown that GRASP65 is phosphorylated by cdc2 and plk1 during mitosis and by the MAP kinase ERK2 in interphase (Lin et al., 2000; Preisinger et al., 2005; Yoshimura et al., 2005). To identify the kinase(s) responsible for phosphorylation of GRASP65 at different sites of the protein, we treated purified rat liver Golgi membranes with mitotic cytosol, or with recombinant cdc2, plk1 or a combination of both kinases at various concentrations (Fig. 4A,B). Phosphorylation of GRASP65 was then analyzed by Western blot using LX108 and 7E10. 7E10 recognizes GRASP65 regardless of its phosphorylation, though phospho-GRASP65 can be detected by a mobility shift of the protein. Treatment with cdc2 alone caused GRASP65 phosphorylation as indicated by the band shift on the 7E10 blot and by the immunoreactivity to LX108. The addition of plk1 increased the phosphorylation state in a dose dependent manner (Fig. 4B, lanes 3–7 and 9). Incubation with plk1 alone induced slow migration of GRASP65 compared to non-treated GRASP65 on the 7E10 blot; but the shifted band was not recognized by LX108 (Fig. 4A,B, lane 8). These results suggest that GRASP65 is phosphorylated on T220/T224 by cdc2. Plk1 enhances cdc2 activity on this site; and plk1 may, in addition, phosphorylate other sites on GRASP65.
Fig. 4. The LX108 recognition site, T220/T224, is phosphorylated by cdc2.

(A) Purified rat live Golgi membranes (20 µg each reaction) were incubated with buffer (lanes 1 and 11), 500 µg mitotic cell cytosol (MC, lanes 2 and 10), or 30 µg plk1 (lanes 3–8) in the presence of increasing amount of cdc2 (lanes 3–7, 0.3, 0.9, 3, 9, and 30 µg, respectively; lane 9, 30 µg). Proteins were solubilized in SDS buffer and analyzed by Western blot for GRASP65 using the LX108 and 7E10 antibodies. Note that LX108 immuno-reactivity is detected only when the membranes were treated with MC or cdc2 (in a dose dependent manner), but not with plk1. (B) As in A, but with increasing amount of plk1 (lanes 3–8, 0.3, 0.9, 3, 9, 30, and 30 µg, respectively) and 30 µg cdc2 (lanes 3–7 and 9). Note that plk1 alone does not cause GRASP65 phosphorylation, but adding plk1 increases cdc2 activity towards GRASP65 (lanes 3–7 vs 8). (C) Interphase (int) or mitotic (mit) HeLa cells stably expressing WT GRASP65-GFP were treated with staurosporine (Stau, a general kinase inhibitor), Roscovitine (Ros, a cdk-specific inhibitor), BI2536 (a plk1 inhibitor) or U0126 (a MAPK inhibitor), lysed and analyzed by Western blot for indicated antibodies. Binding is abolished with the general kinase inhibitor (Stau) and reduced with the cdk-specific inhibitor (Ros). (D,E) Purified rat liver Golgi membranes (D) or recombinant wild type His-GRASP65 full length protein (E) were treated with indicated combinations of kinases and analyzed by Western blot for GRASP65 using the indicated antibodies. 20 µg Golgi membranes or 50 ng recombinant GRASP65 per lane were loaded and Western blot performed using LX108, R3G3 and R3F2; while 5 µg/lane Golgi membranes or 10 ng/lane recombinant His-GRASP65 was used when 7E10 was used for Western blot. Note that cdc2 phosphorylated all three sites during mitosis, while plk1 enhances the activity.
We next confirmed these results in cells. HeLa cells transfected with a GRASP65-GFP plasmid were synchronized to mitosis by 18 h nocodazole treatment and further treated with different kinase inhibitors. As shown in Fig. 4C, treatment of the mitotic cells with the general kinase inhibitor staurosporine totally abolished GRASP65 phosphorylation on T220/T224 on the LX108 blot. The cdc2 inhibitor, roscovitine, largely reduced T220/T224 phosphorylation, but the plk1 inhibitor BI2536 and MEK inhibitor U0126 had no detectable effect on inhibition of T220/T224 phosphorylation. This indicates again that T220/T224 is phosphorylated by cdc2 during mitosis.
To determine the enzymes that phosphorylate residues S277 and S376, we treated purified Golgi membranes or recombinant GRASP65 with different combinations of kinases, as indicated in Fig. 4D,E. When analyzing Golgi membranes, cdc2 alone phosphorylated both T220/T224 and S277, as indicated by LX108 and R3G3 signals, correspondingly, though only at a relatively low level (Fig. 4D, lane 2). However, a combination of cdc2 and plk1 phosphorylated all three sites to a much larger degree than either enzyme alone (Fig. 4D, lane 4), suggesting that plk1 enhances GRASP65 phosphorylation on Golgi membranes. The S277 site was phosphorylated by ERK2 at a low level (Fig. 4D, lane 5). Adding active MEK1, an activator of ERK2, significantly enhanced S277 phosphorylation. Strong activation of ERK2 also led to phosphorylation of all other sites (Fig. 4D, lane 7). When purified recombinant GRASP65 protein was used in the phosphorylation assay, T220/224, S277 and S376 were all phosphorylated by cdc2 but not plk1, although a combination of both kinases increased the phosphorylation level (Fig. 4E, lanes 2–4). S277 was phosphorylated by ERK2, and the activity was enhanced by adding constitutively active MEK1 (Fig. 4E, lane 5 vs 7). A combination of MEK1 with ERK2 could phosphorylate all the three sites. However, since ERK2 activity is relatively low during mitosis (Colanzi et al., 2000), these results may only be found in vitro with a high level of ERK2 activity. In conclusion, cdc2 can phosphorylate all three sites during mitosis, while plk1 enhances the activity of cdc2.
The phosphorylation cycle of each site on GRASP65 during cell division
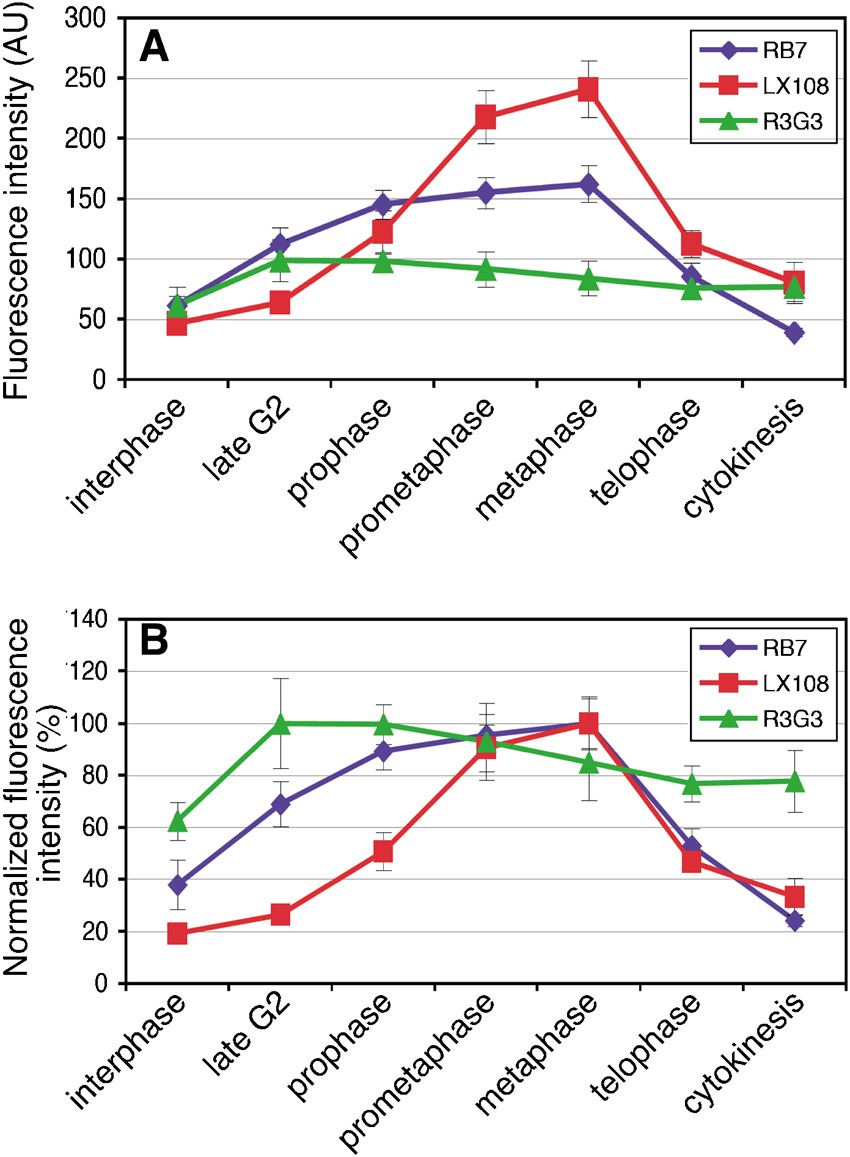
To analyze at what point each site on GRASP65 is phosphorylated and dephosphorylated during the cell cycle, we next examined NRK cells progressing through mitosis using immunofluorescence microscopy. Cells were co-stained with one of the GRASP65 phospho-specific antibodies (LX108, R3G3 or R3F2), with an antibody to phospho-Histone H3 (p-Histone H3), and for DNA to clearly identify the cell cycle phases. Histone H3 phosphorylation, which starts late in G2 phase and remains present until anaphase, was nicely visualized with p-Histone H3 (Fig. 5). All three phospho-specific GRASP65 antibodies stained the Golgi at some point during mitosis, but the exact timing of appearance of the signal was different for each of them. The LX108 (T220/T224) signal started to appear in prophase, when the Golgi fragmentation was already discernible in light microscopy, and lasted until telophase, albeit with a much reduced signal at this point (Fig. 5A). The R3G3 (S277) signal was weakly detected in interphase cells, consistent with previous reports that S277 is activated in interphase by ERK2 in addition to its mitotic phosphorylation by cdc2 (Preisinger et al., 2005; Yoshimura et al., 2005). The R3G3 signal became clearly visible already in late G2 phase and remained strong until telophase (Fig. 5B). In fact, the enhanced R3G3 signal appeared earlier and lasted longer throughout mitosis when compared with that of the other phospho-specific antibodies. R3F2 (S376) had a weak signal during late G2 phase, became stronger from prophase and was reduced at telophase (Fig. 5C). Quantitation of the fluorescence images confirmed these results (supplementary material Fig. S2A,B). Overall, these results suggest that GRASP65 phosphorylation on different sites occurs sequentially.
Fig. 5. Phosphorylation of GRASP65 at three different sites during cell cycle progression.

NRK cells of indicated cell cycle phases were stained for DNA, phosphorylated Histone H3 (Ser10) and GRASP65 with LX108 (T220/T224) (A), R3G3 (S277) (B), or R3F2 (S376) (C) antibodies. Note that in late G2 phase, the staining of R3G3 was much stronger than in interphase and much stronger than that of the other two antibodies. Scale bar in all panels: 10 µm.
Sequential phosphorylation and dephosphorylation of GRASP65 during the cell cycle
To confirm our observation that GRASP65 is phosphorylated and dephosphorylated sequentially at the three studied sites as cells progress through mitosis, we performed double staining of NRK cells with LX108 (T220/T224), and one of the two scFv antibodies, R3G3 (S277) or RB7 (S376). Furthermore, in order to better study the chronology of S277 and S376 phosphorylation during the cell cycle, we obtained an anti-phospho-S277 rabbit polyclonal antibody from Dr Nobuhiro Nakamura (Yoshimura et al., 2005), here referred to as pS277. This collection of antibodies allowed us to study the dynamics of phosphorylation and dephosphorylation of all three phosphorylation sites in pairs using LX108 vs R3G3, LX108 vs RB7, and pS277 vs RB7, at different stages of the cell cycle (Fig. 6; supplementary material Fig. S2). In late G2 phase, S277 was the first residue to be mitotically phosphorylated as indicated by the enhanced R3G3 and pS277 signals. No LX108 signal was detected during this phase, while RB7 showed a weak signal. In early prophase, R3G3 and pS277 signals remained strong, sometimes even increased slightly. At the same time RB7 signal increased significantly and LX108 signal started to appear. During late prophase, LX108 signal became more intense. In prometaphase, LX108 signal reached its highest level, while the fluorescence intensity of the other antibodies remained unchanged. Quantification and graphic depiction of the observed distinct differences in fluorescence intensity for each antibody over time is graphically depicted in supplementary material Fig. S2. In summary, these results strongly suggest that during early mitosis GRASP65 is first phosphorylated on residue S277, followed by modification of the S376 residue, and eventually activation of T220/T224. Upon careful analysis of the fluorescence images we observed that from late prophase to telophase, the R3G3-, pS277- and RB7-signals (indicating phosphorylation of S277 and S376) were largely localized on the Golgi clusters (the Golgi remnants during mitosis) while the LX108-signal (phosphorylated T220/224) appeared in a more dispersed fashion, likely representing the Golgi haze (e.g. dispersed mitotic Golgi vesicles and tubules). Since GRASP65 is a membrane-associated protein that does not detach from the membranes during mitosis (Barr et al., 1997), and since LX108 antibody staining has little if any background in interphase cells (Fig. 3B), the more homogeneous signal of LX108 likely marks dispersed mitotic Golgi vesicles and tubules at a size below the resolution of light microscopy.
Fig. 6. Different phosphorylation sites of GRASP65 are phosphorylated during mitosis and dephosphorylated in telophase at distinct times.

NRK cells of indicated cell cycle phases were double stained by the following antibodies: (A) LX108 (T220/T224) and R3G3 (S277); (B) LX108 and RB7 (S376); (C) a polyclonal antibody pS277 that recognized phosphorylated S277 and RB7. Cell cycle stage was determined by the morphology of the DNA. Note that the signals for R3G3 and RB7 were detected earlier than that of LX108, indicating that the three recognized sites are activated sequentially during mitosis. The intensity of the LX108 signal correlated with the extent of Golgi fragmentation, suggesting that T220/T224 phosphorylation is critical for mitotic Golgi disassembly. Scale bar in all panels: 10 µm.
Dephosphorylation also appeared to be sequential (Fig. 6; supplementary material Fig. S2). In telophase, both LX108 and RB7 signals became reduced significantly, while the signals for R3G3 and pS277 remained strong. During cytokinesis, no more RB7 signal was discernible, while a faint LX108-signal remained. R3G3 and pS277 staining was reduced but remained relatively strong. These results indicate that dephosphorylation at the end of mitosis is a coordinated process similar to that seen for phosphorylation at the onset of cell division. The S376 residue is inactivated first, followed by T220/T224, and eventually S277 partial dephosphorylation.
Phosphorylation of T220/T224 is critical for Golgi vesiculation
LX108 preferentially stained Golgi vesicles, while R3G3, pS277 and RB7 signals, respectively, were stronger on the mitotic Golgi clusters (Figs 5, 6; supplementary material Fig. S2). For a better overall characterization we examined the Golgi morphology using an antibody that recognizes the total GRASP65 pool regardless of its phosphorylation state and then evaluated the phosphorylation of the protein at different sites. We performed triple labeling of NRK cells with LX108, R3G3 or RB7, and “Mary”. Mary is a rabbit polyclonal antibody that recognizes rat GRASP65 in both its phosphorylated and non-phosphorylated forms (Tang et al., 2010b; Wang et al., 2005). Our observation that phosphorylation of T220/224 is more critical for complete Golgi disassembly during mitosis than activation of other sites (see above) was verified. As shown in Fig. 7, the T220/T224 residues began to be modified in prophase, at the time when the Golgi apparatus started to disassemble; R3G3 and RB7 signals were already easily discernible. In the ensuing prometaphase, T220/T224 phosphorylation reached a higher level; at this stage the Golgi apparatus was already significantly fragmented. In metaphase, T220/T224 was highly phosphorylated; Golgi disassembly was completed. Toward the end of mitosis, during telophase, RB7 but not LX108 signal significantly decreased, Golgi reassembly was initiated. Finally during cytokinesis, T220/T224 was dephosphorylated and Golgi reassembly was completed. These results suggest a correlation between T220/T224 dephosphorylation and complete Golgi reassembly.
Fig. 7. Sequential phosphorylation and dephosphorylation of GRASP65 orchestrate mitotic Golgi disassembly and post-mitotic reassembly.

NRK cells of indicated cell cycle phases were triple labeled by (A) LX108 (T220/T224), R3G3 (S277) and Mary (total GRASP65 regardless of the phosphorylation state) and (B) LX108, RB7 (S376) and Mary. Cell cycle stage was determined by the morphology of the DNA. Note that the intensity of LX108 signal correlates with the progress of Golgi disassembly. Scale bar in all panels: 10 µm.
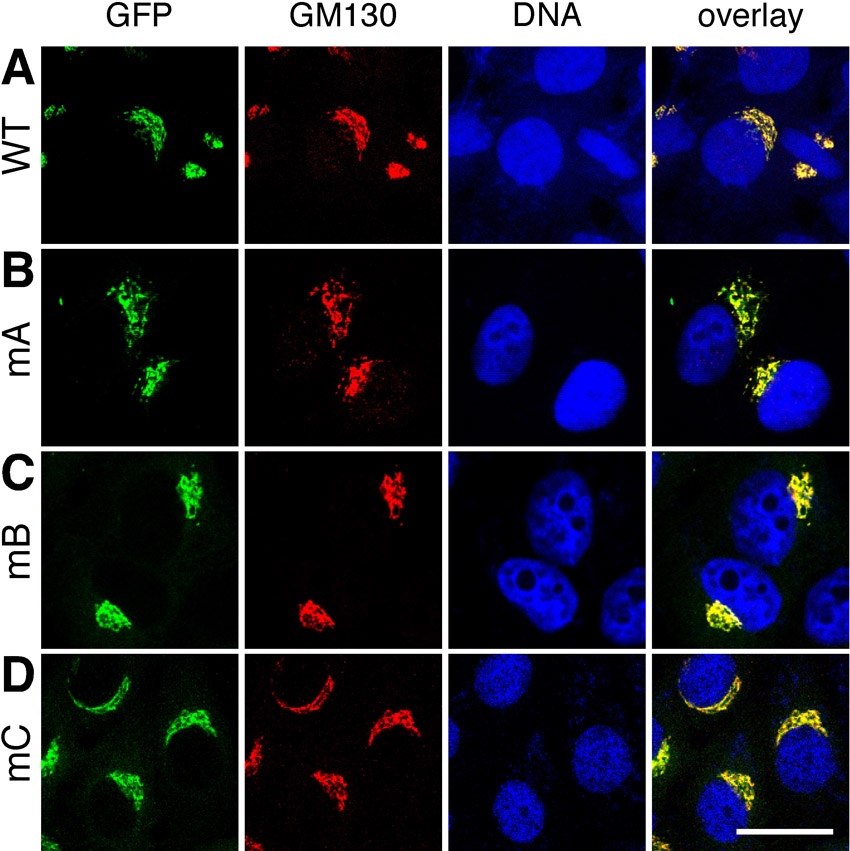
To provide further evidence for the critical role of T220/T224 phosphorylation in Golgi vesiculation, we established HeLa cell lines stably expressing GFP-tagged GRASP65 mutants mA (S216A/S217A/T220A/T224A), mB (S277A), and mC (S367A/S376A), respectively, under a tet-inducible promoter (Tang et al., 2010b; Xiang and Wang, 2010). All proteins correctly localize to the Golgi apparatus (supplementary material Fig. S3) and none of the mutants are recognized by their corresponding phospho-specific antibodies as expected (supplementary material Fig. S4). To determine the effects caused by these mutant proteins on the Golgi architecture, we analyzed these cells using fluorescence microscopy. Metaphase cells expressing WT GRASP65-GFP or the mA, mB, mC mutants were immunostained with GM130. As shown in Fig. 8A, the Golgi membranes were extensively fragmented in cells expressing wild type GRASP65-GFP. Expression of mB-GFP or mC-GFP mutants had only a minor if any effect (Fig. 8C,D). However, in cells expressing the mA-GFP mutant (Fig. 8B), the Golgi was observed as large punctae, or mitotic Golgi clusters, similar to what we previously reported with cells expressing the GRASP65 mG mutant (Tang et al., 2010b; Wang et al., 2005). Quantification of the fluorescence images showed that in mitotic mA-GFP expressing cells, 21.34±3.00% of total GM130 fluorescent signal in the cell was found on mitotic Golgi clusters. This was significant higher than the percentage seen in WT GRASP65-expressing cells (4.36±0.77%), suggesting that mA expression reduced mitotic Golgi disassembly. The intensity of the GM130 signal on mitotic Golgi clusters was not significantly affected by the expression of mB-GFP and mC-GFP mutants, leading to 2.91±0.62% and 5.43±1.20% of the total GM130 signal on the clusters, respectively (Fig. 8E), suggesting that phosphorylation at other sites is less important for this process.
Fig. 8. Mutation of the phosphorylation sites especially of the residue T220/T224 inhibits mitotic Golgi fragmentation.

(A–D) Expression of the mA mutant of GRASP65 inhibits Golgi fragmentation in mitosis. Cells expressing indicated GFP-tagged GRASP65 mutants were stained for GM130 and DNA. Metaphase cells were selected. Note the large remaining clusters, likely representing Golgi structures in the mA-expressing cell line. Scale bar in all panels: 10 µm. (E) Quantification of fluorescence intensity of GM130 on mitotic Golgi clusters as compared to whole-cell fluorescence. The percentage was expressed as the mean ± SEM. Statistical significance was assessed by student's t-test between the indicated pairs. **, P<0.01; ***, P<0.001. (F) Quantification of electron microscopy images counting the number of remaining mitotic Golgi clusters per mitotic cell in cells expressing either GRASP65 WT or mutants as indicated. The number of observed mitotic Golgi clusters per cell was expressed as the mean ± SEM. Statistical significance was assessed by comparison to the cell line expressing WT GRASP65. **, P<0.01; ***, P<0.001.
Finally, analysis by electron microscopy showed that mA-GFP expressing cells contained significantly greater amounts of large membranes that remained as clusters of vesicles, short cisternae and tubular structures (supplementary material Fig. S5). Quantification of the electron microscopy images showed that the number of mitotic Golgi clusters per mitotic cell in cells expressing the mA mutant (8.1±1.1) was significantly higher than in cells expressing WT GRASP65 (2.2±0.4). In contrast, the number of mitotic Golgi clusters per cell was only mildly increased in mB and mC expressing cells (4.2±0.5 or 4.1±0.5 respectively), significantly lower than that in the mA expressing cells (P<0.01) (Fig. 8F). These results confirmed that T220/T224 is the key regulation site for Golgi vesiculation during mitosis.
Discussion
In this study we generated and utilized phospho-specific antibodies, and dissected GRASP65 phosphorylation in the cell cycle in more detail by determining the biological significance of modification of each of the sites. The particular importance of T220/T224 phosphorylation for mitotic Golgi fragmentation was shown using the phospho-specific antibody LX108. Using this and four other GRASP65 phospho-specific antibodies, we were able to follow the temporal sequence of phosphorylation of three sites, T220/T224, S277 and S376, throughout cell cycle progression. These phosphorylation sites had been predicted in previous studies according to the amino acid sequence (Wang et al., 2005) and some had been confirmed by analysis of in vitro phosphorylated recombinant GRASP65 by mass spectrometry (Preisinger et al., 2005). In this study, we confirmed their phosphorylation in single cells. In combination with an in vitro phosphorylation assay, these antibodies also allowed us to determine the mitotic kinases responsible for each site. Furthermore, kinase inhibitors were used as a complementary approach to ensure specificity of our findings. We confirmed that cdc2 phosphorylates the T220/T224 residues required for Golgi disassembly during mitosis. Our in vitro phosphorylation assay using recombinant GRASP65 protein also suggested that S277 and S376 are phosphorylated by cdc2 during mitosis.
The fact that we were able to use phospho-specific antibodies against GRASP65 residues from different species (human, rabbit and mouse) was particularly important. It allowed us to perform double or triple labeling for immunofluorescence microscopy and thus to determine the sequence of phosphorylation on each site in the same cell and with exact knowledge of the stage of the cell cycle by monitoring DNA morphological changes and phosphorylation of Histone H3. Analyses could also be performed looking at Golgi morphology changes by co-staining cells with an antibody that recognized GRASP65 regardless of its phosphorylation state. Two major observations were made. First, GRASP65 is not phosphorylated at the T220/T224 site until prophase i.e. until after the beginning of Golgi disassembly. In contrast, residues S277 and S376 are phosphorylated in late G2 phase, a time when Histone H3 is phosphorylated but the Golgi is not yet significantly fragmented. Eventually GRASP65 is highly phosphorylated at the T220/T224 site from prophase to anaphase, at the time when the Golgi is severely fragmented. Secondly we observed that the phospho-GRASP65 pool, modified on T220/T224 localizes onto the Golgi vesicles. In NRK cells, from prometaphase to anaphase, the Golgi apparatus disassembles into thousands of vesicles distributed throughout the cytosol that were observed as “Golgi haze” under the fluorescence microscopy. Some parts of the Golgi membranes, however, remain as tubulovesicular remnants or “mitotic Golgi clusters” that appear to be more concentrated near the spindle poles (Seemann et al., 2002; Shima et al., 1998). Unlike the phospho-S277 or phospho-S376 signal, which was mainly observed on the Golgi clusters, the LX108 signal for phospho-T220/T224 appeared on both Golgi clusters and Golgi haze. Furthermore, expressing the mA, i.e. T220A/T224A, mutant in HeLa cells inhibited Golgi vesiculation during mitosis, while mB or mC mutants had a much smaller effect. These results suggest that phosphorylation of T220/T224 is the key site needed for mitotic Golgi fragmentation. Previous studies have shown that GRASP65 exerts its function through oligomerization. This is mediated by the GRASP-domain but is regulated by phosphorylation at the C-terminus (Wang et al., 2005; Wang et al., 2003). Of all the characterized phosphorylation sites, the T220/T224 residue is in closest proximity to the GRASP domain, and we speculate that it thus is likely the most important for regulating GRASP65 oligomerization.
The residue S277 is phosphorylated in mitosis (from late G2 phase to cytokinesis) as well as in interphase when cells are stimulated by EGF, although by different kinases (Bisel et al., 2008; Yoshimura et al., 2005) and to a lesser degree (Fig. 5B). Phosphorylation of S277 during interphase is directed by the MEK/ERK pathway. Mitotic phosphorylation of GRASP65 has been shown to regulate its oligomeric state and thereby Golgi cisternal stacking and ribbon linking (Giuliani et al., 2011; Vinke et al., 2011; Wang, 2008; Wang and Seemann, 2011). In interphase cells, partial phosphorylation of GRASP65 is required for dynamic Golgi remodeling in migrating cells (Bisel et al., 2008). Phosphorylation of S277 by MAP kinase partially de-oligomerizes GRASP65 complexes, accompanied by partial disassembly of the Golgi apparatus, thus allowing the organelle to reorganize and migrate towards the leading edge of the cell. Unlike what is observed in mitotic cells, massive Golgi vesiculation has not been observed in migrating cells suggesting that the phosphorylation of S277 is required for continuous reorganization of the Golgi during interphase, but that their modification may not be enough for a complete organelle disassembly during mitosis. Instead, complete collapse of the ribbon requires phosphorylation of T220/T224. Expressing the S277A (mB) or S367A/S376A (mC) mutant somewhat raised the number of Golgi clusters in mitotic cells compared to expressing the WT protein (Fig. 8F). This indicates that S277 phosphorylation facilitates Golgi fragmentation, but not to the same dramatic extent as activation of T220/T224. GRASP65 phosphorylation at different sites leads to different effects and thus serves different functions. This elaborate phosphorylation system allows the cell and its organelle to adapt to different situations and at the same time to effectively divide the single Golgi apparatus to its two daughter cells during cell division.
At the end of mitosis, S376 is the first of the three studied residues to be dephosphorylated, followed by T220/T224, and eventually S277, the latter remaining partially phosphorylated until interphase. It is likely that S277 is constitutively phosphorylated because of the need of the Golgi apparatus to undergo constant remodeling in interphase cells (Gaietta et al., 2006). The role of S376 phosphorylation is the least well understood to date. In addition to the three phosphorylation sites characterized in this study, GRASP65 is also modified by kinases on S189, S216, S217 and S367 at least in vitro (Preisinger et al., 2005; Sengupta and Linstedt, 2010). Characterization of these sites and their relationships may provide further insight in understanding the function of GRASP65 and Golgi biogenesis during cell division.
Materials and Methods
Reagents
All reagents were purchased from Sigma–Aldrich, Roche or Calbiochem, unless otherwise stated. The following antibodies were used: monoclonal antibodies against GM130 (Transduction Laboratories) and α-tubulin (Developmental Studies Hybridoma Bank University of Iowa); polyclonal antibodies against GFP (Tang et al., 2011), GRASP65 called “Mary” (Wang et al., 2003) and phospho-Histone H3 (Upstate Biotechnology). The monoclonal phospho-specific antibody LX108 was generated by immunizing mice with recombinant WT His-GRASP65 full-length protein treated with mitotic cytosol; antibodies were collected from the medium of cultured hybridoma and subsequently enriched by protein G sepharose. The human single chain (scFv) monoclonal antibodies against phosphorylated GRASP65 (R3G3, R3F2 and RB7) were described previously (Vielemeyer et al., 2009). A mouse monoclonal antibody against rat GRASP65, called 7E10, was generously provided by Dr Graham Warren (Max F. Perutz Laboratories, Austria); a rabbit polyclonal antibody for GRASP65 phosphorylated at S277 (pS277) was generously provided by Dr Nobuhiro Nakamura (Kyoto Sangyo University, Japan). Secondary antibodies for indirect immunofluorescence and immunoblotting were purchased from Jackson Laboratories and Invitrogen.
Preparation of GRASP65 recombinant proteins
The rat GRASP65 cDNA was cloned into the pQE9 (Qiagen) vector; correct insertion was confirmed by DNA sequencing. GRASP65 point mutations mA–mJ were introduced using the QuikChange mutagenesis kit (Stratagene) and changes were confirmed by DNA sequencing. Proteins were expressed in BL21-CodonPlus(DE3)RILP bacteria and purified on Ni-NTA agarose (Qiagen).
Characterization of phospho-specific antibodies by Western blot
For epitope mapping, the purified His-tagged GRASP65 full-length protein and various phosphorylation deficient mutants (potential phosphorylation sites mutated to alanines) were incubated with interphase cytosol or mitotic cytosol (buffers were used as negative control) at 37°C for 1 h and purified with Ni-NTA beads. Bound proteins were analyzed by SDS-PAGE and Western blot using antibodies as indicated.
To determine the specificity of GRASP65 phospho-specific antibodies, HeLa cell lines stably expressing wild type rat GRASP65 or its phosphorylation deficient mutants as indicated were either synchronized with 100 ng/ml nocodazole for 20 h or incubated without synchronization. Subsequently, cells were collected, lysed and analyzed by SDS-PAGE and Western blot with antibodies as indicated.
In vitro and in vivo GRASP65 phosphorylation assay
Recombinant GRASP65 proteins or purified rat liver Golgi membranes (RLG) (Wang et al., 2006) were incubated with mitotic cytosol (Tang et al., 2010a) or with different combinations of kinases (Wang et al., 2003). Typically, 20 µg RLG (or 50 ng recombinant GRASP65) were treated with 500 µg HeLa cytosol, or equivalent activity of kinases, in MEB buffer (50 mM Tris-HCl, pH 7.4, 0.2 M sucrose, 50 mM KCl, 20 mM β-glycerophosphate, 15 mM EGTA, 10 mM MgCl2, 2 mM ATP, 1 mM GTP, 1 mM glutathione, and protease inhibitors) at 37°C for 60 min. The samples were subsequently analyzed by Western blot (Chen et al., 2010; Xiang et al., 2007).
To identify the kinases that phosphorylate GRASP65 in vivo, HeLa cells stably expressing wild type rat GRASP65-GFP were first incubated with 100 ng/ml nocodazole for 18 h. Cells were then treated with one of the following chemicals: 1) a general kinase inhibitor staurosporine (at 2 µM) for 30 min (Talasz et al., 2009), 2) a cdk inhibitor roscovitine (at 40 µM) for 2 h (Gwinn et al., 2010), 3) a plk1 inhibitor BI2536 (at 0.1 µM) for 2 h (Lénárt et al., 2007; Park et al., 2011), and 4) a MAPK inhibitor U0126 (at 20 µM) for 2 h (Hayne et al., 2000; Jesch et al., 2001). Cells were subsequently lysed and GRASP65-GFP was immunoprecipitated using an anti-GFP antibody. The phosphorylation of rat GRASP65-GFP was revealed via Western blot using the indicated phospho-specific GRASP65 antibodies.
Cell lines and microscopy
To establish stable cell lines expressing various rat GRASP65 constructs, rtTAm2 HeLa cells were infected with retroviral particles encoding indicated exogenous genes cloned in the pRevTRE2 vector. Wild type rat GRASP65 and the related mA, mB, mC and mG mutants were first cloned into the pEGFP-N2 vector via the EcoRI and ApaI sites. cDNA encoding GFP alone, or GRASP65-GFP, mA-GFP, mB-GFP, or mG-GFP were then excised by Hind III and NotI and religated into the pRevTRE2 vector. Viral preparation, infection and cell selection were performed as previously described (Tang et al., 2010b; Xiang and Wang, 2010). GFP-positive cells were enriched by flow cytometry. The expression of exogenous proteins was induced with 1 µg/ml doxycycline.
Immunofluorescence microscopy was performed as previously reported (Tang et al., 2010b). To enrich mitotic cells, NRK cells, or HeLa cell lines expressing rat GRASP65-GFP, were first synchronized with 2 mM thymidine for 24 h and then incubated in regular growth medium without thymidine for 8 h prior to fixation. Images were obtained with a Leica SP5 confocal laser scanning microscope using a 100× oil objective. For metaphase cells, images were captured using fixed parameters with 0.3 µm intervals at the z-axis and then processed for maximum value projection. For triple labeling of phosphorylated GRASP65 in mitotic NRK cells, three secondary antibodies, goat-anti-mouse IgG-FITC, donkey-anti-human IgG-Cy3 and goat-anti-rabbit IgG-Cy5 were used to stain LX108, R3G3 or RB7, and Mary, respectively.
Quantification of immunofluorescence intensity
Whole-cell fluorescence intensity, generated by the respective phospho-antibody at different cell cycle stages, was quantified using the NIH ImageJ software. For better comparison of the relative changes between the three antibodies, the maximum total cellular intensity of each antibody detected throughout the cell cycle was normalized to 100%. The cell cycle stages were defined based on the chromosome shape, Golgi structure and phospho-Histone H3 staining. Late G2 phase cells were distinguished from interphase cells based on phospho-Histone H3 staining. Cells in cytokinesis were identified as cell pairs, which were not fully separated and had relatively intact nuclei and Golgi structure.
To determine the proportion of fluorescence signal attributed to specific recognition of Golgi clusters, mitotic cells expressing various GRASP65 constructs as indicated were additionally immunostained for GM130. Whole-cell fluorescence intensity was quantified using the NIH ImageJ software and using an adjacent cell free region as background. The light intensity on mitotic Golgi clusters (larger bright dots on fluorescence images) was quantified using the Analyze Particle function of ImageJ. The percentage of the total fluorescence intensity emitted from mitotic Golgi clusters was then calculated.
Electron microscopy
Electron microscopy analysis was performed as described previously (Tang et al., 2010b), mitotic Golgi clusters were identified using morphological criteria and quantified by standard stereological techniques (Wang et al., 2005). For a cell to be considered mitotic, the profile had to contain one or more condensed chromosomes and lack a nuclear envelope. Often multiple condensed chromosomes were aligned in the center of the cell. A low magnification (normally 1,600×) image and serial images of higher magnification (normally 11,000×) were taken to cover the entire cell. At least 15 cells were examined and quantified for each cell line.
Supplementary Material
Acknowledgments
We thank Nobuhiro Nakamura and Graham Warren for antibodies, Yi Xiang and other members of the Wang Lab for suggestions and reagents. This work was supported by the National Institute of Health (GM087364) and American Cancer Society (RGS-09-278-01-CSM) (Y.W.). D.T. was supported by the Rackham Predoctoral Fellowship from the University of Michigan. Author contributions: Y.W., D.T. and H.Y. designed the research; O.V. and F.P. prepared the three scFv antibodies; D.T., H.Y. and Y.W. performed the experiments; Y.W., D.T. and H.Y. analyzed the data; Y.W. and D.T. prepared the manuscript; Y.W., D.T., H.Y. and O.V. edited the paper.
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- Barr F. A., Puype M., Vandekerckhove J., Warren G. (1997). GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 91, 253–262 10.1016/S0092-8674(00)80407-9 [DOI] [PubMed] [Google Scholar]
- Bisel B., Wang Y., Wei J. H., Xiang Y., Tang D., Miron–Mendoza M., Yoshimura S., Nakamura N., Seemann J. (2008). ERK regulates Golgi and centrosome orientation towards the leading edge through GRASP65. J. Cell Biol. 182, 837–843 10.1083/jcb.200805045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Simon E. S., Xiang Y., Kachman M., Andrews P. C., Wang Y. (2010). Quantitative proteomics analysis of cell cycle-regulated Golgi disassembly and reassembly. J. Biol. Chem. 285, 7197–7207 10.1074/jbc.M109.047084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A., Deerinck T. J., Ellisman M. H., Malhotra V. (2000). A specific activation of the mitogen-activated protein kinase kinase 1 (MEK1) is required for Golgi fragmentation during mitosis. J. Cell Biol. 149, 331–339 10.1083/jcb.149.2.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo G., Prencipe L., Iodice L., Beznoussenko G., Savarese M., Marra P., Di Tullio G., Martire G., De Matteis M. A., Bonatti S. (2009). GRASP65 and GRASP55 sequentially promote the transport of C-terminal valine-bearing cargos to and through the Golgi complex. J. Biol. Chem. 284, 34849–34860 10.1074/jbc.M109.068403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J. M., Kinseth M., Bossard C., Rose D. W., Polishchuk R., Wu C. C., Yates J., Zimmerman T., Malhotra V. (2008). The role of GRASP55 in Golgi fragmentation and entry of cells into mitosis. Mol. Biol. Cell 19, 2579–2587 10.1091/mbc.E07-10-0998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein T. N., Linstedt A. D. (2008). GRASP55 regulates Golgi ribbon formation. Mol. Biol. Cell 19, 2696–2707 10.1091/mbc.E07-11-1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G. M., Giepmans B. N., Deerinck T. J., Smith W. B., Ngan L., Llopis J., Adams S. R., Tsien R. Y., Ellisman M. H. (2006). Golgi twins in late mitosis revealed by genetically encoded tags for live cell imaging and correlated electron microscopy. Proc. Natl. Acad. Sci. USA 103, 17777–17782 10.1073/pnas.0608509103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee H. Y., Noh S. H., Tang B. L., Kim K. H., Lee M. G. (2011). Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760 10.1016/j.cell.2011.07.021 [DOI] [PubMed] [Google Scholar]
- Giuliani F., Grieve A., Rabouille C. (2011). Unconventional secretion: a stress on GRASP. Curr. Opin. Cell Biol. 23, 498–504 10.1016/j.ceb.2011.04.005 [DOI] [PubMed] [Google Scholar]
- Gwinn D. M., Asara J. M., Shaw R. J. (2010). Raptor is phosphorylated by cdc2 during mitosis. PLoS ONE 5, e9197 10.1371/journal.pone.0009197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayne C., Tzivion G., Luo Z. (2000). Raf-1/MEK/MAPK pathway is necessary for the G2/M transition induced by nocodazole. J. Biol. Chem. 275, 31876–31882 10.1074/jbc.M002766200 [DOI] [PubMed] [Google Scholar]
- Jesch S. A., Lewis T. S., Ahn N. G., Linstedt A. D. (2001). Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol. Biol. Cell 12, 1811–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinseth M. A., Anjard C., Fuller D., Guizzunti G., Loomis W. F., Malhotra V. (2007). The Golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell 130, 524–534 10.1016/j.cell.2007.06.029 [DOI] [PubMed] [Google Scholar]
- Kondylis V., Spoorendonk K. M., Rabouille C. (2005). dGRASP localization and function in the early exocytic pathway in Drosophila S2 cells. Mol. Biol. Cell 16, 4061–4072 10.1091/mbc.E04-10-0938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A., Zhong C., Lane W. S., Derynck R. (2000). Transmembrane transforming growth factor-α tethers to the PDZ domain-containing, Golgi membrane-associated protein p59/GRASP55. EMBO J. 19, 6427–6439 10.1093/emboj/19.23.6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky M. S., Mastronarde D. N., McIntosh J. R., Howell K. E., Staehelin L. A. (1999). Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J. Cell Biol. 144, 1135–1149 10.1083/jcb.144.6.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lénárt P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J. J., Hoffmann M., Rettig W. J., Kraut N., Peters J. M. (2007). The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17, 304–315 10.1016/j.cub.2006.12.046 [DOI] [PubMed] [Google Scholar]
- Lin C. Y., Madsen M. L., Yarm F. R., Jang Y. J., Liu X., Erikson R. L. (2000). Peripheral Golgi protein GRASP65 is a target of mitotic polo-like kinase (Plk) and Cdc2. Proc. Natl. Acad. Sci. USA 97, 12589–12594 10.1073/pnas.220423497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. E., Erikson R. L., Lee K. S. (2011). Feed-forward mechanism of converting biochemical cooperativity to mitotic processes at the kinetochore plate. Proc. Natl. Acad. Sci. USA 108, 8200–8205 10.1073/pnas.1102020108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisinger C., Körner R., Wind M., Lehmann W. D., Kopajtich R., Barr F. A. (2005). Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 24, 753–765 10.1038/sj.emboj.7600569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu M. A., Bachert C., Puri S., Lanni F., Linstedt A. D. (2006). GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 8, 238–248 10.1038/ncb1366 [DOI] [PubMed] [Google Scholar]
- Seemann J., Pypaert M., Taguchi T., Malsam J., Warren G. (2002). Partitioning of the matrix fraction of the Golgi apparatus during mitosis in animal cells. Science 295, 848–851 10.1126/science.1068064 [DOI] [PubMed] [Google Scholar]
- Sengupta D., Linstedt A. D. (2010). Mitotic inhibition of GRASP65 organelle tethering involves Polo-like kinase 1 (PLK1) phosphorylation proximate to an internal PDZ ligand. J. Biol. Chem. 285, 39994–40003 10.1074/jbc.M110.189449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D., Truschel S., Bachert C., Linstedt A. D. (2009). Organelle tethering by a homotypic PDZ interaction underlies formation of the Golgi membrane network. J. Cell Biol. 186, 41–55 10.1083/jcb.200902110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima D. T., Cabrera–Poch N., Pepperkok R., Warren G. (1998). An ordered inheritance strategy for the Golgi apparatus: visualization of mitotic disassembly reveals a role for the mitotic spindle. J. Cell Biol. 141, 955–966 10.1083/jcb.141.4.955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J., Watson R., Giannakou M. E., Clarke M., Warren G., Barr F. A. (1999). GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 18, 4949–4960 10.1093/emboj/18.18.4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sütterlin C., Lin C. Y., Feng Y., Ferris D. K., Erikson R. L., Malhotra V. (2001). Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc. Natl. Acad. Sci. USA 98, 9128–9132 10.1073/pnas.161283998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sütterlin C., Polishchuk R., Pecot M., Malhotra V. (2005). The Golgi-associated protein GRASP65 regulates spindle dynamics and is essential for cell division. Mol. Biol. Cell 16, 3211–3222 10.1091/mbc.E04-12-1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talasz H., Sarg B., Lindner H. H. (2009). Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma 118, 693–709 10.1007/s00412-009-0228-2 [DOI] [PubMed] [Google Scholar]
- Tang D., Mar K., Warren G., Wang Y. (2008). Molecular mechanism of mitotic Golgi disassembly and reassembly revealed by a defined reconstitution assay. J. Biol. Chem. 283, 6085–6094 10.1074/jbc.M707715200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Xiang Y., Wang Y. (2010a). Reconstitution of the cell cycle-regulated Golgi disassembly and reassembly in a cell-free system. Nat. Protoc. 5, 758–772 10.1038/nprot.2010.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Yuan H., Wang Y. (2010b). The role of GRASP65 in Golgi cisternal stacking and cell cycle progression. Traffic 11, 827–842 10.1111/j.1600-0854.2010.01055.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Xiang Y., De Renzis S., Rink J., Zheng G., Zerial M., Wang Y. (2011). The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle. Nat. Commun. 2, 501 10.1038/ncomms1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truschel S. T., Sengupta D., Foote A., Heroux A., Macbeth M. R., Linstedt A. D. (2011). Structure of the membrane-tethering GRASP domain reveals a unique PDZ ligand interaction that mediates Golgi biogenesis. J. Biol. Chem. 286, 20125–20129 10.1074/jbc.C111.245324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truschel S. T., Zhang M., Bachert C., Macbeth M. R., Linstedt A. D. (2012). Allosteric regulation of GRASP protein-dependent Golgi membrane tethering by mitotic phosphorylation. J. Biol. Chem. 287, 19870–19875 10.1074/jbc.M111.326256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielemeyer O., Yuan H., Moutel S., Saint–Fort R., Tang D., Nizak C., Goud B., Wang Y., Perez F. (2009). Direct selection of monoclonal phosphospecific antibodies without prior phosphoamino acid mapping. J. Biol. Chem. 284, 20791–20795 10.1074/jbc.M109.008730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinke F. P., Grieve A. G., Rabouille C. (2011). The multiple facets of the Golgi reassembly stacking proteins. Biochem. J. 433, 423–433 10.1042/BJ20101540 [DOI] [PubMed] [Google Scholar]
- Wang Y. (2008). Golgi apparatus inheritance. The Golgi Apparatus: State Of The Art 110 Years After Camillo Golgi's Discovery (ed. Mironov A, Pavelka M, Luini A.), pp. 580–607 New York: Springer. [Google Scholar]
- Wang Y., Seemann J. (2011). Golgi biogenesis. Cold Spring Harb. Perspect. Biol. 3, a005330 10.1101/cshperspect.a005330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Seemann J., Pypaert M., Shorter J., Warren G. (2003). A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J. 22, 3279–3290 10.1093/emboj/cdg317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Satoh A., Warren G. (2005). Mapping the functional domains of the Golgi stacking factor GRASP65. J. Biol. Chem. 280, 4921–4928 10.1074/jbc.M412407200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Taguchi T., Warren G. (2006). Purification of rat liver golgi stacks. Cell Biology: A Laboratory Handbook, 3rd edition, (ed. Celis J E.), pp. 33–39 San Diego: Elsevier Academic Press. [Google Scholar]
- Xiang Y., Wang Y. (2010). GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J. Cell Biol. 188, 237–251 10.1083/jcb.200907132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Wang Y. (2011). New components of the Golgi matrix. Cell Tissue Res. 344, 365–379 10.1007/s00441-011-1166-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Seemann J., Bisel B., Punthambaker S., Wang Y. (2007). Active ADP-ribosylation factor-1 (ARF1) is required for mitotic Golgi fragmentation. J. Biol. Chem. 282, 21829–21837 10.1074/jbc.M611716200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura S., Yoshioka K., Barr F. A., Lowe M., Nakayama K., Ohkuma S., Nakamura N. (2005). Convergence of cell cycle regulation and growth factor signals on GRASP65. J. Biol. Chem. 280, 23048–23056 10.1074/jbc.M502442200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}