

Summary
Bacterial invasion results in the rapid induction of an acute state of cytosolic amino acid (AA) starvation, provoked by host membrane damage. Bacteria-induced AA starvation, in turn, down-regulates mTOR signaling while triggering autophagy and the integrated stress response pathway dependent on GCN2, eIF2α and ATF3. In Salmonella-infected cells, we now demonstrate that the host AA starvation response program depended on the Salmonella pathogenicity island (SPI)-1, the activity of which was required to damage the Salmonella-containing vacuole (SCV) in the early stage of infection. At a later stage (3–4 hour post-infection), the progressive recruitment of mTOR to the surface of the SCV appeared to be independent of the activity of SPI-2 and of SCV positioning in the cell. Instead, mTOR localization to the SCV required the activity of host AA transporters SLC1A5, SLC3A2 and SLC7A5, resulting in bacterial escape from autophagy. These results expand our understanding of the mechanisms underlying the AA starvation response in Salmonella-infected cells.
Key words: Amino acid starvation, Salmonella, mTOR
Introduction
All prokaryotic and eukaryotic cells have evolved intrinsic mechanisms to monitor nutrient availability in both the extra- and intracellular environment. In turn, these nutrient-sensing systems regulate key cellular processes, such as cell growth, cell division, gene expression and most, if not all, biosynthetic pathways. In eukaryotes from yeast to mammals, the serine/threonine kinase TOR (Target of Rapamycin) is a fundamental metabolic checkpoint kinase (Zoncu et al., 2011). In mammals, this kinase (termed mammalian TOR, or mTOR) integrates multiple pathways triggered by glucose, growth factors, oxygen tension and ATP levels (Sengupta et al., 2010), which all converge to activate the Rheb GTPase at the surface of late endosomes/lysosomes (LE/Ly), where Rheb is constitutively located (Laplante and Sabatini, 2009). Upon engagement of Rheb, a complex comprised of mTOR, Raptor and mLST8 (together, known as the mTOR complex 1 or mTORC1) is activated, which controls key cellular functions, such as mRNA translation, cell growth and ribosomal biogenesis, in part through the phosphorylation of S6K1 and 4EBP1 (Laplante and Sabatini, 2009; Sengupta et al., 2010; Zoncu et al., 2011).
Another critical role of mTOR is to repress autophagy. Autophagy (literally “self-eating”) is a fundamental cellular process, conserved from yeast to mammals, which results in the degradation by lysosomes and the recycling of cellular constituents (such as defective organelles or large macromolecular complexes) that have been engulfed into an isolation membrane (Klionsky, 2007). While a basal level of autophagy is important to sustain constitutive scavenging functions in normally growing, nutrient-replete cells, autophagy activity increases in conditions of nutrient or energy deprivation, thus allowing nutrient recycling to sustain essential metabolic functions during starvation (Klionsky, 2007). The fundamental role of mTOR as a repressor of autophagy is well characterized (Ganley et al., 2009; Hosokawa et al., 2009; Jung et al., 2009), and rapamycin is one of the most commonly used drugs to experimentally induce autophagy.
In addition to its critical role in nutrient recycling, autophagy has recently emerged as an important arm of the host innate immune arsenal against invasive bacterial pathogens (Birmingham et al., 2007; Birmingham et al., 2006; Campoy and Colombo, 2009; Deretic and Levine, 2009; Huang and Brumell, 2009; Ogawa and Sasakawa, 2006; Py et al., 2007; Rich et al., 2003). Indeed, a number of studies recently demonstrated that intracellular bacteria are rapidly trapped into double membranes characteristic of autophagosomes, following recruitment of proteins of the autophagic machinery. This promotes the delivery of the cargo to lysosomes, resulting in the destruction of the engulfed bacteria (this process is often referred to as “xenophagy”). An important question in the field of bacterial autophagy is to understand the mechanisms responsible for the specific targeting of bacteria, over other cellular constituents, by the autophagy system. It was recently shown that peri-bacterial accumulation of ubiquitin serves as a signal to recruit host proteins such as p62 and NDP52, which could in turn favor the accumulation of the autophagy protein LC3 around intracellular bacteria (Cemma et al., 2011; Deretic, 2010; Dupont et al., 2010; Ivanov and Roy, 2009; Randow, 2011; Thurston et al., 2009; Zheng et al., 2009). In addition, membrane damage inflicted by bacteria was shown to promote the peri-bacterial accumulation of galectins 3 and 8, which could also recruit NDP52 (Thurston et al., 2012). Finally, Toll-like receptors (TLRs) and Nod-like receptors (NLRs) contribute to the targeting of invasive bacteria by the autophagy system (Cooney et al., 2010; Travassos et al., 2010; Xu et al., 2007).
Salmonella enterica serovar Typhimurium is a Gram-negative bacterial human pathogen that infects the intestinal tract. In non-myeloid cells, Salmonella uses a type three secretion system (TTSS) encoded by the Salmonella pathogenicity island 1 (SPI-1) to invade the host cytosol, where the bacterium resides in a specific Salmonella containing vacuole (SCV) (Ramsden et al., 2007a; Steele-Mortimer, 2008). The progressive maturation of the SCV is a complex process that requires the action of specific bacterial effectors delivered sequentially into the host cytosol, and encoded by the SPI-1 locus in the initial 1–2 h post-infection (p.i.) phase, and later on by the SPI-2 locus (Ramsden et al., 2007a; Steele-Mortimer, 2008). Overall, the SCV maturation is characterized by the progressive acquisition of early then late endosomal markers, as well as the precise sub-cellular trafficking of the SCV in the host cell: in a first phase (approx. 1–2 h p.i.), the SCV displays a centripetal movement towards the nucleus, while the action of SPI-2 effectors later promotes the relocalization of the SCV to the periphery of the Golgi apparatus (Ramsden et al., 2007a; Ramsden et al., 2007b; Steele-Mortimer, 2008).
We have recently demonstrated that cellular invasion with Salmonella triggered an acute and transient state of amino acid (AA) starvation, which altered mTOR sub-cellular localization and activity, thereby impacting on host defense pathways, including anti-bacterial autophagy (Tattoli et al., 2012). However, whether AA starvation-dependent responses to bacterial invasion are directly modulated by Salmonella remains unclear. In addition, the mechanism underlying the cellular adaptation to AA starvation in infected cells is also unexplored. Here, we demonstrate that the rapid induction of AA starvation in Salmonella-infected cells depends on the activity of the SPI-1, likely as a result of early damage to the SCV membrane. However, normalization of cytosolic AA levels and reactivation of mTOR signaling during the SCV maturation phase appeared to be unrelated to either the activity of the bacterial SPI-2 system or the SCV positioning in infected cells. Instead, mTOR reactivation and targeting to the surface of the SCV were dependent on the active transport of AA by the host AA transporter systems composed of SLC1A5, SLC3A2 and SLC7A5. Notably, infection with Salmonella resulted in a transient redistribution of SLC7A5 sub-cellular localization during the peak phase of AA starvation. These results delineate the critical interplay between bacterial and host determinants in the regulation of AA starvation-dependent mTOR signaling alterations in Salmonella-infected cells.
Materials and Methods
Antibodies and reagents
Mouse anti-LAMP2 (ab25631) and Rabbit anti-Phospho-GCN2 (ab75836), anti-calcoco2 (NDP52) (ab68588), SLC7A5 (AB18493) were from Abcam; mouse anti-tubulin clone DM1A (T9026), Sigma; p62 (human), (BML-pw9860-0100) Enzo Life Sciences; rabbit anti-Phospho-p70S6 Kinase (Thr389) (no. 92345), rabbit anti-S6K1 (no. 9202), rabbit anti-Myc (no. 2278), Cell Signaling Technology; mouse anti-ubiquitin (clone FK2), Millipore; mouse anti-Golgin-97 (clone CDF4), Invitrogen; Goat anti-rabbit IgG and Goat anti-mouse IgG peroxidase conjugated, Thermo Scientific USA; FITC-conjugated Goat anti-rabbit and Cy3-conjugated Goat anti-mouse, Jackson ImmunoResearch Laboratories, Canada. 4′, 6-diamidino-2-phenylindole (DAPI), Vector Laboratories; Dynasore (no. D7693), Brefeldin A (no. B7651), D-Phenylalanine (no. P1751), L-Isoleucine (no. I2752) and were from Sigma. L-Leucine (no. Leu222.100) from BioShop Life Science Products; L-Glutamine was from GIBCO Invitrogen.
Bacterial strains and cell culture
Salmonella typhimurium SL1344, ΔSPI-2 (from K. Tedin, Freie Universität, Berlin) and ΔSPI-1/Inv (from J. Galan, Yale University) strains were grown in Luria–Bertani broth (LB; Invitrogen by life Technology). Human breast carcinoma epithelial cells (MDAMC cells) stably transfected with GFP-LC3 were from Dr Yoshimori (Osaka Univ.). The human epithelial HeLa cell line (American Type Culture Collection) and MDAMC cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% Fetal Calf Serum (FCS), 2 mM L-glutamine, 50 IU of penicillin and 50 µg/ml streptomycin. Cells were maintained in 95% air, 5% CO2 at 37°C. Endotoxin-free FCS and Phosphate Buffer Saline (PBS) were from Wisent (St-Bruno, Quebec).
Bacterial infection
Overnight bacterial cultures Salmonella WT, ΔSPI-2 and ΔSPI-1/Inv strains were diluted 100-fold and grown to exponential phase (OD600 = 0.4 to 0.6) in aerobic conditions, collected by centrifugation 5,000 g for 5 min, washed in saline buffer (150 mM NaCl) and resuspended in DMEM. Cells cultured in antibiotic-free medium were infected at an MOI of 100, centrifuged (2,000 g for 15 min at 37°C), and incubated at 37°C/5% CO2 for 30 min. Cells were washed 3× with PBS and fresh medium containing gentamicin (50 µg/ml) added. Where indicated, Dynasore, Brefeldin and D-Phenylalanine were added to the cells 30 min after HeLa cells being infected in order to avoid potential side effects on bacteria entry.
Buffer for amino acid starvation
Cells rinsed three times with PBS were incubated in Krebs Ringer Bicarbonate (KRB) buffer (118.5 mM NaCl, 4.74 mM KCl, 1.18 mM KH2PO4, 23.4 mM NaHCO3, 5 mM glucose, 2.5 mM CaCl2, 1.18 mM MgSO4, adjusted to pH 7.6 by titration with 1 N NaOH).
Immunofluorescence microscopy
HeLa cells placed on glass coverslips were processed for IF as previously described (Travassos et al., 2010). Samples were visualized on a Carlo Zeiss Axiovert 200 microscope with a 63× oil fluorescence objective, and images analyzed using Volocity software (Quorum Technologies). Nuclei and bacteria were visualized using DAPI staining.
Western blotting
Cells were washed twice in cold PBS, lysed in ice-cold lysis buffer: 40 mM HEPES [pH 7.4], 120 mM NaCl, 1 mM EDTA, 0.3% CHAPS, EDTA-free protease inhibitors and Phosphatase inhibitor cocktail (Roche). Soluble fractions of lysates were isolated by centrifugation at 12,000 rpm for 10 min at 4°C. Protein concentration was determined using Bradford (Pierce).
shRNA lentivirus packaging and transduction
The following shRNA sequences 5′-CCGGTCCTGTACCGTCCTCAA-3′ from Human SLC1A5; 5′-CAGATCCTGAGCCTACTCGAA-3′ from Human SLC3A2; 5′-TGCTAACGTCTTACTAATTTA-3′ from Human SLC7A5, were inserted into the pLKO.1 vector (Addgene). Packaging and purification of shRNA-expressing lentivirus, using the lentiviral packaging/envelope vectors psPAX2 and pMD2.G, were performed according to procedures previously described (Benko et al., 2010), with few adjustments: cells were systematically analyzed 3–4 days after lentiviral transduction, and neomycin selection was omitted.
Expression vectors and transfection
Expression vector for Myc-SLC7A5 was from Origene. GFP-RILP was a gift from S. Grinstein (Hospital for Sick Children, Toronto). Transfection was performed using Fugene (Roche) according to the manufacturer's instructions.
Quantitative PCR
Real-time PCR analysis of ATF3 (Forward 5′-CTGGGTCACTGGTGTTTGAGGATT-3′, Reverse 5′AGGTGCTTGTTCTGGATGGCAAAC3′), IL-8 (Forward 5′-ACTGAGAGTGATTGAGAGTGGAC-3′, Reverse 5′-AACCCTCTGCACCCAGTTTTC-3′), expression was performed using SYBR green reagents, and normalized to the endogenous housekeeping control gene, TBT, as previously described (Benko et al., 2010).
Statistical analysis and experimental representation
Significant differences between mean values were evaluated with a one-sample or unpaired t test. All the experiments presented in this manuscript have been performed at least three times with similar results and representative illustrations are provided.
Results
SPI-1-dependent SCV membrane damage causes early host AA starvation responses
NDP52 was recently shown to target damaged vesicular membranes through a mechanism involving galectin proteins and ubiquitin (Thurston et al., 2009; Thurston et al., 2012). Accordingly, NDP52 accumulates at the surface of the SCV at 1 h p.i. (Cemma et al., 2011; Tattoli et al., 2012; Thurston et al., 2009), at a time when the activity of the SPI-1 system induces transient damage to the SCV membrane (Birmingham et al., 2006). We recently observed that membrane damage (either aseptic or bacteria-induced) was sufficient to trigger an AA starvation response in epithelial cells, which provided correlative evidence that Salmonella-induced early AA starvation response was likely caused by SPI-1-dependent membrane damage (Tattoli et al., 2012). We aimed to obtain a direct proof of this hypothesis, but a complication comes from the fact that SPI-1-deficient Salmonella fails to invade non-phagocytic cells altogether, making a direct analysis of the impact of SPI-1 activity on early SCV membrane impossible. To circumvent this problem, we used a Salmonella mutant strain (ΔSPI-1/Inv) deficient for SPI-1 but for which invasion capacity was functionally complemented by ectopic expression of the Yersinia invasin protein (Inv), thereby allowing efficient Inv-dependent invasion through a “zipper” mechanism. We observed by immunofluorescence (IF) that infection of human HeLa epithelial cells with wild type (WT) Salmonella resulted in the accumulation of NDP52 at the surface of the SCV at 2 h p.i. (as well as at 1 h p.i., data not shown), but no longer at 4 h p.i., illustrating the transient nature of the SCV membrane damage (Fig. 1A), in agreement with our previous study (Tattoli et al., 2012). Interestingly, although the Salmonella mutant strain ΔSPI-1/Inv invaded HeLa cells as efficiently as WT Salmonella (data not shown), NDP52 did not accumulate at the surface of the SCV at any time following infection (Fig. 1A), thus supporting the notion that early damage to the SCV was driven by the activity of the SPI-1 system, in agreement with a previous study (Birmingham et al., 2006).
Fig. 1. SPI-1-dependent SCV membrane damage causes early host AA starvation responses.

(A,B) HeLa cells were infected with wild type (WT) or ΔSPI-1/Inv Salmonella strain for 2 h or 4 h, analyzed by IF using antibodies against NDP52 and LAMP2 (A) or mTOR and LAMP2 (B). (C) qPCR analysis of AFT3 and IL-8 induction in HeLa cells infected with WT or ΔSPI-1/Inv Salmonella strains. Values are means s.e.m. n = 3.
Using this experimental system, we then aimed to provide a direct demonstration that Salmonella-induced early AA starvation and mTOR inhibition were SPI-1-dependent. Infection with WT Salmonella resulted in a transient and reversible dispersion of mTOR away from LAMP2-positive LE/Ly vesicles (Fig. 1B), correlating with an AA starvation state, as we previously demonstrated (Tattoli et al., 2012). Interestingly, infection with the Salmonella mutant strain ΔSPI-1/Inv did not result in the cytosolic dispersion of mTOR staining, as we observed strong colocalization between mTOR and LAMP2 at the surface of the SCVs at both 2 h and 4 h p.i. (Fig. 1B). This result strongly suggests that, although Salmonella ΔSPI-1/Inv could efficiently invade host cells, the induction of cytosolic AA starvation and inhibition of mTOR targeting to host endomembranes by Salmonella was a process that fully depended on the activity of the SPI-1.
We recently demonstrated that, in addition to the inhibition of mTOR signaling via cytosolic dispersion of mTORC1 away from host endomembranes, bacteria-induced AA starvation also resulted in induction of the AA stress response pathway dependent on GCN2 and ATF3 (Tattoli et al., 2012). We followed the regulation of this pathway by measuring the induction of ATF3 by quantitative PCR (qPCR). In the same samples, we also measured IL-8 induction as a way to analyze an ATF3-independent, NF-κB-dependent signaling cascade, which is triggered by infection. Interestingly, while infection with WT Salmonella triggered a massive yet reversible induction of ATF3 mRNA at 1 h p.i., as we previously reported (Tattoli et al., 2012), infection with the Salmonella ΔSPI-1/Inv resulted in a considerably blunted induction of ATF3 (Fig. 1C). This was not explained by a general impairment in host detection of the Salmonella ΔSPI-1/Inv strain since IL-8 induction was comparable between the two strains, at least in the early phase of infection (Fig. 1C). Together, these results provide evidence that the early induction of a host AA starvation response in Salmonella-infected cells was dependent on the activity of the SPI-1, and was likely caused by SPI-1-dependent damage to the SCV membrane.
Membrane recruitment of NDP52, p62 and ubiquitin is not influenced by AA starvation
The results presented above, together with our previous observations (Tattoli et al., 2012), highlight the fact that the damage to the SCV (as observed by the targeting of this vacuole by NDP52) and AA starvation are two synchronous and transient events. The fact that aseptic damage to host membranes can cause NDP52 accumulation (Tattoli et al., 2012; Thurston et al., 2012) also strongly suggests that membrane damage directly causes AA starvation through an undefined pathway, and thus implying that SPI-1-dependent insult to the SCV is transient in nature, resulting in normalization of NDP52 staining and cytosolic AA pools at later times p.i. Nevertheless, it is also possible that a reverse scenario takes place and that, in Salmonella-infected cells, cytosolic AA starvation is the primary event that causes SCV membrane instability and NDP52 recruitment. In this second hypothesis, the replenishment of cytosolic AA pools observed at later stages of infection (3–4 h p.i.), which we previously showed to be dependent on internalization of extracellular AAs (Tattoli et al., 2012), would favor the normalization of SCV membrane integrity and detachment of NDP52 from these vacuoles. To test these hypotheses, we followed by IF the temporal recruitment of not only NDP52, but also p62 and ubiquitin, in cells infected with Salmonella in either normal AA-rich DMEM medium or in AA starvation Krebs–Ringer Buffer (KRB). Interestingly, we observed that NDP52 targeting to the SCV occurred at 2 h p.i. but not 4 h p.i. when cells were infected in both media (Fig. 2), thus supporting the contention that SCV damage is likely a transient phenomenon that peaks during the first 2 h p.i. Similar to NDP52, infection in either DMEM or KRB did not affect the dynamics of p62 or ubiquitin recruitment to the SCV (Fig. 2). However, we also noticed that, contrary to NDP52, these markers remained strongly associated with the SCV even at 4 h p.i. (Fig. 2), suggesting that NDP52 is likely a better marker of the dynamic regulation of membrane damage/healing at the surface of the SCVs than p62 or ubiquitin accumulation.
Fig. 2. Membrane recruitment of NDP52, p62 and ubiquitin is not influenced by AA starvation.

HeLa cells were infected with Salmonella in either a normal AA-rich DMEM medium or in an AA-depleted medium (KRB) for 1 h, 2 h or 4 h. Next, cells were fixed and analyzed by IF using antibodies against NDP52, p62 and ubiquitin.
Enforced peri-nuclear clustering of SCVs does not impact on the recruitment of mTOR
We next aimed to gain a better understanding of the mechanisms underlying the normalization of mTOR endomembrane localization at late stages of infection (3–4 h p.i.) and the accumulation of this protein to the surface of the maturing SCVs, as we previously reported (Tattoli et al., 2012). We previously reported that such mTOR accumulation to the SCV correlated with normalization of cytosolic AA pools and required the presence of extracellular AAs, thus suggesting that the internalization of AAs was a required step (Tattoli et al., 2012). However, it remains to be defined if AA internalization is not only necessary but also sufficient to allow mTOR localization to the SCV. Indeed, critical events occur during SCV maturation at 3–4 h p.i.; SCVs migrate in a centripetal fashion along microtubules in an early phase, followed by centrifugal movement and clustering occurring in the vicinity of the Golgi apparatus. Also during this time-point, genes from SPI-2 are expressed, which are critical for SCV maturation at later stages of infection (Bakowski et al., 2008; Fass and Groisman, 2009; Gorvel and Méresse, 2001; Ramsden et al., 2007a; Ramsden et al., 2007b; Steele-Mortimer, 2008). We first analyzed mTOR targeting to the SCV in conditions where normal SCV trafficking was impaired. To do so, cells were transfected with a plasmid encoding GFP-tagged Rab7 interacting lysosomal protein (RILP). RILP drives endosomal targeting to the microtubule organizing center (MTOC) by linking Rab7, which is expressed at the surface of LE/Ly and also SCVs, with the microtubule motor molecule dynein (Harrison et al., 2004; Marsman et al., 2004). As expected, over-expression of GFP-RILP resulted in the massive accumulation of mTOR to GFP-RILP+ LE/Ly vesicles to the nuclear periphery and in particular in the concave curvature of bean-shaped nuclei where the MTOC localizes, in uninfected conditions (Fig. 3). From these control experiments, we also noted that the massive peri-nuclear accumulation of LE/Ly did not seem to inhibit mTOR localization to these vesicles because mTOR was found to colocalize very strongly with GFP-RILP in these conditions (Fig. 3). Next, cells were infected with Salmonella WT for 2 h or 4 h. Interestingly, at the 2 h p.i. time-point, we observed that mTOR localization to GFP-RILP+ LE/Ly was severely blunted overall (Fig. 3 and inset no. 1), similar to the general decrease of mTOR targeting to LE/Ly in non-transfected cells infected for 2 h with Salmonella that we had observed previously (Tattoli et al., 2012). This suggests that Salmonella-induced transient AA starvation affects mTOR localization to LE/Ly independently from the positioning of these vesicles in infected cells. Three important observations could be made at the 4 h p.i. time-point. First, at this stage corresponding to cytosolic AA pool normalization in infected cells (Tattoli et al., 2012), mTOR was found to relocalize to GFP-RILP+ LE/Ly as expected (Fig. 3 and inset no. 2). Second, we noted that Salmonella remained localized mainly at the vicinity of the MTOC at 4 h p.i. in GFP-RILP-expressing cells (Fig. 3 and inset no. 2), in agreement with previous studies demonstrating that the centrifugal movement of the SCV requires the action of the late bacterial effector SifA to uncouple Rab7 from RILP (Guignot et al., 2004; Harrison et al., 2004). Third, we observed that mTOR also efficiently relocalized to the SCVs in these conditions (Fig. 3 and inset no. 2), thus showing that the centrifugal movement of these vacuoles, typically occurring in the second phase of the infection, was not a prerequisite for mTOR targeting to the SCV at 3–4 h p.i.
Fig. 3. Enforced peri-nuclear clustering of SCVs does not impact on the recruitment of mTOR.
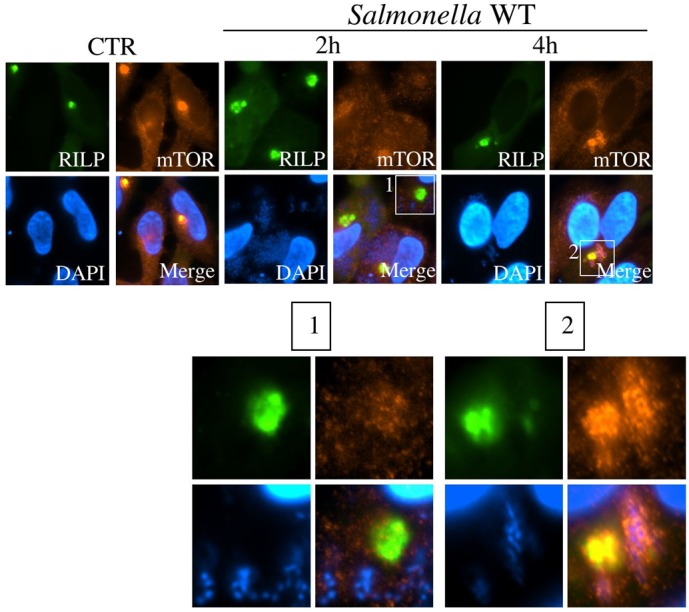
HeLa cells were transfected with an expression vector encoding for RILP-GFP, infected with Salmonella for 2 h or 4 h, and mTOR sub-cellular localization was identified by IF using an anti-mTOR antibody.
AA starvation responses in Salmonella-infected cells are SPI-2-independent
Because normalization of AA starvation and mTOR localization to host endomembranes was shown to occur at 3–4 h p.i. in Salmonella-infected cells (Tattoli et al., 2012), a time which coincides with the initial expression of the genes from SPI-2, we aimed to directly determine the influence of SPI-2 in mTOR targeting to the SCV. Using a Salmonella strain functionally deficient for the expression of SPI-2 genes (ΔSPI-2), we observed that mTOR relocalization to the maturing SCV at 4 h p.i. was similar in cells infected with WT or ΔSPI-2 strains of Salmonella (Fig. 4A). Moreover, GCN2 and S6K1 phosphorylation profiles were comparable in cells infected with WT and ΔSPI-2 Salmonella (Fig. 4B), and so were also the induction levels of ATF3 and IL-8 in qPCR (Fig. 4C). Together, the results obtained with GFP-RILP over-expression and by comparing the host response to Salmonella WT or ΔSPI-2 strains, support the contention that normalization of host cytosolic AA starvation and mTOR targeting to host endomembranes do not depend on SCV sub-cellular positioning or bacterial manipulation through the SPI-2 system. Therefore, these observations strongly suggest that host-mediated internalization of extracellular AA into the host cytosol, which we previously showed to be critical for the normalization of cytosolic AA pools (Tattoli et al., 2012), is the principal event underlying mTOR localization to the maturing SCV in Salmonella-infected cells.
Fig. 4. AA starvation responses in Salmonella-infected cells are SPI-2-independent.

(A) HeLa cells were infected with wild type (WT) or ΔSPI-2 Salmonella strains for 4 h, analyzed by IF using antibodies against mTOR and LAMP2. (B) HeLa cells were infected with the WT or ΔSPI-2 Salmonella strain for 1 h or 3 h, analyzed by blotting using the antibodies indicated. (C) qPCR analysis of AFT3 and IL-8 induction in Hela cells infected with WT or ΔSPI-2 Salmonella strain. Values are means ± s.e.m.
Essential role of LNAAs, L-glutamine and LNAA transporters in mTOR targeting to the SCV
In light of the above results, we next aimed to better understand the mechanisms underlying the progressive normalization of cytosolic AA pools that takes place in Salmonella-infected cells at 3–4 h p.i. (Tattoli et al., 2012). While a role for internalization of extracellular AAs was demonstrated (Tattoli et al., 2012), it remains unclear whether specific AAs contribute to this effect. A recent study demonstrated that the heterodimeric bidirectional transporter SLC7A5/SLC3A2, which mediates the cytosolic internalization of Large Neutral AAs (LNAAs) such as L-leucine and L-isoleucine, in exchange for L-glutamine, was a critical regulator of mTOR activity (Nicklin et al., 2009). As expected, in cells that were AA-starved in KRB buffer, addition of L-leucine or L-isoleucine, but not L-glutamine, was sufficient to restore mTOR localization to LAMP2+ LE/Ly vesicles in uninfected cells (Fig. 5A). Similarly, mTOR relocalized to the SCV when infection was performed in AA-free starvation buffer KRB supplemented with either L-leucine or L-isoleucine but not L-glutamine (Fig. 5B), although reconstitution was not complete and did not reach the levels observed when infection was performed in DMEM (Fig. 5C).
Fig. 5. Essential role of LNAAs and L-glutamine in mTOR targeting to the SCV.

(A,B) HeLa cells were placed in AA-depleted medium (KRB) supplemented with the indicated AA, left uninfected (A) or infected with Salmonella for 4 h (B), and analyzed by IF using antibodies against mTOR and LAMP2. (C) Percentage of cells displaying mTOR targeting to the SCV following infection by Salmonella in AA-depleted medium supplemented with various AA or DMEM. Values are means s.e.m. n = 3. *P<0.05, **P<0.01 over infected in KRB (−AA). (D) HeLa cells were placed in various minimal media (−AA, −AA/L-Leu, −AA/L-Gln, −AA/L-Gln+L-Leu) as indicated, and infected with Salmonella for 4 h. At 2 h p.i., medium was removed and replaced for an additional 2 h. Next, cells were fixed and analyzed by IF using antibodies against mTOR and LAMP2. (E) HeLa cells were incubated in various media as indicated, left uninfected or infected with Salmonella for 4 h, analyzed by blotting using the antibodies indicated. In the “DMEM → KRB 2 h” condition, cells were incubated for 2 h in DMEM before medium changed to KRB for an additional 2 h.
Although supplementation of KRB with L-glutamine was not sufficient to restore mTOR targeting to SCVs in Salmonella-infected cells, we speculated that if this process were dependent on SLC7A5/SLC3A2, it would nonetheless require cellular pools of L-glutamine to allow for LNAA/L-glutamine exchange (Nicklin et al., 2009). In support for this, we noticed that while addition of L-leucine to KRB was sufficient to direct sub-optimal recruitment of mTOR to the SCV, medium replacement during the course of infection resulted in a strong dependency on supplementation with L-glutamine, suggesting that this AA was rapidly exported to the extracellular milieu in Salmonella-infected cells (Fig. 5D). Moreover, L-glutamine addition potentiated mTOR recruitment to the SCV that was supported by L-leucine in KRB (Fig. 5C), suggesting that L-glutamine is a limiting factor for mTOR recruitment to the SCV. Finally, supplementation of KRB with L-glutamine + L-leucine resulted in substantial restoration of S6K1 activation in Salmonella-infected cells, to levels similar to cells infected in KRB supplemented with L-glutamine plus all essential AA (Fig. 5E).
The above results suggest that mTOR targeting to SCVs and S6K1 hyper-activation might depend on the action of LNAA transporters, such as SLC7A5/SLC3A2. In support of this, D-Phenylalanine (Fig. 6A) and 2-aminobicyclo-(2,2,1)heptane-2-carboxylic acid (BCH) (data not shown), two molecules that specifically inhibit the activity of LNAA transporters (Nicklin et al., 2009), potently blocked both mTOR recruitment to LE/Ly in uninfected cells and the targeting to the SCV in Salmonella-infected cells (Fig. 6A). Next, we observed that lentiviral-mediated knockdown of SLC7A5 expression resulted in poor recruitment of mTOR to LE/Ly membranes in non-infected cells (Fig. 6B, top panels), and to the SCV when infection was performed in either DMEM (Fig. 6B, bottom panels) or in AA-starvation buffer supplemented with L-glutamine and L-leucine (data not shown). Accordingly, knockdown of either SLC7A5 or SLC3A2 expression severely blunted S6K1 phosphorylation in Salmonella-infected cells (Fig. 6C). Finally, knocking down the expression of SLC1A5, which transports L-glutamine and thus functionally acts upstream of SLC7A5/SLC3A2, also resulted in reduced Salmonella-dependent activation of S6K1 (Fig. 6C).
Fig. 6. Essential role of LNAA transporters in mTOR targeting to the SCV.

(A) HeLa cells either uninfected or infected with Salmonella for 4 h, in the presence or absence of the LNAA transporter inhibitor D-Phenylalanine (D-Phe), were fixed and analyzed by IF using antibodies against mTOR and LAMP2. (B) HeLa cells were transduced with lentiviruses targeting a scramble sequence or SLC7A5, left unstimulated (CTR) or infected with Salmonella for 4 h, analyzed by IF using antibodies against mTOR and LAMP2. (C) HeLa cells were transduced with lentiviruses targeting a scramble sequence (Scr), SLC1A5, SLC3A2, or SLC7A5, left unstimulated (CTR) or infected with Salmonella for 4 h, analyzed by blotting using the antibodies indicated.
Sub-cellular localization of SLC7A5 in Salmonella-infected cells
To further explore the potential role of SLC7A5 in inducing mTOR recruitment to the SCV, we sought to determine the sub-cellular localization of this transporter. Transiently transfected Myc-SLC7A5 localized predominantly to the Golgi apparatus (Fig. 7A), and in indirect support for the importance of SLC7A5 in mTOR endomembrane targeting in infected cells, we also noted that disruption of the Golgi apparatus using brefeldin A resulted in severely blunted recruitment of mTOR to the SCV in cells infected with Salmonella for 4 h (Fig. 7B). Using an antibody against the endogenous form of SLC7A5, we next observed that, while endogenous SLC7A5 also localized to the Golgi apparatus in resting conditions (Fig. 7C), the sub-cellular localization of the protein was transiently altered in cells infected for 2 h with Salmonella (thus at the peak of the AA starvation period). Indeed, at this time, SLC7A5 appeared to localize to intracellular vesicles that likely budded from the Golgi apparatus (Fig. 7C). Importantly, localization of endogenous SLC7A5 to the host plasma membrane remained marginal, suggesting that SLC7A5-dependent internalization of LNAAs likely occurred in intracellular vesicles rather than at the plasma membrane. At 4 h p.i., corresponding to a normalization of cytosolic AA pools, SLC7A5 was found to be again mainly associated with the Golgi apparatus (Fig. 7C). The observation that SLC7A5 might mediate AA cytosolic internalization from intracellular vesicular compartments is also supported by the fact that mTOR targeting to the SCV was inhibited by Dynasore (Fig. 7D), which blocks clathrin-mediated endocytosis. Thus, AAs from the extracellular milieu are likely transported to the cytosol by SLC7A5 from an intracellular vesicular compartment, rather than from the plasma membrane.
Fig. 7. Sub-cellular localization of SLC7A5 in Salmonella-infected cells.

(A) HeLa cells were transfected overnight with an expression vector encoding for Myc-SLC7A5 and analyzed by IF using antibodies against Myc and Golgin-97. (B) HeLa cells were infected with Salmonella in the absence or presence of the Golgi dissassembly promoting drug Brefeldin A, added 30 min after HeLa cells were infected with Salmonella, in order to avoid potential side-effects on bacterial entry. Next, cells were fixed and analyzed by IF using antibodies against mTOR and LAMP2. (C) HeLa cells infected with Salmonella WT for 1 h or 4 h were analyzed by IF using antibodies against the Golgi marker protein Golgin-97 and SLC7A5 (D) HeLa cells were infected with Salmonella in the absence or presence of the inhibitor of clathrin-dependent endocytosis, Dynasore, added 30 min after HeLa cells were infected with Salmonella, in order to avoid potential side-effects on bacterial entry. Next, cells were fixed and analyzed by IF using antibodies against mTOR and LAMP2.
Sustained targeting of Salmonella by the autophagic machinery in SLC7A5-silenced cells
Finally, we aimed to determine the functional impact of SLC7A5 expression on host-mediated targeting of Salmonella by the autophagic machinery, since bacterial autophagy is regulated by mTOR signaling. Interestingly, silencing of SLC7A5 expression resulted at 4 h p.i. in a dramatic increase (3.5%±0.6% in scramble knockdown versus 20.8%±3.7% in SLC7A5 knockdown cells) in the targeting of intracellular Salmonella by GFP-LC3, a marker of autophagy, at 4 h p.i. (Fig. 8), at a time when targeting of Salmonella to autophagosomes is normally very limited. Indeed, autophagy of Salmonella is typically maximal at 1–2 h p.i. (Birmingham and Brumell, 2006; Tattoli et al., 2012), which corresponds to the peak of the AA starvation phase (Tattoli et al., 2012). This observation supports the notion that the progressive decline in autophagic targeting of Salmonella at 3–4 h p.i. previously observed (Birmingham and Brumell, 2006; Tattoli et al., 2012) results from the normalization of cytosolic AA pools, and is an event controlled by LNAA transporters.
Fig. 8. Sustained targeting of Salmonella by the autophagic machinery in SLC7A5-silenced cells.

MDAMC cells stably expressing GFP-LC3, transduced with a lentivirus targeting either a scramble sequence or SLC7A5, were infected with Salmonella for 4 h and analyzed by fluorescence microscopy.
Discussion
The links between microbial infection, host cell metabolism and gene expression regulation have been widely documented in the case of viruses. It is noteworthy that several viruses have been shown to target mTOR signaling (Buchkovich et al., 2008), and critical host responses to viruses implicate the kinase PKR. Interestingly, like the AA starvation sensor, GCN2, PKR similarly triggers phosphorylation of the translation regulator eIF2α, thus resulting in translation regulation under stress (Dauber and Wolff, 2009). In addition, recent evidence also demonstrated that mTOR signaling is altered in cells infected with parasites, such as Toxoplasma and Leishmania (Jaramillo et al., 2011; Wang et al., 2009a; Wang et al., 2009b). In the case of bacterial pathogens, our recent investigations uncovered the critical interplay between mTOR signaling and host responses to intracellular bacteria (Tattoli et al., 2012). We provided evidence that invasive bacterial pathogens, such as Shigella and Salmonella, trigger a host AA starvation program that disarms mTOR signaling while inducing stress responses dependent on the AA sensor GCN2 (Tattoli et al., 2012). However, little is known about the mechanisms underlying the modulation of mTOR signaling and AA starvation responses in bacteria-infected cells. Using Salmonella as a model organism, we show here that Salmonella-induced membrane damage and mTOR inhibition required the action of the SPI-1 system in the early stages of infection. In contrast, we found that the AA normalization, which occurs later p.i., was independent of SPI-2 or the control of SCV positioning in infected cells, but depended on the active uptake of extracellular AA by the LNAA transporters SLC7A5/SLC3A2, likely from an intracellular vesicular compartment.
In this study, we provide evidence that the activity of the SPI-1 system is crucial for (i) the early recruitment of NDP52 to the SCV membrane in the early stage of infection, (ii) the inhibition of mTOR association with host endomembranes and (iii) the induction of the ATF3-dependent AA stress response pathway. These results are in agreement with previous results that proposed a crucial role for SPI-1-dependent damage to the SCV in the induction of bacterial autophagy (Birmingham and Brumell, 2006). Although it is not clear what factor from the SPI-1 system is required for these effects, and indeed, if specific bacterial effectors play a role, a likely possibility is that the insertion of the TTSS apparatus across the membrane of the early SCV could itself cause a host membrane damage response. The exact nature of this host innate response to membrane damage remains elusive. However, one can speculate that the accumulation of diacylglycerol, which was shown to occur at the SCV membrane (Shahnazari et al., 2010) and also at the surface of damaged lysosomes (Shaughnessy et al., 2007), together with the recruitment of host factors such as NDP52, p62 and ubiquitinated proteins, likely contributes to drive the recruitment of host signaling and repair machineries to damaged endomembranes.
The mechanism through which membrane damage results in AA starvation, inhibition of mTOR and induction of the GCN2/ATF3 signaling axis remains unclear. Nevertheless, it is noteworthy that a recent study demonstrated similar induction of GCN2 and phosphorylation of eIF2α in cells treated with several bacterial pore-forming toxins, which are known to induce significant damage to host endomembranes (Kloft et al., 2010). Because we previously demonstrated that similar effects could be obtained with digitonin and glycyl-L-phenylalanine 2-naphthylamide, two drugs that induce aseptic membrane damage (Tattoli et al., 2012), it is likely that both SPI-1- and bacterial toxin-mediated membrane damage depend on biophysical alterations of host membranes rather than on the action of specific bacterial factors. With regards to the potential mechanisms linking membrane damage to AA starvation responses, studies in yeast have demonstrated that high concentrations of sodium chloride induced phosphorylation of GCN2 and eIF2α (Goossens et al., 2001; Zhan et al., 2004), which suggests that perturbations in transmembrane electrochemical or ionic gradients could result in AA starvation, likely caused by altered activity of key AA transporters. Further work is needed to understand how membrane damage causes AA starvation responses.
Relocalization of mTOR to the SCV membrane at 3–4 h p.i. in Salmonella-infected cells coincides with the activation of the SPI-2 system and the tightly regulated sub-cellular rerouting of the SCV towards the Golgi apparatus (Bakowski et al., 2008; Ramsden et al., 2007a; Ramsden et al., 2007b). Our results strongly suggest that none of these events significantly contribute to the regulation of mTOR sub-cellular localization. This observation contrasts with a recent study, which demonstrated that the sub-cellular positioning of lysosomes could condition mTOR recruitment to these organelles, thereby contributing to the fine-tuning of mTOR activity in the presence or absence of nutrients (Korolchuk et al., 2011). In particular, mTOR-negative lysosomes clustered towards the peri-nuclear region in starved cells, which was shown to favor the fusion of lysosomes with autophagosomes and the recycling of nutrients by autophagy (Korolchuk et al., 2011). Our results suggest that this level of regulation does not take place in Salmonella-infected cells, likely highlighting the fact that vesicular trafficking is potently hijacked by the bacterium.
Normalization of cytosolic AA pools and mTOR signaling at 3–4 h p.i. in Salmonella-infected cells is likely beneficial to the pathogen, because it results in severe blunting of the anti-bacterial autophagic response. Our results suggest that the transient induction of AA starvation responses is explained by the fact that the SPI-1-dependent damage to the host membranes is also a transient event that is followed by a rapid membrane healing process. In this scenario, it remains surprising that the activation of the SPI-2 system, which also results in the perforation of host membranes by a second SPI-2-encoded TTSS, does not provoke the generation of a detectable second wave of AA starvation responses. A first explanation could be that in the cells used for our study (HeLa), the induction of SPI-1 and SPI-2 occur in rapid succession, resulting in overlapping responses. Indeed, the exact time at which the SPI-2 is activated is matter of debate and might be cell type specific. Alternatively, it is possible that a specific bacterial effector, delivered by the SPI-2-dependent TTSS, would counteract the effect of membrane damage, in order to inhibit host AA starvation responses.
Together, the results presented in this manuscript provide new insights into the complex interplay between bacterial infection, mTOR regulation and AA starvation responses, which might pave the way towards the development of novel approaches aiming at boosting host defenses against bacterial pathogens.
Acknowledgments
This work was supported by grants from the Burroughs Wellcome Fund (to S.E.G.) and the Canadian Institutes for Health Research (to D.J.P.).
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- Bakowski M. A., Braun V., Brumell J. H. (2008). Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 9, 2022–2031 10.1111/j.1600-0854.2008.00827.x [DOI] [PubMed] [Google Scholar]
- Benko S., Magalhaes J. G., Philpott D. J., Girardin S. E. (2010). NLRC5 limits the activation of inflammatory pathways. J. Immunol. 185, 1681–1691 10.4049/jimmunol.0903900 [DOI] [PubMed] [Google Scholar]
- Birmingham C. L., Brumell J. H. (2006). Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2, 156–158. [DOI] [PubMed] [Google Scholar]
- Birmingham C. L., Smith A. C., Bakowski M. A., Yoshimori T., Brumell J. H. (2006). Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281, 11374–11383 10.1074/jbc.M509157200 [DOI] [PubMed] [Google Scholar]
- Birmingham C. L., Canadien V., Gouin E., Troy E. B., Yoshimori T., Cossart P., Higgins D. E., Brumell J. H. (2007). Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3, 442–451. [DOI] [PubMed] [Google Scholar]
- Buchkovich N. J., Yu Y., Zampieri C. A., Alwine J. C. (2008). The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 6, 266–275 10.1038/nrmicro1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campoy E., Colombo M. I. (2009). Autophagy in intracellular bacterial infection. Biochim. Biophys. Acta 1793, 1465–1477 10.1016/j.bbamcr.2009.03.003 [DOI] [PubMed] [Google Scholar]
- Cemma M., Kim P. K., Brumell J. H. (2011). The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 7, 341–345 10.4161/auto.7.3.14046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney R., Baker J., Brain O., Danis B., Pichulik T., Allan P., Ferguson D. J., Campbell B. J., Jewell D., Simmons A. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97 10.1038/nm.2069 [DOI] [PubMed] [Google Scholar]
- Dauber B., Wolff T. (2009). Activation of the antiviral kinase PKR and viral countermeasures. Viruses 1, 523–544 10.3390/v1030523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. (2010). Autophagy in infection. Curr. Opin. Cell Biol. 22, 252–262 10.1016/j.ceb.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V., Levine B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549 10.1016/j.chom.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N., Temime–Smaali N., Lafont F. (2010). How ubiquitination and autophagy participate in the regulation of the cell response to bacterial infection. Biol. Cell 102, 621–634 10.1042/BC20100101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass E., Groisman E. A. (2009). Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 12, 199–204 10.1016/j.mib.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley I. G., Lam D. H., Wang J., Ding X., Chen S., Jiang X. (2009). ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens A., Dever T. E., Pascual–Ahuir A., Serrano R. (2001). The protein kinase Gcn2p mediates sodium toxicity in yeast. J. Biol. Chem. 276, 30753–30760 10.1074/jbc.M102960200 [DOI] [PubMed] [Google Scholar]
- Gorvel J. P., Méresse S. (2001). Maturation steps of the Salmonella-containing vacuole. Microbes Infect. 3, 1299–1303 10.1016/S1286-4579(01)01490-3 [DOI] [PubMed] [Google Scholar]
- Guignot J., Caron E., Beuzón C., Bucci C., Kagan J., Roy C., Holden D. W. (2004). Microtubule motors control membrane dynamics of Salmonella-containing vacuoles. J. Cell Sci. 117, 1033–1045 10.1242/jcs.00949 [DOI] [PubMed] [Google Scholar]
- Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S., Natsume T., Takehana K., Yamada N.et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Brumell J. H. (2009). Autophagy in immunity against intracellular bacteria. Curr. Top. Microbiol. Immunol. 335, 189–215 10.1007/978-3-642-00302-8_9 [DOI] [PubMed] [Google Scholar]
- Ivanov S., Roy C. R. (2009). NDP52: the missing link between ubiquitinated bacteria and autophagy. Nat. Immunol. 10, 1137–1139 10.1038/ni1109-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo M., Gomez M. A., Larsson O., Shio M. T., Topisirovic I., Contreras I., Luxenburg R., Rosenfeld A., Colina R., McMaster R. W.et al. (2011). Leishmania repression of host translation through mTOR cleavage is required for parasite survival and infection. Cell Host Microbe 9, 331–341 10.1016/j.chom.2011.03.008 [DOI] [PubMed] [Google Scholar]
- Jung C. H., Jun C. B., Ro S. H., Kim Y. M., Otto N. M., Cao J., Kundu M., Kim D. H. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J. (2007). Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 10.1038/nrm2245 [DOI] [PubMed] [Google Scholar]
- Kloft N., Neukirch C., Bobkiewicz W., Veerachato G., Busch T., von Hoven G., Boller K., Husmann M. (2010). Pro-autophagic signal induction by bacterial pore-forming toxins. Med. Microbiol. Immunol. (Berl.) 199, 299–309 10.1007/s00430-010-0163-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk V. I., Saiki S., Lichtenberg M., Siddiqi F. H., Roberts E. A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F. M.et al. (2011). Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460 10.1038/ncb2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M., Sabatini D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594 10.1242/jcs.051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman M., Jordens I., Kuijl C., Janssen L., Neefjes J. (2004). Dynein-mediated vesicle transport controls intracellular Salmonella replication. Mol. Biol. Cell 15, 2954–2964 10.1091/mbc.E03-08-0614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P., Bergman P., Zhang B., Triantafellow E., Wang H., Nyfeler B., Yang H., Hild M., Kung C., Wilson C.et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534 10.1016/j.cell.2008.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M., Sasakawa C. (2006). Shigella and autophagy. Autophagy 2, 171–174. [DOI] [PubMed] [Google Scholar]
- Py B. F., Lipinski M. M., Yuan J. (2007). Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3, 117–125. [DOI] [PubMed] [Google Scholar]
- Ramsden A. E., Holden D. W., Mota L. J. (2007a). Membrane dynamics and spatial distribution of Salmonella-containing vacuoles. Trends Microbiol. 15, 516–524 10.1016/j.tim.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Ramsden A. E., Mota L. J., Münter S., Shorte S. L., Holden D. W. (2007b). The SPI-2 type III secretion system restricts motility of Salmonella-containing vacuoles. Cell. Microbiol. 9, 2517–2529 10.1111/j.1462-5822.2007.00977.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F. (2011). How cells deploy ubiquitin and autophagy to defend their cytosol from bacterial invasion. Autophagy 7, 304–309 10.4161/auto.7.3.14539 [DOI] [PubMed] [Google Scholar]
- Rich K. A., Burkett C., Webster P. (2003). Cytoplasmic bacteria can be targets for autophagy. Cell. Microbiol. 5, 455–468 10.1046/j.1462-5822.2003.00292.x [DOI] [PubMed] [Google Scholar]
- Sengupta S., Peterson T. R., Sabatini D. M. (2010). Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40, 310–322 10.1016/j.molcel.2010.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahnazari S., Yen W. L., Birmingham C. L., Shiu J., Namolovan A., Zheng Y. T., Nakayama K., Klionsky D. J., Brumell J. H. (2010). A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe 8, 137–146 10.1016/j.chom.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy L. M., Lipp P., Lee K. D., Swanson J. A. (2007). Localization of protein kinase C ε to macrophage vacuoles perforated by Listeria monocytogenes cytolysin. Cell. Microbiol. 9, 1695–1704 10.1111/j.1462-5822.2007.00903.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele–Mortimer O. (2008). The Salmonella-containing vacuole—moving with the times. Curr. Opin. Microbiol. 11, 38–45 10.1016/j.mib.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I., Sorbara M. T., Vuckovic D., Ling A., Soares F., Carneiro L. A., Yang C., Emili A., Philpott D. J., Girardin S. E. (2012). Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575 10.1016/j.chom.2012.04.012 [DOI] [PubMed] [Google Scholar]
- Thurston T. L., Ryzhakov G., Bloor S., von Muhlinen N., Randow F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221 10.1038/ni.1800 [DOI] [PubMed] [Google Scholar]
- Thurston T. L., Wandel M. P., von Muhlinen N., Foeglein A., Randow F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travassos L. H., Carneiro L. A., Ramjeet M., Hussey S., Kim Y. G., Magalhães J. G., Yuan L., Soares F., Chea E., Le Bourhis L.et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62 10.1038/ni.1823 [DOI] [PubMed] [Google Scholar]
- Wang Y., Weiss L. M., Orlofsky A. (2009a). Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J. Biol. Chem. 284, 1694–1701 10.1074/jbc.M807890200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Weiss L. M., Orlofsky A. (2009b). Intracellular parasitism with Toxoplasma gondii stimulates mammalian-target-of-rapamycin-dependent host cell growth despite impaired signalling to S6K1 and 4E-BP1. Cell. Microbiol. 11, 983–1000 10.1111/j.1462-5822.2009.01305.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Jagannath C., Liu X. D., Sharafkhaneh A., Kolodziejska K. E., Eissa N. T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144 10.1016/j.immuni.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan K., Narasimhan J., Wek R. C. (2004). Differential activation of eIF2 kinases in response to cellular stresses in Schizosaccharomyces pombe. Genetics 168, 1867–1875 10.1534/genetics.104.031443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. T., Shahnazari S., Brech A., Lamark T., Johansen T., Brumell J. H. (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- Zoncu R., Efeyan A., Sabatini D. M. (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]