

Abstract
c-Jun-N-terminal Kinase (JNK) is a mitogen-activated protein kinase (MAPK) family member that is activated by diverse stimuli, including cytokines (such as tumor necrosis factor and interleukin-1), reactive oxygen species (ROS), pathogens, toxins, drugs, endoplasmic reticulum stress, free fatty acids, and metabolic changes. Upon activation, JNK induces multiple biologic events through the transcription factor AP-1 and transcription-independent control of effector molecules. JNK isozymes regulate cell death and survival, differentiation, proliferation, ROS accumulation, metabolism, insulin signaling, and carcinogenesis in the liver. The biologic functions of JNK are isoform, cell-type, and context dependent. Recent studies using genetically engineered mice showed that loss or hyper-activation of the JNK pathway contributes to the development of inflammation, fibrosis, cancer growth, and metabolic diseases that include obesity, hepatic steatosis, and insulin resistance. We review the functions and pathways of JNK in liver physiology and pathology, and discuss findings from pre-clinical studies with JNK inhibitors.
Keywords: MAPK, hepatocellular carcinoma, insulin resistance, c-Jun, TNF, acetaminophen
Introduction
c-Jun-N-terminal Kinase (JNK) is a mitogen-activated protein kinase (MAPK) family member. There are 3 isoforms of JNK in mammals: JNK1, JNK2, and JNK3 (encoded by MAPK8, MAPK9, and MAPK10, respectively). JNK1 and JNK2 are expressed in almost all cells, including liver parenchymal cells, whereas JNK3 is mainly expressed in brain, heart, and testis1,2. At least 10 alternative splicing variants are known, which increase the diversity of JNK proteins, but their functional significance is unclear. The JNK proteins, including splicing variants, range from 46 kDa to 55 kDa in size. JNK's enzymatic activity is induced in response to diverse stimuli, such as cytokines (tumor necrosis factor [TNF], interleukin-1 [IL-1], transforming growth factor β [TGFβ], platelet-derived growth factor [PDGF], and epidermal growth factor [EGF]), intra- and extracellular pathogens (lipopolysaccharide [LPS], peptidoglycan, and bacterial unmethylated CpG-DNA that activates Toll-like receptors [TLRs]), reactive oxygen species (ROS), pathologic and environmental stress (ischemia, hypoxia, and ultraviolet and ionizing radiation), toxins, drugs, endoplasmic reticulum (ER) stress, and metabolic changes, including obesity and hyperlipidemia.
The JNKs are activated via 3-tiered signaling modules comprising MAP kinase kinase kinases (MAP3Ks or MKKKs), MAP kinase kinases (MAP2Ks or MKKs), and MAP kinases (MAPKs, in this case the JNKs). At least, 14 different MAP3Ks have been found to activate JNK (see Figure 1A). The best-characterized MAP3Ks are mixed-linage kinase 3 (MLK3), MEKK1, and TAK1. The 2 MAP2Ks (MKK4 and MKK7) phosphorylate JNKs at threonine and tyrosine residues within a conserved dual phosphorylation Thr-Pro-Tyr motif in their activation loop3. MKK7 primarily activates JNK, but MKK4 activates JNK and p384. MKK7 is specifically associated with cytokine-induced JNK activation, through phosphorylation of the Thr residue of JNK4. The JNKs have a common substrate docking site in their C-terminus and a glutamateaspartate domain in their N-terminus that is the site of protein–protein interactions with MAP2Ks, phosphatases (e.g. JNK dual-specificity phosphatases), and substrates (Figure 1B)3,4.
Figure 1. The JNK Activation Pathway and Domain Structure and.

A. number of stimuli, including growth factors, cytokines, PAMPs, ROS, and environmental stresses activate the JNK signaling pathway through a 3-tier kinase cascade that includes MAP3Ks (ASK1, TAK1, MEKKs and MLKs), MAP2Ks (MKK4 and MKK7), and JNKs. Activated JNKs phosphorylate their substrates, which include transcription factors (c-Jun, JunB, JunD, p53, and c-Myc) and mitochondrial proteins (Bcl-2, Bid, Bim, Bax, and Mcl-1), to induce various biological responses.
B. The JNK protein contains a conserved dual phosphorylation Thr-Pro-Tyr motif in its activation loop and has a common substrate docking site in C-terminus and a glutamate/aspartate domain in its N-terminus to mediate interactions with MAP2Ks, phosphatases, and substrates.
At least 50 proteins have been identified as JNK substrates. These proteins control multiple cellular processes, acting either as transcription factors or by controlling protein degradation, localization, and signaling. JNK substrates include c-Jun, JunB, JunD, activating transcription factor 2 (ATF2), p53, c-Myc, serum response factor (SRF), Itch, insulin receptor substrate-1 (IRS-1), JNK interacting protein 1 (JIP1), 14-3-3, Sab (SH3BP5), Bcl-2, Bcl-xL, Bid, Bim, Bad, Bax, and Mcl-11,5. Among these substrates, c-Jun is a representative target of JNKs. c-Jun dimerizes with JunB, JunD, or Fos to form the transcription factor activator protein (AP)-1, whereas SRF controls expression of the Fos proteins that dimerize with the Jun proteins6.
One of the best-studied pathways that leads to JNK activation is TNF signaling via TNF receptor1 (TNFR1) (Figure 2). Upon binding of a TNF trimer to trimerized TNFR1, the intracellular potion of TNFR1 recruits the adaptor TRADD through homotypic interaction between their death domains, leading to formation of an intracellular signaling complex that includes RIP1, cellular inhibitor of apoptosis 1 (cIAP1), cIAP2, and TRAF2, termed complex I7,8. With K63-polyubiquitination of RIP1 by cIAP1 and cIAP2, complex I recruits, phosphorylates, and ubiquitinates the MAP3Ks TAK1, MEKK1, or MLK3, which in turn activate MKK4 and MKK7, leading to activation of JNK and other downstream effectors including IκB kinase (IKK)1,7. In the second step, K63-polyubiquitination of RIP1 is removed by the deubiquitinases cylindromatosis (CYLD) or A20, leading to dissociation of the TRADD–RIP1–TRAF2 complex from TNFRI8. The dissociated cytosolic complex binds to FADD, caspase-8, RIP1, and RIP3 to form complex II, which contributes to programmed cell death, including apoptosis and necrosis8–10. In complex II, caspase-8 cleaves RIP1 and RIP3 to prevent necrotic death, promoting the induction of apoptosis8–11. When caspase-8 or FADD is inhibited, RIP1 and RIP3 prevent apoptotic death, shifting complex II activity towards necrosis8,11. TNF-induced ROS is generated by NADPH oxidase 1 (NOX1) and Rac1, which are recruited to complex I in a TRADD- and RIP1-dependent manner and by mitochondrial respiratory complex I. ROS accumulation promotes prolonged activation of JNK by inactivating JNK phosphatases and TNF-mediated necrosis12,13. TNF-induced ROS accumulation and prolonged JNK activation are suppressed by nuclear factor (NF)-κB–mediated sequestration of ROS and induction of c-FLIP12,14,15. Initial TNFRI-mediated JNK activation is transient and associated with cell survival and proliferation through AP-1, whereas sustained JNK activation and ROS accumulation are associated with apoptotic and necrotic cell death1,7,16,17. Moreover, interactions among JNK, p38, and IKK–NF-κB pathways regulate transient and sustained activation of JNK. Upon inactivation of p38 or IKKs, TNF stimulation induces prolonged activation of JNK17–21.
Figure 2. JNKs in TNF Signaling.

(1) Binding of TNF to the TNFR type I leads to the rapid formation of complex I, comprising TRADD, RIP1, TRAF2, cIAP1, cIAP2, and Ubc13. cIAP-mediated K63-ubiquitination of RIP1 recruits and activates TAK1. (2) MAP3Ks (TAK1 and ASK1) activate JNK1 and 2 through MKK4 and 7. JNK activates AP-1, which comprises c-Jun and c-Fos. Simultaneously, JNK1 phosphorylates ITCH to ubiquitinate c-FLIP, which promotes caspase-8 dependent apoptosis. JNKs can also induce mitochondria-dependent apoptosis through Bax and degradation of Bim. (3) TAK1 phosphorylates and activates the IKK complex, which leads to phosphorylation, unbiquitination, and degradation of IκBα, resulting in nuclear translocation and activation of NF-κB, which is comprises the p50 and p65 subunits. NF-κB induces the transcription of SOD2 and c-FLIP to prevent ROS production and caspase-8 activation, respectively. (4) Complex I also contributes to ROS production through NOX1 and Rac1. (5) Following formation of complex I, RIP1 is deubiquitinated by CYLD or A20 to form complex II, comprising TRADD, FADD, RIP1, RIP3, and caspase-8. Normally, caspase-8 induces apoptosis. (6) However, if caspase-8 or FADD is blocked, RIP1 and RIP3 are phosphorylated and cause necrosis.
JNK signaling is associated with cell death, survival, differentiation, proliferation, and tumorigenesis in hepatocytes. In nonparenchymal liver cells, such as hepatic macrophages (Kupffer cells) and hepatic stellate cells (HSCs), JNK is involved in inflammation and fibrosis. Studies of cell-specific ablation (by the Cre/lox-P strategy) and bone marrow chimeras have identified the specific functions of JNK in distinct cell types and organs and in the interactions between the liver and other organs22–24. Moreover, the distinct functions of the isoforms JNK1 and JNK2 in pathogenesis of liver diseases have been characterized in Jnk1−/− and Jnk2−/− mice. We review recent advances in our understanding of the role of the JNK and/or c-Jun pathways in different types of liver injury (TNF-induced liver injury, fibrosis and carcinogenesis, and steatohepatitis) from studies of genetically engineered mice and human cells and tissues.
TNF-Mediated Hepatocyte Death and Liver Injury
In hepatocytes, TNF rapidly activates JNK, leading to phosphorylation and activation of AP-1/c-Jun25. Simultaneously, TNF activates NF-κB via the IKK complex and induces expression of anti-apoptotic genes that are regulated by NF-κB and block caspase-8–dependent cell death and prolonged activation of JNK25. Inhibition of de novo protein synthesis or NF-κB activation sensitizes hepatocytes to TNF-induced hepatocyte death as a result of sustained JNK activation12,26. Caspase or JNK inhibitors prevent TNF-mediated hepatocyte apoptosis26. Sustained JNK activation increases ROS accumulation, which further promotes JNK activation through oxidative inhibition of JNK dual-specificity phosphatases12. JNK-mediated apoptosis seems to depend on ROS production but is independent of AP-126. Conversely, NF-κB activation inhibits ROS accumulation and excessive ROS attenuate NF-κB activation12,14. TNF-induced hepatocyte death is prevented by Jnk1 disruption 15. TNF-mediated activation of JNK1 phosphorylates and activates the E3 ubiquitin ligase Itch, which contributes to the K48-linked ubiquitination of c-FLIP, an NF-κB–induced anti-apoptotic molecule (an endogenous caspase-8 inhibitor), to promote activation of caspases-8 and -3 and apoptosis of hepatocytes (Figure 2). Loss of Itch prevents ubiquitination and degradation of inducible c-FLIP, which abrogates TNF-induced liver injury15. However, mice deficient in only JNK2 are resistant to D-galactosamine(GalN)/LPS or GalN/TNF-mediated liver injury, indicating that JNK2 activation is involved in caspase-8 activation, Bid cleavage, and mitochondrial cytochrome release in hepatocytes27. A subsequent study demonstrated that in Jnk2−/− hepatocytes, TNF overactivates JNK1, indicating that JNK1 activation protects Jnk2−/− mice from GalN/TNF-induced liver injury28. JNK1 activation in Jnk2−/− hepatocytes stabilizes the anti-apoptotic protein Mcl-1 to prevent its degradation and block TNF-mediated apoptosis28. The function of Mcl-1 in Jnk2−/− mice has been confirmed by induction of TNF-mediated liver injury in Mcl-1−/−Jnk2−/− (double knockout) mice28. In wild-type hepatocytes, JNK1 is proapoptotic, but in Jnk2−/− cells, JNK1 might be anti-apoptotic, via stabilization of Mcl-1.
Recruitment of activated JNK to the outer membrane of mitochondria is another important step in induction of JNK-mediated hepatocyte death. Mitochondrial Bcl-XL, Mcl-1, and Sab are substrates for JNK29. TNF induces recruitment of phosphorylated JNK and MKK4 and the Bcl-2 family member Bax to mitochondrial outer membrane, which promotes generation of mitochondrial ROS and sustained activation of JNK, inducing hepatocyte death29. Knockdown of Sab initially activates JNK, but prevents accumulation of JNK, MKK4, and Bax at the mitochondrial outer membrane and sustained activation of JNK, thereby inhibiting TNF-induced hepatocyte death. These findings indicate the important roles of Sab and recruitment of JNK to mitochondrial outer membrane in sustaining activation of JNK29. They also indicate that a mitochondrial activation loop that produces ROS, rather than only continuous TNFR signaling, sustains JNK activation. Upon TNF stimulation, Bid, a BH3-only protein, is cleaved by caspase-8 in a JNK2-dependent manner27,30. The cleaved Bid translocates to the mitochondrial outer membrane to induce cytochrome-c release and caspase-9/caspase-3 activation, resulting in TNF-mediated hepatocyte death27,31. The requirement for Bid in hepatocyte death has been demonstrated by the resistance of Bid−/− mice to TNF-induced liver injury30.
Although many biological functions induced by JNK are mediated through c-Jun activation, TNF-mediated hepatocyte apoptosis is JNK-dependent, but c-Jun- and transcription-independent26,32. c-Jun seems to protect against rather than induce cell death33.
In concanavalin A (ConA)-induced liver injury, membrane bound TNF, rather than soluble TNF, is important. Membranous TNF binds to TNFRI and TNFRII, and, given the ability of TNFRII to activate JNK and not NF-κB, leads to strong activation of JNK, thereby promoting hepatocyte death. Jnk1−/− and Jnk2−/− mice are protected from ConA-induced liver injury19. Because IKK signaling to NF-κB prevents JNK activation, ConA-induced liver injury is exacerbated in mice that lack hepatocyte IKKβ (IkkβΔhep), in which JNK activation is potentiated19. However, a recent study demonstrated that compound deletion of Jnk1 and Jnk2 from hepatocytes did not impair ConA- or LPS-induced liver injury22. Notably, hematopoietic deficiency in JNK1 and JNK2 prevented ConA-induced liver injury, suppressing TNF production22. These findings indicate that under certain conditions, JNK activation in hematopoietic cells is required for optimal production of TNF, which is essential for ConA-induced hepatitis. It is possible that JNK2 is required to protect Jnk1−/− hepatocytes from cell death, so hepatocytes with combined disruption of Jnk1 and Jnk2 are no longer protected. Interestingly, c-Jun is important in hepatocytes, rather than hematopoietic cells, where it negatively regulates ConA-induced hepatitis34. Loss of c-Jun aggravated ConA-induced liver injury and suppressed Nos2 expression. Liver-specific supplementation of NO attenuated overt ConA-induced liver injury in mice with hepatocyte-specific deletion of c-Jun (c-JunΔhep)34. c-JunAA mice, which have mutations in c-Jun at sites phosphorylated by JNK, did not have this phenotype, indicating that inducible expression of Nos2 requires c-Jun but does not require phosphorylation of c-Jun34.
TRAIL induces cell death in cancer cells that express its receptor, and JNK inhibition sensitizes HCC cells to TRAIL-induced cell death35. In contrast, primary hepatocytes do not undergo apoptosis by TRAIL, but TRAIL amplifies Fas-induced death36. In mice, Fas-mediated hepatitis is associated with strong activation of JNK, leading to phosphorylation of Bim and its translocation to the mitochondrial outer membrane, causing release of cytochrome c and activation of the mitochondrial caspase cascade36. Trail−/− mice are protected from Fas-mediated fulminant hepatitis, and have reduced activation of JNK and no translocation of Bim to mitochondria36. Bim deficiency also prevented Fas-induced hepatitis36. These findings indicate that TRAIL is required for activation of JNK and Bim in Fas-mediated liver injury. In summary, JNK has dual roles in TRAIL signaling. In normal hepatocytes, JNK is required for TRAIL-mediated cell death. On the contrary, in HCC cells, JNK prevents TRAIL-mediated cell death.
Ischemia/Reperfusion (I/R) Liver Injury
I/R liver injury is a serious clinical complication following liver transplantation, surgical resection of liver tumors, and circulation shock. Hepatic I/R injury is characterized by hepatocyte necrosis and apoptosis induced by multiple mediators, including TNF, ROS, and intracellular signaling via JNK-dependent pathways. I/R liver injury causes JNK1 activation and subsequent increases in AP-1 activity during the reperfusion phase. Overexpression of superoxide dismutase 2 (SOD2) decreases AP-1 activity, indicating that I/R-mediated oxidative stress contributes to activation of JNK and AP-1 and I/R liver injury37. Specific inhibitors of JNK prevented c-Jun phosphorylation, AP-1 activation, Bak induction, Bid degradation, caspase-3 activation, and mitochondrial cytochrome c release, eventually attenuating hepatocyte necrosis and apoptosis after I/R or liver transplantation38,39. Absence of JunD, another AP-1 component, increased I/R liver injury, along with phosphorylation of c-Jun, activity of AP-1, and expression of NOX2 and NOX4, which was suppressed by overexpression of a dominant negative form of JNK140. JunD therefore appears to regulate c-Jun activity and ROS generation in I/R liver injury; this pathway is controlled by JNK1. Moreover, Jnk2−/− mice had reduced I/R liver injury and increased expression of HO-1. Inhibition of HO-1 blocked the protective effect of Jnk2 disruption, indicating a role for HO-1 in protecting Jnk2−/− mice from I/R injury41. JNK2 also increases the mitochondrial permeability transition, resulting in hepatocellular injury after I/R42,43. Interactions between the IKK–NF-κB and JNK–AP-1 pathways are also important in I/R injury. Hepatocyte-specific deficiency in IKKγ/NEMO (NemoΔhep mice) have a greater level of I/R liver injury and stronger activation of JNK, indicating that lack of protective IKK–NF-κB signaling and increases in the apoptotic JNK pathway promote I/R liver injury44.
Acetaminophen-Induced Liver Injury
Acetaminophen overdose is the most common cause of drug-induced acute liver failure in the United States. Acetaminophen is converted to N-acetyl-p-benzoquinone imine (NAPQI) by cytochrome P-450-CYP2E1, which is expressed in zone 3 hepatocytes. Metabolic inactivation of NAPQI requires its binding to glutathione. However, excess NAPQI generates ROS that further deplete glutathione and cause mitochondrial dysfunction, DNA damage, and large amounts of necrosis and apoptosis among hepatocytes.
Exposure of hepatocytes to acetaminophen activates JNK and leads to subsequent translocation of JNK and Bax to mitochondrial outer membrane, induction of the mitochondrial permeability transition, generation of ROS, and cell death (Figure 3)45. Disruption of Jnk2, but not Jnk1, prevented acetaminophen-induced liver injury, indicating the importance of JNK2 in this form of hepatoxicity45. JNK translocation to the mitochondria is required for acetaminophen-induced hepatotoxicity. Phosphorylated JNK and MKK4, but not apoptosis signal-regulating kinase 1 (ASK1) or MKK7, translocate to mitochondria when hepatocytes are exposed to acetaminophen 29. Blocking this translocation by silencing Sab suppressed prolonged activation of JNK and acetaminophen-induced death of hepatocytes. Therefore, MKK4-mediated activation of JNK and Sab-mediated recruitment of JNK to the mitochondrial outer membrane (which sustains JNK activation) are required for acetaminophen-induced hepatocyte death (Figure 3)29.
Figure 3. Role of JNK in Acetaminophen-Induced Liver Injury.
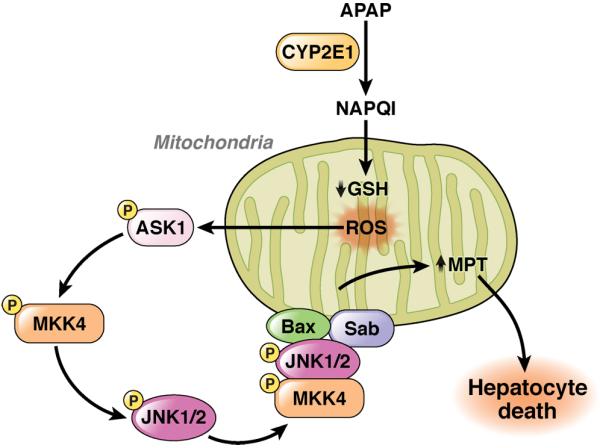
Acetaminophen is metabolized to NAPQI through CYP2E1, which reduces glutathione levels in mitochondria. Excessive NAPQI induces ROS, which activates ASK1, MKK4, and JNK. JNK, MKK4 and Bax translocate to the outer membrane of mitochondria through binding of Sab to increase the mitochondrial permeability transition, resulting in induction of massive hepatocyte death.
Bax−/− mice have reductions in only the early phase of acetaminophen-induced liver injury, indicating that additional factors are required for complete acetaminophen-induced liver injury46. Acetaminophen exposure induced JNK-dependent expression of the pro-apoptotic factor Bim in hepatocytes. Bim−/− mice are protected from acetaminophen liver injury47. Bim expression is amplified by TRAIL, and acetaminophen-induced liver injury is inhibited in Trail−/− mice, indicating a role for a TRAIL–JNK–Bim pathway in acetaminophen-induced hepatocyte death47. Although JNK activation is required for mitochondrial dysfunction, the initial activation of JNK is induced by mitochondrial generation of ROS, which results from mitochondrial depletion of glutathione in response to acetaminophen48. Although ASK1 does not translocate to mitochondria, this factor is important for JNK activation in acetaminophen-induced liver injury49. ASK1 deficiency protected mice from acetaminophen-induced liver injury and reduced JNK activation49, indicating that ASK1 acts upstream of MAP3K in this process.
The specific JNK inhibitors SP600125 and leflunomide rescued mice from acetaminophen-induced liver injury45,50. These reagents might therefore be developed for treatment of patients with acetaminophen-induced liver injury.
ER Stress and Hepatocyte Death
The ER is the major site of protein folding, maturation, and trafficking. When unfolded or misfolded proteins accumulate in the ER, the ER becomes stressed and the protective unfolded protein response (UPR) is activated. The UPR involves activation of 3 ER membrane sensors: ATF6α, inositol-requiring enzyme (IRE)1α, and PKR-like ER localized kinase (PERK)51,52. These sensors activate signaling pathways that induce molecules that reduce ER stress. Among the ER sensors, IRE-1α and the PERK-associated metaflammasome (which comprises PKR and eIF2) contribute to activation of JNK and IKK–NF-κB. IRE-1α interacts with TRAF2 to activate JNK through ASK1, which is implicated in ER stress-induced apoptosis51, 52. However, it is not clear whether ER stress-mediated activation of JNK is involved in hepatocyte death. In response to thapsigargin, an inducer of ER stress, c-Jun−/− hepatocytes had exacerbated and sustained ER stress, characterized by increased expression of CHOP, sXBP-1, and GRP7853. c-Jun−/− hepatocytes are more susceptible to ER-stress–induced cell death and have a defect in autophagy, indicating that c-Jun couples ER stress and autophagy to promote cell survival.
Distinct Roles of JNK1 and JNK2 in Liver Regeneration
The liver can regenerate to its original size after a substantial loss of its mass. The JNK isoforms and AP-1 are activated within 1 hour of partial hepatectomy. JNK activates AP-1, which promotes expression of cyclin D and initiates the G0–G1 transition54,55. In regenerating liver, JNK is activated by mediators such as TNF and EGF. Mice that lack TNFR1 and rats injected with an antibody against TNF have reduced liver regeneration and activation of JNK and AP-156–58. Exogenous administration of ATP activates JNK and induces hepatocyte proliferation; this effect is potentiated by EGF59. Upon partial hepatectomy, pharmacologic inhibition of JNK blocks c-Jun phoshorylation, AP-1 activation, and cyclin D1 expression, delaying regeneration58. Consistently, Jnk1−/− mice have decreased liver regeneration following partial hepatectomy, with increased p21 and decreased c-Myc expression. Interestingly, p21 deficiency reversed defective regeneration in Jnk1−/− livers, indicating that JNK1 promotes hepatocyte proliferation by inhibiting p2123,60. The role of JNK2 in liver regeneration is less clear. One study demonstrated that the loss of JNK2 accelerated liver regeneration61. The authors of this study suggested that compensatory upregulation of JNK1 might accelerate regeneration in Jnk2−/− livers. Another reported no role for JNK2 in liver regeneration23.
c-Jun−/− mice die during embryogenesis; the fetal livers have high levels of hepatocyte apoptosis62. Conditional or hepatocyte-specific deletion of c-Jun (c-JunΔhep) does not cause abnormalities in liver development, but causes a severe defect in liver regeneration after partial hepatectomy63. This defect results from increased accumulation of p21 protein that requires p53 and overactivation of p3864. Disruption of p21 or p53 rescued liver regeneration in c-JunΔhep mice64. Interestingly, the high level of phosphorylation of p38 observed after partial hepatectomy in c-JunΔhep mice was reduced by disruption of p21 or p5364. Hepatic disruption of p38α also increased proliferation of hepatocytes, because it reduced expression of p2164. IkkβΔhep mice, however, have accelerated regeneration of liver, which correlates with increased activation of JNK65.
In summary, the combined activities of JNK–AP-1, IKK–NF-κB, p38, and p53 signaling to p21 control liver regeneration. One study used Jnk1Δhep/JNK2−/− mice to assess the specific roles of hepatocyte JNKs in liver regeneration and exclude the effects of compensatory activation of JNK223. Although findings from the study agreed with those from previous reports of defects in liver regeneration in Jnk1−/− mice, it concluded that compensatory activation of JNK2 inhibits regeneration in livers of Jnk1−/− mice.
In contrast to the role of JNK in promoting liver regeneration, persistent JNK activation attenuates liver regeneration. Mice deficient in growth arrest and expression of the DNA-damage-inducible gene 45β (GADD45β) have reduced proliferation and increased death of hepatocytes, and sustained activation JNK following partial hepatectomy66. Disruption of Jnk2 increased liver regeneration in Gadd45β−/− mice66, indicating that the magnitude and duration of JNK activation is important.
Metabolic Syndrome and Hepatic Steatosis
Non-alcoholic fatty liver disease (NAFLD) is a hepatic manifestation of the metabolic syndrome, which is characterized by obesity and insulin resistance. The spectrum of NAFLD ranges from simple steatosis to steatosis with hepatic inflammation and fibrosis, known as non-alcoholic steatohepatitis (NASH). NASH is a high risk factor for cirrhosis. Excessive fat intake is believed to cause simple steatosis as a first hit, and then a second hit, such as oxidative stress, adipose tissue-derived cytokines, or translocation of endotoxin from the intestinal lumen, leads to hepatic inflammation. Alternatively, simple steatosis and NASH could have separate etiologies, such that steatosis is not a preliminary condition for NASH. Because obesity and insulin resistance are risk factors for NAFLD, activities of JNK in the liver, along with the adipose tissues and muscles, must be important in the development of NAFLD. Strong activation of JNK has been observed in the liver, fat, and muscle tissues in mice placed on a high fat diet (HFD) and genetically (ob/ob) obese mice67,68. We discuss how systemic activation of JNK affects development of hepatic steatosis and insulin resistance, based on the experiments in Jnk1−/− and Jnk2−/− mice.
The HFD causes obesity in wild-type, but not Jnk1−/− mice67–69. Jnk1−/− mice have decreased phosphorylation of IRS-1 at Ser 307 (an inhibitory site), compared with wild-type mice, and increased insulin-induced tyrosine phosphorylation of IRS-1, which ameliorates insulin resistance67. Moreover, HFD-induced hepatocyte injury and steatosis were suppressed in Jnk1−/− mice. Jnk2−/− mice placed on the HFD have a similar degree of hepatic steatosis as wild-type mice, but have increases in hepatocyte injury, obesity, and insulin resistance69. Interestingly, livers from Jnk2−/− mice have greater JNK activity, indicating that JNK1 overcompensation damages the liver and contributes to insulin resistance69. The effect of JNK1 overcompensation was confirmed in Jnk2−/− mice with Jnk1 haploinsufficiency, which reduced obesity and hepatic steatosis and increased insulin sensitivity70. To avoid JNK1-mediated compensation, another study attempted to knockdown JNK1 or JNK2 in a model of established steatosis. Although acute knockdown of JNK1 or JNK2 increased insulin sensitivity, only JNK1 knockdown attenuated hepatocyte damage and steatosis. JNK2 knockdown actually increased liver injury, and increased levels of Bim, because its degradation is JNK2 dependent69. The effect of Bim accumulation in Jnk2−/− mice was demonstrated by a reduction in liver injury upon Bim ablation69. So, JNK1 and JNK2 each participate in hepatic injury, steatosis, insulin resistance, and obesity (Figure 4).
Figure 4. Roles of JNKs in the Pathogenesis of NAFLD.

Obesity and hyperlipidemia increase plasma levels of FFA, which activate MLK3 and JNK in hepatocytes. JNK1 contributes to hepatic insulin resistance by phosphorylating IRS-1 at serine 307, and mitochondria-mediated hepatocyte death through Bax and PUMA. Saturated FFAs activate JNK through oxidative stress, ER stress, and lipid peroxidation in hepatocytes. Saturated FFAs also activate JNK in inflammatory cells to contribute to production of inflammatory cytokines. TLR4 on inflammatory cells might be involved in FFA-induced JNK activation.
The HFD and genetically induced obesity activate JNK in the liver. Hyperglycemia in mice with streptozotocin- or alloxan-induced, insulin-dependent diabetes does not activate hepatic JNK. Therefore, in obese mice, hyperglycemia is not a cause of hepatic JNK activation. However, other sugars, such as fructose, activate hepatic JNK and attenuate insulin signaling, by increasing phosphorylation at Ser307 and decreased tyrosine phosphorylation of IRS-171. Treatment with saturated free fatty acids (FAA) activates JNK and leads to insulin resistance68,72. In adipocytes and fibroblasts, saturated FAA causes aggregation of the membrane-anchored tyrosine kinase c-Src within lipid rafts, leading to MLK3 activation and subsequent JNK activation72. However, this mechanism does not operate in hepatocytes72. JNK activation is also associated with ER stress-mediated insulin resistance. ER stress causes JNK activation through IRE1α, and insulin resistance occurs as a consequence of JNK-mediated IRS-1 phosphorylation73. In mice with mutations in XBP-1, abnormal ER functions cause insulin resistance through JNK activation73. However, mice with hepatocyte specific deficiency in XBP-1 did not develop insulin resistance, in spite of increased ER stress and JNK activation in the liver74. The ER stress induced by hepatic XBP-1 deficiency is therefore not associated with JNK-mediated insulin resistance.
JIP1 is a scaffold proteins involved in JNK activation that contributes to insulin resistance and hepatic steatosis75. Double-stranded RNA-dependent protein kinase (PKR) is another upstream regulator of JNK that has been associated with metabolic disease. PKR senses high levels of nutrients and obese states to activate JNK, which causes insulin resistance and hepatic steatosis76.
Hepatocyte Lipoapoptosis and NASH
What are the specific roles of JNK in hepatocytes during lipoapoptosis and development of NASH? Saturated FFA cause apoptosis in hepatocytes—an effect known as lipoapoptosis. In hepatocytes, saturated FFA induce direct interaction between the small GTPase Cdc 42/Rac1 and MLK3 to activate JNK1, independent of the ER stress transducer IRE1α (Figure 4)77. This pathway promotes lipoapoptosis. GSK-3β is also involved in FFA-induced JNK activation, independently of FFA-induced ER stress responses78. The saturated FFA palmitate upregulates the p53-upregulated mediator of apoptosis (PUMA) and Bax in a JNK1-dependent manner (Figure 4)79. Puma−/− hepatocytes are resistance to FFA-induced lipoapoptosis, indicating the importance of this death regulation pathway. In addition, FFA cause degradation of Mcl-1 through JNK1, which contributes to lipoapoptosis28,80. Steatotic hepatocytes are more susceptible to TNF-induced apoptosis, which involves activation of ASK1 and JNK81. Moreover, TRAIL–DR5-mediated hepatocyte lipoapoptosis is mediated via JNK in steatotic hepatocytes82. However, these findings were all obtained through in vitro experiments with high concentrations of FFA, which causes high rates of cell death; the importance of lipoapoptosis and its relation to JNK activation in vivo are not clear.
The roles of JNK in NASH have been studied using mice placed on methione-choline deficient (MCD) diets. The MCD diet causes hepatic steatosis, injury, and inflammation, but does not induce extrahepatic metabolic features, such as obesity and insulin resistance. The MCD diet-induced model of NASH has been used to assess liver-specific functions of JNK. The diet was found to activate JNK and AP-1 in the liver83. JNK1 deficiency protected mice from the effects of the MCD diet, but JNK2 deficiency had no effect83. JNK1 therefore promotes induction of NASH by the MCD diet.
There has been debate over what liver cell type is involved in JNK-mediated metabolic effects. Studies in radiation chimeras showed that JNK1 functions within radiation-resistant stroma cells and in radiation-sensitive hematopoietic cells. Activation of JNK1 in radiation-resistant stroma, including hepatocytes, adipocytes and muscles, is required for HFD-induced obesity24. Yet, JNK1 activation in hematopoietic cells, including tissue macrophages, is also involved in HFD-induced insulin resistance24,84. In myeloid cells exposed to FFA, JNK contributes to production of inflammatory cytokines, such as TNF, IL-1, and IL-6, which induce insulin resistance in other cells24,84,85. A study that used a choline-deficient amino acid-defined (CDAA) diet to induce NASH in mice found that activities of JNK1 in hematopoietic cells were more important in resident liver cells for induction of hepatocellular injury, inflammation, and fibrosis86.
Intriguingly, hepatocyte-specific deletion of JNK1 does not protect mice from HFD-induced insulin resistance87. However, Jnk1Δhep mice have increased lipogenesis and attenuated insulin resistance on normal diets and importantly, spontaneously develop hepatic steatosis87. These findings indicate an anti-steatotic and anti-diabetic function of JNK1 in hepatocytes, and that the hepatic phenotypes of Jnk1−/− mice could result from JNK1 functions in non-parenchymal cells and extra-hepatic tissues (e.g. adipose tissues).
Disruption of the combination of Jnk1 and Jnk2 in adipocytes reduced obesity in mice placed on the HFD, along with hepatic steatosis and insulin resistance, indicating that adipocyte JNK affects metabolic states in other tissues, including liver and muscle88. Surprisingly, deletion of JNK1 from skeletal muscle worsened the metabolic state in liver and adipose tissues89. Jnk1 ablation in the central nervous system reduced food intake and body weight gain in mice fed the HFD90. The main effects of JNK1 in the central nervous system are mediated through thyroid stimulating hormone and thyroid hormone90.
Although JNK1 is important for FFA-induced lipoapoptosis and inhibition of insulin signaling by phosphorylation of IRS-1 at Ser 307 in hepatocytes, findings from studies in genetically engineered mice indicate that the activities of JNK1 in hematopoietic cells, adipocytes, and the nervous systems promote, (but in hepatocytes and muscles might prevent) NASH and metabolic syndrome. So, inhibitors of JNK1 might be used to treat patients with NASH or insulin resistance. BI-78D3, a small molecule JIP1 mimics that inhibits JNK activity, increased insulin sensitivity in mouse models of type 2 diabetes91. Cell- or tissue-specific delivery of JNK1 inhibitors might improve efficacy and reduce adverse effects.
HSC Activation and Fibrosis
Fibrosis is a wound-healing response following chronic liver damage caused by hepatitis B or C viruses, cholestatic liver inflammation, alcoholic steatohepatitis, NASH, and autoimmune hepatitis. The pathogenesis of liver fibrosis is characterized by excessive production and deposition of extracellular matrix proteins, including type I and III collagen, and requires activation of HSC. JNK is involved in HSC activation. Blocking JNK activity with SP600125 inhibited HSC activation, demonstrated by decreased expression of α smooth muscle actin (SMA), and reduced proliferation92. Interestingly, SP600125 increased production of collagen, indicating complexity in control of HSC activation, proliferation, and collagen production92. TGF-β and angiotensin II upregulate expression of αSMA through activation of JNK. PDGF-mediated HSC proliferation is also JNK dependent93. Both TGF-β and PDGF activate SMAD2 and SMAD3 through JNK, resulting in HSC migration94. TLR4 signaling potentiates TGF-β signaling by downregulating bone morphogenetic protein and activin membrane bound inhibitor (Bambi), which is partly dependent on JNK (Seki E, unpublished data)95. Endothelin promotes HSC activation in an autocrine manner. TNF also induces endothlin-1 production through JNK96.
Among the AP-1 components, JunD is expressed and regulates TIMP-1 expression in activated HSCs97. However, JNK is dispensable for JunD-mediated TIMP-1 expression in HSCs, and JunD activation is mediated by ERK1/297. JNK is strongly activated in HSCs following induction of liver fibrosis induced by bile duct ligation or chronic administration of carbon tetrachloride, and in patients with liver fibrosis from HCV infection or NASH93. Jnk1−/− mice are protected from liver fibrosis, but Jnk2−/− mice develop more liver fibrosis than wild-type mice—another example for the pathogenic effects of JNK1 overactivation in Jnk2−/− mice. Another study reported are requirement for hematopoietic JNK1, but not JNK2, in mice that develop liver fibrosis from the CDAA diet86. JNK therefore mediates many mechanisms that contribute to the pathogenesis of liver fibrosis.
Hepatocellular Carcinoma (HCC)
HCC is the third-leading cause of cancer-related deaths. HCC develops in individuals with liver diseases such as chronic HBV or HCV infection, NASH, or alcoholic cirrhosis. The incidence of HCC in the United States has doubled in recent decades, due to the increased prevalence of hepatitis C and NASH, which both involve JNK activity 98, 99. Studies in mice with diethylnitrosamine (DEN)-induced HCC indicated a role for JNK1 in pathogenesis. A single injection of DEN to young mice induces HCC at the age of 8–10 months. JNK1 deficiency protects mice from DEN-induced hepatocyte death, resulting in reduced compensatory proliferation and HCC formation100. In a study that used the DEN-phenobarbital protocol (in which DEN is the initiator and phenobarbital is the promoter of hepatocarcinogenesis)60, Jnk1−/− mice developed fewer and smaller liver tumors than wild-type or Jnk2−/− mice60,100. Livers from the Jnk1−/− had decreased levels of c-Myc and increased levels of p21; c-Myc overexpression or disruption of p21 restored HCC development to levels observed in wild-type mice60.
JNK1 downregulates transcription of p21 via c-Myc but not p53. JNK1 regulates transcription of c-Myc expression, through phosphorylation of c-Jun and other mechanisms60. c-Jun is important in hepatocarcinogenesis101. Deletion of c-Jun from hepatocytes of mice reduced development of HCC following administration of DEN33. Disruption of c-Jun in hepatocytes increased their expression of p53 and sensitivity to TNF-induced death33. Interestingly, the activities of c-Jun effects in DEN-induced tumor formation do not require Jun phosphorylation by JNK, based on mutational analyses (in c-JunAA mice)33.
JNK also phosphorylates SMAD3 in its linker region to antagonize TGF-β–induced carboxy-terminal phosphorylation of SMAD3, which would lead to p21 induction and tumor suppression102. JNK phosphorylation of SMAD3 therefore interferes with p21 upregulation by SMAD3 and c-Myc102. JNK also promotes apoptosis of HCC cells, as well as hepatocytes103.
JNK1 therefore contributes to the initiation of HCC by inducing hepatocyte apoptosis and compensatory proliferation. JNK1-dependent downregulation of p21 is mediated by c-Myc upregulation, independently of c-Jun and its phosphorylation. JNK also regulates p21 expression by inhibiting TGF-β-induced activation of SMAD3. Independent of JNK, c-Jun is required for liver carcinogenesis, by suppressing p53 (Figure 5).
Figure 5. Functions of JNKs in HCC.

In HCC, JNK1 is overactivated either by overactivation of the upstream kinases MKK4 and 7 or the inactivation of DUSP1. Activated JNK1 induces c-Myc, which suppresses p21 expression and promotes HCC proliferation via cyclin D1 expression. JNK1 also upregulates MLL3 and EZH2, 2 histone H3 methyl transferases to increase expression of the cell cycle-associated genes, such as cyclins, and inhibit the transcription of tumor suppressors, respectively. c-Jun inhibits p53 activity post-transcriptionally. JNK1 increase generation of ROS, which is inhibited by p38α and IKKβ activation of NF-κB, through induction of SOD2. JNK1 also inhibits TGF-β–induced Smad3 activation, thereby inducing p21 to suppress HCC promotion. All of these pathways regulate hepatocyte death, which results in the release of IL-6, TNF, and TGF-β through JNK1 in non-parenchymal cells, including Kupffer cells. IL-6 induces HCC proliferation via activation of STAT3in hepatocytes. JNK1 promotes (but TAK1, p38α, and IKKβ activation of NF-κB prevent) hepatocyte death and HCC formation.
Inactivation of IKK and NF-κB promotes activation of JNK and increases hepatocarcinogenesis following administration of DEN104. In IkkβΔhep mice, disruption of Jnk1 reduced development of HCC100. Increased JNK activation was also observed during spontaneous hepatocarcinogenesis in NemoΔhep and Tak1Δhep mice, in which NF-κB is inactivated105–107. Administration of anti-oxidants to NemoΔhep mice or disruption of TnfrI in Tak1Δhep mice prevented activation of JNK and development of HCC, indicating that overactivation of JNK promotes HCC in these mice (Seki, unpublished data)105,106. Similarly, hepatocyte-specific disruption of p38α increased generation of ROS, activation of JNK, and hepatocarcinogenesis following a single injection of DEN or administration of DEN-phenobarbital. The increased formation of HCC was reversed when c-Jun was disrupted or JNK activity was inhibited with an anti-oxidant18,108.
Mdr-2−/− mice develop inflammation-dependent spontaneous HCC; NF-κB and JNK are each induced through TNF overproduction109. In Mdr-2−/− mice, inactivation of NF-κB prevented formation of HCC, and antagonists of TNF also inhibited HCC development, by decreasing JNK phosphorylation109. TNF receptor signaling and ROS might be the major upstream mediators of JNK activation, whereas NF-κB and p38 suppress JNK activity, through inhibition of ROS. Importantly, JNK1 has a larger role than JNK2 in development of HCC.
The cell-specific functions of JNK in hepatocarcinogenesis were determined from studies of Jnk1−/−Jnk2−/− mice and Jnk1Δhep/JNK2−/− mice. Conditional Jnk1−/−Jnk2−/− mice develop less numbers of HCCs, and have reduced activities of c-Myc and c-Jun and increased expression of p2123. Intriguingly, Jnk1Δhep/JNK2−/− mice developed more HCCs, with increased c-Myc and c-Jun expression, hepatocyte apoptosis, and compensatory proliferation, but paradoxically increased p21 expression23. This puzzling finding indicates that HCC formation depends on c-Myc and c-Jun, but is independent of JNK. The carcinogenic effects of JNK might be restricted to non-parenchymal cells, rather than hepatocytes, and induce an inflammatory response, such as production of IL-6, TNF, and TGF-β. Alternatively, hepatocyte JNK might inhibit non-parenchymal, cell-mediated liver inflammation that contributes to hepatocarcinogenesis.
There are additional mechanisms for the paradoxical roles of JNK in HCC formation. Studies in different models of HCC could determine where JNK functions to promote hepatoccarcinogenesis, such as in non-parenchymal cells vs hepatocytes. Hui et al. studied induction of HCC with the DEN-phenobarbital protocol, whereas Das et al. study tumor formation following a single injection of DEN in young mice; Tak1Δhep mice develop spontaneous HCC23,60,106,107. Alternatively, deletion of a combination of JNK1 and JNK2 from hepatocytes increases their susceptibility to DEN-induced death and transformation; these effects are not observed in hepatocytes with disruption of only Jnk1. The presence or absence of JNK2 in hepatocytes could determine the contradictory functions of JNK1 in these cells. One study reported that Ask1−/− mice develop HCC following administration of DEN, with less activation of JNK than in other models110. Further investigations are required to determine the paradoxical roles of JNK in hepatocytes and hepatocarcinogenesis.
Studies support the importance of JNK in pathogenesis of human HCC. JNK1 and JNK2 are phosphorylated in primary HCC samples from patients; 50%–60% of HCC samples had higher levels of phospho-JNK1 than non-neoplastic lesions, whereas levels of phospho-JNK2 did not differ between HCC and non-tumor tissues60,111. JNK1 activation was correlated with tumor size, and knockdown of JNK1, but not JNK2, reduced proliferation of a human HCC cell line, indicating that JNK1 promotes proliferation of human HCCs60,111. Moreover, expression of dual-specific phosphatase (DUSP)1 is reduced in human HCCs, leading to overactivation of JNK111. In human HCCs, JNK1 upregulates c-Myc and downregulates p21; JNK1 also regulates expression of factors that control the cell cycle, proliferation and metabolism, through methylation of histone H3 lysine 4 and 9 by MLL3 and EZH260,111.
HCV is a major cause of HCC. The HCV core protein activates JNK through ROS112. Mice that express an HCV core transgene develop spontaneous HCC with high frequencies of p53 mutations, which are amplified by administration of DEN; hepatocyte-specific disruption of c-Jun reduces HCC in these mice112. The role of JNK in HCV-induced HCC development remains to be examined.
Reagents designed to target JNK1 might be developed to treat patients with HCC. D-JNKI1, a specific inhibitor of JNK1 and 2 (a dual inhibitor), suppressed JNK activity in HCC cells, decreasing levels of c-Myc and increasing levels of p21. This reagent suppressed growth of human HCC cells in culture and xenograft tumors in mice1,60. D-JNK1 also suppressed DEN-induced HCC in mice60. The JNK inhibitor SP600125 blocked HCC growth in rats with DEN-induced liver tumors102,113. The JNK inhibitor sensitizes HCC cells to TRAIL35, indicating that JNK inhibitors might be given in combination with TRAIL as therapeutic agents.
Future Directions
The JNKs are important signaling molecules in multiple pathways in liver physiology and disease pathogenesis. JNKs regulate transcription by phosphorylating and activating transcription factors (such as cJun, JunB, and ATF2), or independently of transcription, by phosphorylating signaling molecules (such as IRS-1, Itch, Mcl-1, and Bid). When appropriately activated, JNKs regulate important biologic functions, such as liver regeneration. However, the same signaling pathway can also be detrimental, such in carcinogenesis. The liver expresses 2 JNK isoforms. Most pathologic processes are associated with JNK1; the severe phenotypes observed following deletion of JNK2 probably result from compensatory JNK1 activation.
The metabolic syndrome, which includes obesity, insulin resistance, and NASH is a significant public health issue worldwide. JNK activity regulates various pathophysiologic processes, including hepatocyte death, steatosis, inflammation, and insulin resistance, which are associated with NASH, fibrosis and HCC. Pre-clinical studies in animal models or human cells have indicated that reagents that inhibit JNK might be used to treat patients with liver diseases, including acute liver failure, I/R injury, fibrosis, HCC, and NASH1,60,91. Several JNK inhibitors, such as SP600125, D-JNKI1, and BI-78D3 have been tested in pre-clinical studies. CC-930 is currently being tested in Phase II clinical trial for idiopathic pulmonary fibrosis114, and dual inhibitors of JIP and JNK have been identified115. To identify the most effective approach to block JNK signaling in patients with liver disease, in addition to the established reagents, new JNK inhibitors must also be developed and their specificity assessed for different JNK isoforms; the reagents must then be tested in different cells, tissues, and systems. Clinical trials are also required to assess new JNK inhibitors, alone or in combination with other therapeutics116.
Acknowledgments
Grant support: NIH grant R01AA020172 (ES), R01DK085252 (ES), and R01GM041804 (DAB).
Abbreviations
- AP
activator protein
- ASK
apoptosis signal-regulating kinase
- ATF
activating transcription factor
- Bambi
bone morphogenetic protein and activin membrane bound inhibitor
- cDISC
cytosolic death-initiating signaling complex
- cIAP
cellular inhibitor of apoptosis
- ConA
concanavalin A
- CYLD
cylindromatosis
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- FAA
free fatty acid
- GADD
growth arrest and DNA-damage-inducible gene
- GalN
D-galactosamine
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HFD
high fat diet
- HSC
hepatic stellate cells
- IKK
IκB kinase
- IL-1
interleukin-1
- I/R
ischemia and reperfusion
- IRE
inositol requiring enzyme
- IRS
insulin receptor substrate
- JIP
JNK interacting protein 1; c-Jun-N-terminal Kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MCD
methione-choline deficient
- MLK
mixed linage kinase
- NAFLD
non-alcoholic fatty liver disease
- NAPQI
N-acetyl-p-benzoquinone imine
- NASH
non-alcoholic steatohepatitis
- NOX
NADPH oxidase
- PERK
PKR-like ER localized kinase
- PDGF
platelet-derived growth factor
- PKR
double-stranded RNA-dependent protein kinase
- PUMA
p53-up-regulated mediator of apoptosis
- ROS
reactive oxygen species
- SMA
smooth muscle actin
- SRF
serum response factor
- SOD
superoxide dismutase
- TGF
transforming growth factor
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- UPR
unfolded protein response
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contribution of authors: each author was involved with this manuscript.
1. Ekihiro Seki : study concept and design, acquisition of data, analysis and interpretation of data, drafting and critical revision of the manuscript for important intellectual content, obtained funding, study supervision
2. David A. Brenner : analysis and interpretation of data, critical revision of the manuscript for important intellectual content, obtained funding
3. Michael Karin : analysis and interpretation of data, critical revision of the manuscript for important intellectual content
References
- 1.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 2.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 3.Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–39. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 4.Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol. 2011;90:536–44. doi: 10.1016/j.ejcb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–95. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond B Biol Sci. 1996;351:127–34. doi: 10.1098/rstb.1996.0008. [DOI] [PubMed] [Google Scholar]
- 7.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 8.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 9.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 10.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 11.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 13.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26:675–87. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–42. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–13. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr, Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–15. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 18.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–65. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–37. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 20.Geisler F, Algul H, Paxian S, Schmid RM. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–503. doi: 10.1053/j.gastro.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 21.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–7. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 22.Das M, Sabio G, Jiang F, Rincon M, Flavell RA, Davis RJ. Induction of hepatitis by JNKmediated expression of TNF-alpha. Cell. 2009;136:249–60. doi: 10.1016/j.cell.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011;25:634–45. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM, Karin M. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–97. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Lo CR, Czaja MJ. NF-kappaB inhibition sensitizes hepatocytes to TNF-induced apoptosis through a sustained activation of JNK and c-Jun. Hepatology. 2002;35:772–8. doi: 10.1053/jhep.2002.32534. [DOI] [PubMed] [Google Scholar]
- 26.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–2. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–67. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology. 2009;136:1423–34. doi: 10.1053/j.gastro.2008.12.064. [DOI] [PubMed] [Google Scholar]
- 29.Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem. 2011;286:35071–8. doi: 10.1074/jbc.M111.276089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni HM, Chen X, Shi YH, Liao Y, Beg AA, Fan J, Yin XM. Genetic delineation of the pathways mediated by bid and JNK in tumor necrosis factor-alpha-induced liver injury in adult and embryonic mice. J Biol Chem. 2009;284:4373–82. doi: 10.1074/jbc.M807259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takamura M, Matsuda Y, Yamagiwa S, Tamura Y, Honda Y, Suzuki K, Ichida T, Aoyagi Y. An inhibitor of c-Jun NH2-terminal kinase, SP600125, protects mice from Dgalactosamine/ lipopolysaccharide-induced hepatic failure by modulating BH3-only proteins. Life Sci. 2007;80:1335–44. doi: 10.1016/j.lfs.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 32.Czaja MJ. The future of GI and liver research: editorial perspectives. III. JNK/AP-1 regulation of hepatocyte death. Am J Physiol Gastrointest Liver Physiol. 2003;284:G875–9. doi: 10.1152/ajpgi.00549.2002. [DOI] [PubMed] [Google Scholar]
- 33.Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–92. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 34.Hasselblatt P, Rath M, Komnenovic V, Zatloukal K, Wagner EF. Hepatocyte survival in acute hepatitis is due to c-Jun/AP-1-dependent expression of inducible nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:17105–10. doi: 10.1073/pnas.0706272104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mucha SR, Rizzani A, Gerbes AL, Camaj P, Thasler WE, Bruns CJ, Eichhorst ST, Gallmeier E, Kolligs FT, Goke B, De Toni EN. JNK inhibition sensitises hepatocellular carcinoma cells but not normal hepatocytes to the TNF-related apoptosis-inducing ligand. Gut. 2009;58:688–98. doi: 10.1136/gut.2008.154625. [DOI] [PubMed] [Google Scholar]
- 36.Corazza N, Jakob S, Schaer C, Frese S, Keogh A, Stroka D, Kassahn D, Torgler R, Mueller C, Schneider P, Brunner T. TRAIL receptor-mediated JNK activation and Bim phosphorylation critically regulate Fas-mediated liver damage and lethality. J Clin Invest. 2006;116:2493–9. doi: 10.1172/JCI27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W, Zhang Y, Hosch MS, Lang A, Zwacka RM, Engelhardt JF. Subcellular site of superoxide dismutase expression differentially controls AP-1 activity and injury in mouse liver following ischemia/reperfusion. Hepatology. 2001;33:902–14. doi: 10.1053/jhep.2001.23073. [DOI] [PubMed] [Google Scholar]
- 38.Uehara T, Xi Peng X, Bennett B, Satoh Y, Friedman G, Currin R, Brenner DA, Lemasters J. c-Jun N-terminal kinase mediates hepatic injury after rat liver transplantation. Transplantation. 2004;78:324–32. doi: 10.1097/01.tp.0000128859.42696.28. [DOI] [PubMed] [Google Scholar]
- 39.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, Brenner DA. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42:850–9. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 40.Marden JJ, Zhang Y, Oakley FD, Zhou W, Luo M, Jia HP, McCray PB, Jr, Yaniv M, Weitzman JB, Engelhardt JF. JunD protects the liver from ischemia/reperfusion injury by dampening AP-1 transcriptional activation. J Biol Chem. 2008;283:687–95. doi: 10.1074/jbc.M705606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devey L, Mohr E, Bellamy C, Simpson K, Henderson N, Harrison EM, Ross JA, Wigmore SJ. c-Jun terminal kinase-2 gene deleted mice overexpress hemeoxygenase-1 and are protected from hepatic ischemia reperfusion injury. Transplantation. 2009;88:308–16. doi: 10.1097/TP.0b013e3181ae3067. [DOI] [PubMed] [Google Scholar]
- 42.Theruvath TP, Czerny C, Ramshesh VK, Zhong Z, Chavin KD, Lemasters JJ. C-Jun N-terminal kinase 2 promotes graft injury via the mitochondrial permeability transition after mouse liver transplantation. Am J Transplant. 2008;8:1819–28. doi: 10.1111/j.1600-6143.2008.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 2008;85:1500–4. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beraza N, Ludde T, Assmus U, Roskams T, Vander Borght S, Trautwein C. Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132:2504–17. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 45.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–78. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 46.Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2008;324:8–14. doi: 10.1124/jpet.107.129445. [DOI] [PubMed] [Google Scholar]
- 47.Badmann A, Keough A, Kaufmann T, Bouillet P, Brunner T, Corazza N. Role of TRAIL and the pro-apoptotic Bcl-2 homolog Bim in acetaminophen-induced liver damage. Cell Death Dis. 2011;2:e171. doi: 10.1038/cddis.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–77. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, Ichijo H, Omata M. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology. 2008;135:1311–21. doi: 10.1053/j.gastro.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 50.Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–21. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- 51.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuest M, Willim K, Macnelly S, Fellner N, Resch GP, Blum HE, Hasselblatt P. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology. 2012 doi: 10.1002/hep.24699. [DOI] [PubMed] [Google Scholar]
- 54.Alcorn JA, Feitelberg SP, Brenner DA. Transient induction of c-jun during hepatic regeneration. Hepatology. 1990;11:909–15. doi: 10.1002/hep.1840110602. [DOI] [PubMed] [Google Scholar]
- 55.Westwick JK, Weitzel C, Leffert HL, Brenner DA. Activation of Jun kinase is an early event in hepatic regeneration. J Clin Invest. 1995;95:803–10. doi: 10.1172/JCI117730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, Diehl AM. Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am J Physiol. 1992;263:G579–85. doi: 10.1152/ajpgi.1992.263.4.G579. [DOI] [PubMed] [Google Scholar]
- 57.Yamada Y, Webber EM, Kirillova I, Peschon JJ, Fausto N. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: requirement for type 1 but not type 2 receptor. Hepatology. 1998;28:959–70. doi: 10.1002/hep.510280410. [DOI] [PubMed] [Google Scholar]
- 58.Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–32. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 59.Thevananther S, Sun H, Li D, Arjunan V, Awad SS, Wyllie S, Zimmerman TL, Goss JA, Karpen SJ. Extracellular ATP activates c-jun N-terminal kinase signaling and cell cycle progression in hepatocytes. Hepatology. 2004;39:393–402. doi: 10.1002/hep.20075. [DOI] [PubMed] [Google Scholar]
- 60.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–53. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–25. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 62.Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, Zenz R, Wagner EF, Zatloukal K. Functions of c-Jun in liver and heart development. J Cell Biol. 1999;145:1049–61. doi: 10.1083/jcb.145.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, Wagner EF. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J. 2002;21:1782–90. doi: 10.1093/emboj/21.7.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, Wagner EF. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006;20:2306–14. doi: 10.1101/gad.390506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malato Y, Sander LE, Liedtke C, Al-Masaoudi M, Tacke F, Trautwein C, Beraza N. Hepatocyte-specific inhibitor-of-kappaB-kinase deletion triggers the innate immune response and promotes earlier cell proliferation during liver regeneration. Hepatology. 2008;47:2036–50. doi: 10.1002/hep.22264. [DOI] [PubMed] [Google Scholar]
- 66.Papa S, Zazzeroni F, Fu YX, Bubici C, Alvarez K, Dean K, Christiansen PA, Anders RA, Franzoso G. Gadd45beta promotes hepatocyte survival during liver regeneration in mice by modulating JNK signaling. J Clin Invest. 2008;118:1911–23. doi: 10.1172/JCI33913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 68.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. 2006;103:16454–9. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2006;103:10741–6. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei Y, Wang D, Pagliassotti MJ. Fructose selectively modulates c-jun N-terminal kinase activity and insulin signaling in rat primary hepatocytes. J Nutr. 2005;135:1642–6. doi: 10.1093/jn/135.7.1642. [DOI] [PubMed] [Google Scholar]
- 72.Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–84. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 74.Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol requiring enzyme (IRE1alpha)-mediated JNK activation from hepatic insulin resistance in conditional X-box binding protein-1 (XBP1) knockout mice. J Biol Chem. 2012 doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morel C, Standen CL, Jung DY, Gray S, Ong H, Flavell RA, Kim JK, Davis RJ. Requirement of JIP1-mediated c-Jun N-terminal kinase activation for obesity-induced insulin resistance. Mol Cell Biol. 2010;30:4616–25. doi: 10.1128/MCB.00585-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, Gorgun CZ, Hotamisligil GS. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140:338–48. doi: 10.1016/j.cell.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharma M, Urano F, Jaeschke A. Cdc42 and Rac1 are major contributors to the saturated fatty acid-stimulated JNK pathway in hepatocytes. J Hepatol. 2012;56:192–8. doi: 10.1016/j.jhep.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ibrahim SH, Akazawa Y, Cazanave SC, Bronk SF, Elmi NA, Werneburg NW, Billadeau DD, Gores GJ. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol. 2011;54:765–72. doi: 10.1016/j.jhep.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, Gores GJ. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masuoka HC, Mott J, Bronk SF, Werneburg NW, Akazawa Y, Kaufmann SH, Gores Gj. Mcl-1 degradation during hepatocyte lipoapoptosis. J Biol Chem. 2009;284:30039–48. doi: 10.1074/jbc.M109.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang W, Kudo H, Kawai K, Fujisaka S, Usui I, Sugiyama T, Tsukada K, Chen N, Takahara T. Tumor necrosis factor-alpha accelerates apoptosis of steatotic hepatocytes from a murine model of non-alcoholic fatty liver disease. Biochem Biophys Res Commun. 2010;391:1731–6. doi: 10.1016/j.bbrc.2009.12.144. [DOI] [PubMed] [Google Scholar]
- 82.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56:1124–31. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–72. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 84.Vallerie SN, Furuhashi M, Fucho R, Hotamisligil GS. A predominant role for parenchymal c-Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS One. 2008;3:e3151. doi: 10.1371/journal.pone.0003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–29. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, Brenner DA. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137:1467–1477. e5. doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, Davis RJ. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10:491–8. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Xu A, Chung SK, Cresser JH, Sweeney G, Wong RL, Lin A, Lam KS. Selective inactivation of c-Jun NH2-terminal kinase in adipose tissue protects against diet-induced obesity and improves insulin sensitivity in both liver and skeletal muscle in mice. Diabetes. 2010;60:486–95. doi: 10.2337/db10-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol. 2010;30:106–15. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK, Davis RJ. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010;24:256–64. doi: 10.1101/gad.1878510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci U S A. 2008;105:16809–13. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schnabl B, Bradham CA, Bennett BL, Manning AM, Stefanovic B, Brenner DA. TAK1/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology. 2001;34:953–63. doi: 10.1053/jhep.2001.28790. [DOI] [PubMed] [Google Scholar]
- 93.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–59. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005;166:1029–39. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 96.Zhan S, Rockey DC. Tumor necrosis factor alpha stimulates endothelin-1 synthesis in rat hepatic stellate cells in hepatic wound healing through a novel IKK/JNK pathway. Exp Cell Res. 2011;317:1040–8. doi: 10.1016/j.yexcr.2010.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smart DE, Green K, Oakley F, Weitzman JB, Yaniv M, Reynolds G, Mann J, Millward-Sadler H, Mann DA. JunD is a profibrogenic transcription factor regulated by Jun N-terminal kinase-independent phosphorylation. Hepatology. 2006;44:1432–40. doi: 10.1002/hep.21436. [DOI] [PubMed] [Google Scholar]
- 98.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 99.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544–51. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yuen MF, Wu PC, Lai VC, Lau JY, Lai CL. Expression of c-Myc, c-Fos, and c-jun in hepatocellular carcinoma. Cancer. 2001;91:106–12. doi: 10.1002/1097-0142(20010101)91:1<106::aid-cncr14>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 102.Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, Narita M, Yanagida A, Tamaki N, Yagi S, Ikai I, Matsuzaki K, Uemoto S. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology. 2009;49:1944–53. doi: 10.1002/hep.22860. [DOI] [PubMed] [Google Scholar]
- 103.Saxena NK, Fu PP, Nagalingam A, Wang J, Handy J, Cohen C, Tighiouart M, Sharma D, Anania FA. Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology. 2010;139:1762–73. 1773, e1–5. doi: 10.1053/j.gastro.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 105.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–32. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 106.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A. 2010;107:844–9. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, Heymann F, Weber A, Weiskirchen R, Liedtke C, Gassler N, Muller M, de Vos R, Wolf MJ, Boege Y, Seleznik GM, Zeller N, Erny D, Fuchs T, Zoller S, Cairo S, Buendia MA, Prinz M, Akira S, Tacke F, Heikenwalder M, Trautwein C, Luedde T. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell. 2010;17:481–96. doi: 10.1016/j.ccr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 108.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H, Wagner EF. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–9. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 109.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 110.Nakagawa H, Hirata Y, Takeda K, Hayakawa Y, Sato T, Kinoshita H, Sakamoto K, Nakata W, Hikiba Y, Omata M, Yoshida H, Koike K, Ichijo H, Maeda S. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology. 2011;54:185–95. doi: 10.1002/hep.24357. [DOI] [PubMed] [Google Scholar]
- 111.Chang Q, Zhang Y, Beezhold KJ, Bhatia D, Zhao H, Chen J, Castranova V, Shi X, Chen F. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J Hepatol. 2009;50:323–33. doi: 10.1016/j.jhep.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Machida K, Tsukamoto H, Liu JC, Han YP, Govindarajan S, Lai MM, Akira S, Ou JH. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology. 2010;52:480–92. doi: 10.1002/hep.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hanajiri K, Mitsui H, Maruyama T, Hashimoto N, Sata M, Omata M. Echographic detection of diethylnitrosamine-induced liver tumors in rats and the effect of the intratumoral injection of an inhibitor of c-Jun N-terminal kinase. J Gastroenterol Hepatol. 2009;24:866–71. doi: 10.1111/j.1440-1746.2008.05722.x. [DOI] [PubMed] [Google Scholar]
- 114.Plantevin Krenitsky V, Nadolny L, Delgado M, Ayala L, Clareen SS, Hilgraf R, Albers R, Hegde S, D'Sidocky N, Sapienza J, Wright J, McCarrick M, Bahmanyar S, Chamberlain P, Delker SL, Muir J, Giegel D, Xu L, Celeridad M, Lachowitzer J, Bennett B, Moghaddam M, Khatsenko O, Katz J, Fan R, Bai A, Tang Y, Shirley MA, Benish B, Bodine T, Blease K, Raymon H, Cathers BE, Satoh Y. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22:1433–8. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 115.Chen T, Kablaoui N, Little J, Timofeevski S, Tschantz WR, Chen P, Feng J, Charlton M, Stanton R, Bauer P. Identification of small-molecule inhibitors of the JIP-JNK interaction. Biochem J. 2009;420:283–94. doi: 10.1042/BJ20081899. [DOI] [PubMed] [Google Scholar]
- 116.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;1804:463–75. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]