

Abstract
We present here the phasor approach to biosensor FRET detection by fluorescence lifetime imaging microscopy (FLIM) and show that this method of data representation is robust towards biosensor design as well as the fluorescence artifacts inherent to the cellular environment. We demonstrate this property on a series of dual and single chain biosensors which report the localization of Rac1 and RhoA activity, whilst performing concomitant ratiometric FRET analysis on the acquired FLIM data by the generalized polarization (GP) approach. We then evaluate and compare the ability of these two methods to quantitatively image biosensor FRET signal as a function of time and space. We find that with lifetime analysis in the phasor plot each molecular species is transformed into a two dimensional coordinate system where independent mixtures of fluorophores can be distinguished from changes in lifetime due to FRET. This enables the fractional contribution of the free and bound state of a dual chain biosensor or the low and high FRET species of a single chain biosensor to be quantified in each pixel of an image. The physical properties intrinsic to each biosensor design are also accurately characterized by the phasor analysis; thus this method could be used to inform biosensor optimization at the developmental stage. We believe that as biosensors become more sophisticated and are multiplexed with other fluorescent molecular tools, biosensor FRET detection by the phasor approach to FLIM will not only become imperative to their use but also their advancement.
Keywords: Fluorescence lifetime, phasor analysis, Förster resonance energy transfer, biosensor
Introduction
The development of fluorescent protein biosensors coupled with live-cell imaging has enabled the visualization and measurement of intracellular molecular dynamics with high spatiotemporal resolution (Gaits and Hahn, 2003; Hahn and Toutchkine, 2002). Fluorescent biosensors designed to probe kinase, phosphatase and GTPase activity, second messenger dynamics, metabolites such as glucose, and receptor–effector coupling are just a few examples of the continuously expanding collection currently available (DiPilato and Zhang, 2010; Wallrabe and Periasamy, 2005). The great majority of biosensors based on fluorescent proteins use an inherent FRET (Förster resonance energy transfer) interaction to respond to the level of cellular activity being probed (Ibraheem and Campbell,; Tsien, 1998). Given that these molecular tools are designed to report on the spatial localization, temporal coordination and biochemical concentrations of specific signaling events, detection and quantitation of FRET as a function of time and space in cells is a matter of great interest.
FRET is a phenomenon where a donor fluorophore in an excited electronic state transfers its excitation energy to a nearby acceptor fluorophore via non-radiative dipole-dipole interaction (Clegg, 1995; Förster, 1965). Distance and orientation determine whether a donor-acceptor fluorophore pair can undergo FRET, and this is the key to differentiating the on and off state (dynamic range) of a FRET biosensor (Tsien, 1998). When a FRET interaction does occur the donor fluorescence intensity and lifetime are quenched and the acceptor fluorescence intensity is enhanced, as well as shifted in lifetime (Lakowicz and Masters, 2008). In theory any one of these changes can be used to detect the spatiotemporal dynamics of a FRET biosensor and thereby generate a FRET image. In practice the technical challenge is to quantify and differentiate a FRET signal from the several other sources of fluorescence that are present in live cells (Periasamy and others, 2008; Wallrabe and Periasamy, 2005).
A FRET biosensor can be constructed using either a dual chain or single chain design (Hodgson and others, 2010; Tsien, 1998). A dual chain design places the donor and acceptor on two separate molecules (chains) which only bind upon activation. This design is advantageous in terms of dynamic range, in that there is no baseline interaction between the two biosensor chains. However by employing two chains, there is a propensity for the donor and acceptor fluorophores to distribute unequally throughout the cell and this causes artifact for intensity based FRET detection (Hodgson and others, 2010). A single chain design places the donor and acceptor on the same molecule and activation of the target causes a conformational change that alters the proximity and/or orientation of the fluorophores (Hodgson and others, 2008; Hodgson and others, 2010). This design is advantageous for ease of image analysis. However by connecting the donor and acceptor, they are in close enough proximity to cause an inherent residual FRET signal that does not originate from the cellular activity being probed.
We present here the phasor approach to biosensor FRET detection by fluorescence lifetime imaging microscopy (FLIM) and show that this method of data representation is robust towards biosensor design and the fluorescent artifacts inherent to a cellular environment. We demonstrate this on a series of dual and single chain GTPase biosensors which report the localization of Rac1 and RhoA activity (Hodgson and others, 2008; Hodgson and others, 2001; Hodgson and others, 2010; Kraynov and others, 2000; Machacek and others, 2009; Pertz and Hahn, 2004). We perform concomitant ratiometric FRET analysis on the acquired FLIM data by the generalized polarization (GP) approach (Parasassi and others, 1991), demonstrating the advantages of the phasor approach.
Rac1 is known to promote membrane protrusion at the leading edge, while RhoA regulates contractility in the cell body and at adhesions throughout the cell (Burridge and Wennerberg, 2004). Recent studies using the biosensors employed here indicate that Rac and Rho interact at the leading edge to regulate protrusion, with RhoA activation being synchronous with edge advancement and Rac1 shifted 2μm behind (Pertz and others, 2006) (Machacek and others, 2009). With these spatiotemporal dynamics in mind we evaluate the ability of each method to quantitatively image a FRET signal as a function of time and space.
Determination of FRET using intensity based ratiometric analysis and the generalized polarization (GP) function
Quantitation of the FRET signal from a biosensor requires knowledge of the relative concentrations of the different states of the biosensor: the free and bound state of a dual chain design and the low FRET (LF) and high FRET (HF) state of a single chain design (Hodgson and others, 2010). In either case the various forms of the biosensor are a linear combination of the spectra of the donor and the acceptor fluorophores; thus an intensity based ratiometric method of analysis should be sufficient to derive the relative concentration of the two species after calibration for instrumental artifacts (e.g. spectral bleed through). In the cellular environment this is not the case, since not all of the biosensor expressed by the cell is optically active. In all cases of fluorescent protein expression there is a fraction of the protein that does not completely mature (Llères and others, 2001). This leads to biosensor fluorescence at donor and/or acceptor wavelengths that are not responsive to the physiological state being measured. Furthermore the two fluorescent proteins (donor and acceptor) invariably experience photo-bleaching at different rates (Hodgson and others, 2006) and this also results in a population of unresponsive donor only and acceptor only FRET pairs which accumulate over the duration of the experiment (Fig. 1A–B).
Figure 1.

Intensity based biosensor FRET detection of a dual and single chain biosensor. (A) The fluorescent species encountered in dual chain biosensor FRET detection: donor chain, acceptor chain and the donor and acceptor chain bound. (B) The fluorescent species encountered in single chain biosensor detection: optically inactive biosensor, low FRET biosensor and high FRET biosensor. (C) Emission spectra of the various fluorescent species present in a FRET experiment that must be linearly unmixed when performing ratiometric analysis of a single chain or dual chain biosensor. (D) Graphical representation of the additive property of GP transformation. If we know the GP of the on and off state of the biosensor, then the fractional contribution of the GP of an unknown mixture can be found by the linear combination of GP1 and GP2. The fractional contribution of a third species with a different spectrum (e.g. cell autofluorescence) can also be determined in each pixel with detection at a third wavelength. (E)–(F) The alignment of the 3 or 4 GP values in the dual or single chain FRET experiment, respectively, prevents the determination of the contribution of the individual species by spectral unmixing: unlike the situation illustrated with the phasor approach (Figure 2).
Determination of the individual concentrations of the two states of the biosensor based on intensity is thus virtually impossible; only relative changes in the population of the on versus off state can be ascertained by a ratiometric method of analysis (Fig. 1C). One way to better determine this relative change is to calculate the normalized FRET ratio of each pixel in an image by use of the generalized polarization (GP) function (defined in equations 10–12 of the materials and methods section). The GP function has been used extensively to quantify the change in spectrum of Laurdan in response to membrane fluidity (Parasassi and others, 1991; Yu and others, 1996). The GP function can also be used to measure the change in emission spectrum due to FRET and has the advantageous property of transforming the concentration of each species into becoming additive, since the denominator of the GP is proportional to the total fluorescence intensity (Jameson and others, 1984). This enables a graphical representation, where if we know the GP of the on and off state of a biosensor, then the relative GP of an unknown mixture can be found from the linear combination of the two known GPs (Fig. 1D). It is simply the alignment of the donor and acceptor only spectra with the on and off state of a biosensor that prevents the determination of the individual species by spectral un-mixing (Fig. 1E–1F).
Determination of FRET using quenching of the donor fluorescence and the phasor approach to FLIM
Determination of FRET in cells at a quantitative level is thought to be best achieved using the quenching of donor lifetime, since this parameter is changed to a value that can be resolved from the on and off state of a dual or single chain biosensor (Bastiaens and Squire, 1999; Suhling and others, 2005; Wallrabe and Periasamy, 2005). Fluorescence lifetime imaging microscopy (FLIM) measures the average lifetime of the donor fluorescence emission in the presence of the acceptor in each pixel of an image and with prior knowledge of the donor lifetime in the absence of the acceptor, assigns those pixels producing a quenched lifetime to the FRET localization. There are different approaches available to detect FRET by FLIM, distinguished by image acquisition in either the time or frequency domain (Chandler and others, 2006; van Munster EB, 2005; Wallrabe and Periasamy, 2005). FLIM data is primarily acquired in the time domain with analysis proceeding by fitting the average fluorescence decay in each pixel using an exponential model. However this mode of analysis presents a formidable computational problem when multiple fluorescent species are present as is always the case in any FRET experiment.
Analysis of FLIM data in the time domain can be simplified by use of the phasor approach, a vector representation conventionally employed to analyze data collected in the frequency domain (Clayton and others, 2004; Digman and others, 2008; Gratton and others, 1984; Jameson and others, 1984; Lakowicz and others, 1984). The phasor approach provides a global view of the fluorescence decay in an image by transforming the histogram of time delays in each pixel into a phasor. The sine-cosine transforms of each phasor are plotted in a two dimensional space termed the universal plot, and each phasor position is characteristic of a particular molecular species and its local environment (Colyer and others, 2008; Digman and others, 2008). In the phasor space you can distinguish a mixture of independent molecular species (which form a linear trajectory) from a change in lifetime due to FRET (which forms a curved trajectory) without having to resolve the decay at each pixel into the individual exponential components (Digman and others, 2008). Figure 2 illustrates this concept and shows that irrespective of biosensor design, analysis of a FRET signal in the phasor plot enables the various fluorescent species present in a FRET experiment to be resolved. The striking difference of the phasor representation compared with the intensity ratio in Figure 1 is that FRET changes the lifetime so that a new phasor position is produced that is not aligned with the on and off state of the biosensor.
Figure 2.
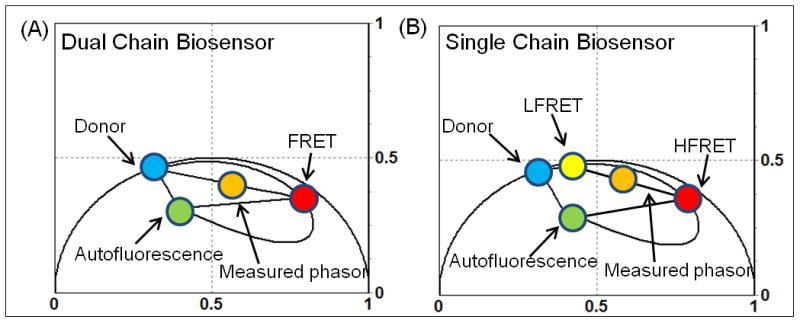
Analysis of the FRET signal from a dual chain and single chain biosensor in the phasor plot. (A) Dual chain design: the phasor location of the donor-only species and autofluorescence form the linear combination from which a phasor distribution due to FRET will originate. By superimposing a FRET trajectory (defined by equation 9 in the materials and methods section) over the most quenched lifetime we can determine the efficiency of the FRET state of the biosensor, and from the line that connects this FRET state back to the donor, quantitate the population of donor undergoing FRET in each pixel. (B) Single chain design: the phasor location of the donor-only species is no longer the off state of the biosensor. Instead it is a low FRET state due to a residual FRET signal inherent to the single chain biosensor design. In this scenario the low FRET state, which is shifted toward a shorter lifetime along the FRET trajectory from the donor phasor is the off state and then further shifted to the most quenched lifetime along the FRET trajectory is the high FRET state. The population of molecules undergoing FRET can be quantified from the line that connects these two phasor positions.
Materials and Methods
Cell culture and treatments
COS7 cells were grown in high glucose medium from Invitrogen, supplemented with10% of Fetal Bovine Serum, 5ml of Pen-Strep and HEPES at 37°C and in 5% CO2. Freshly split cells were plated onto 35-mm glass bottom dishes coated with fibronectin and then after twenty four hours, transiently transfected with the following series of GTPase biosensors sourced from the Hahn Lab, University of North Carolina 27599, USA: (1) Rac1 dual chain biosensor (CyPet-Rac1 and YPet-PBD) (2) Rac1 dominant negative dual chain biosensor (CyPet-T17N-Rac1 and YPet-PBD) (3) Rac1 constitutively active dual chain biosensor of (CyPet-Q61L-Rac1 and YPet-PBD)(4) RhoA single chain biosensor (RBD-Citrine-1L-ECFP-RhoA) (5) RhoA constitutively membrane bound biosensor(RBD-Citrine-1L-ECFP-RhoA-kRas). The plated cells were then left for a further twenty four hours at 37°C/5% CO2 and then serum starved in un-supplemented high glucose medium. The Rac biosensors tested were stimulated with 50ng ml−1 of epidermal growth factor (EGF) (Sigma Aldrich). The Rho biosensors tested were stimulated with 2.3μM of lysophosphatidic acid (LPA) (Cayman).
Microscope
FLIM and ratiometric data were acquired concomitantly with the Zeiss LSM710 META laser scanning microscope, coupled to a 2-photon Ti:Sapphire laser (Spectra-Physics Mai Tai, Newport Beach) producing 80fs pulses at a repetition of 80MHz, and a ISS A320 FastFLIM box to acquire the lifetime data. A 63X water immersion objective 1.2 N.A. (Zeiss, Germany) was used for all experiments. The donor fluorophore of each biosensor was excited at 800nm with the 2-photon laser: this wavelength caused negligible direct excitation of the acceptor fluorophore. A SP 760nm dichroic filter was used to separate the fluorescence signal from the laser light. The fluorescence signal was directed through a 509 LP CFP/YFP filter, and the donor and acceptor signal split between two photomultiplier detectors (H7422P-40 of Hamamatsu), with the following bandwidth filters in front of each: CFP 470/22 and YFP 542/27, respectively. For image acquisition, the pixel frame size was set to 256 ×256 and the pixel dwell time to 25.61μs/pixel. The average laser power at the sample was maintained at the mW level. The FLIM and ratiometric data were acquired and processed by the SimFCS software developed at the Laboratory for Fluorescence Dynamics (www.lfd.uci.edu). Calibration of the system and phasor plot space was performed by measuring fluorescein (pH 9.0), which has a known single exponential lifetime of 4.04ns.
Data analysis
Phasor Transformation
The phasor transformation and data analysis were performed using the SimFCS software available at the www.lfd.uci.edu. Every pixel of the FLIM image is transformed into one pixel in the phasor plot. The s and g coordinates in the phasor plot which correspond to a given decay I(t) are defined by the expressions by:
| (1) |
| (2) |
where ω is the laser repetition angular frequency, and the indexes i and j identify a pixel of the image. If the data are measured in the frequency domain then:
| (3) |
| (4) |
where mi,j and ϕi,j are the modulation and the phase of the emission with respect to the excitation. If the decay is single exponential the coordinates of the phasor are given by:
| (5) |
| (6) |
In the case where a pixel i,j has the contribution of several exponential components, the coordinates g and s of the phasor are given by:
| (7) |
| (8) |
where fk is the intensity weighted fractional contribution of the component with lifetime τk. According to expressions 5 and 6 for the coordinate of a phasor for a single exponential decay , which implies that all single exponential components are represented by a semicircle of center (½,0) and radius ½ in the phasor plot. We name this the “universal circle”. Along this semicircle a phasor corresponding to a very short lifetime (small phase angle) is close to the point (1,0), where as a phasor corresponding to a very long lifetime will be close to the (0,0) point.
Resolution of two components
To resolve the fractional contribution of two phasor components we graphically solve equations 7 and 8 in the phasor plot. All phasors corresponding to the combination of the component phasors are along a segment where the segment extremes correspond to the phasors of the two isolated components. Generally, the locations of the two component phasors are known or are derived from the experimental points of systems in which all possible combinations are realized. The segment is drawn on the phasor plot and the operator selects points along the segment to match the experimental points. The fractional contribution of the two phasors at the cursor location is displayed on the screen. Using the reciprocal property of the cursor, the points in the image corresponding to that particular solution are highlighted.
FRET efficiency calculation
The trajectory is calculated according to the classical definition of FRET efficiency:
| (9) |
The phasor of the donor in the absence of the acceptor is obtained from an independent preparation in which the acceptor is absent. The phasor corresponding to the quenched donor is calculated according the quenching equation 9. The realizations of all possible phasors that are quenched with different efficiencies describe a curved trajectory in the phasor plot. The experimental position of the phasor of a given pixel along the trajectory determines the amount of quenching and therefore the FRET efficiency. The contributions of the background and of the donor without acceptor are evaluated using the rule of the linear combination (equations 7 and 8), with the background phasor and the donor unquenched determined independently. As depicted in Figure 4 the operator moves the cursor along the curved trajectory to the point where there is a cluster of phasors. Using the reciprocal property of the cursor, the pixels in the image corresponding to a given FRET efficiency are highlighted.
Figure 4.

FRET detection of a single chain RhoA biosensor, with normal sub-cellular localization.(A) Intensity images of COS7 cell transfected with (RBD-Citrine)-1L-(ECFP-RhoA) before and after LPA stimulation (donor channel). (B) Phasor plot of (RBD-Citrine)-1L-(ECFP-RhoA) experiment. (C) FRET efficiency calculator of (RBD-Citrine)-1L-(ECFP-RhoA) experiment (D) Painted FLIM image of this (RBD-Citrine)-1L-(ECFP-RhoA) experiment. (E) Painted GP images of an (RBD-Citrine)-1L-(ECFP-RhoA) experiment.
Normalized ratio or GP
For the intensity based ratiometric analysis we employ the normalized ratio approach, which has the property of transforming the concentrations of the various species additively. The normalized ratio function calculates the normalized FRET ratio, which enables visualization of the changes between the two FRET states. The GP function can be used when there is a change in the emission spectrum due to FRET and automatically compensates for bleed through. The GP function is related to the normal ratiometric methods of analysis and defined as follows:
| (10) |
| (11) |
| (12) |
whereI1 and I2 are the fluorescence intensities at the spectral band pass of fluorophores1 and 2, respectively. Mathematically, the GP function is always bound between −1 and +1. The GP function depends on the choice of the band pass filters and given 2 band pass filters, each fluorescent substance has a specific value of the GP. When the fluorescence intensity is measured in a “spectrally corrected” fluorometer, the correct GP value can be calculated directly. If we know the corrected GP value for a molecular species, for example for fluorescein or any other commonly available dye, we can calibrate an unknown system (generally the microscope) using the following expression and correction factor g:
| (13) |
where g is the correction factor calculated from the following expression:
| (14) |
Where GPc is the known value of the GP for the calibration compound and GPr is the value of the raw GP of the same substance measured in the particular instrument that needs to be calibrated. If the denominator of the GP function is proportional to the total fluorescence intensity then the GP function of a mixture of two fluorophores is equal to the intensity weighted average of the GP of the two separate species. This property of the GP function is referred to as the “additive property of the GP” and it will be used throughout. Using the GP, the fraction α of biosensor in the bound form can be directly calculated from the following equation:
| (15) |
where k is the ratio of the quantum yield of the biosensor in the two forms. As mentioned in the introduction, when working with cells, there are a series of problems with the direct application of ratiometric formula like the GP function. Firstly, it is unclear whether the spectra of the two forms of the biosensor (bound and free) remain the same in the cell environment. Secondly, the measured GP contains the contribution arising from autofluorescence. Figure 5 illustrates how to use the GP approach: in the vertical axis we plot the GP of the two species, for example high FRET and Low FRET, and for every GP on the y axis we obtain the fractional contribution of the two species using the linear combination rule. However, for every system with more than two components, the derivation of the fractional intensity given the GP cannot be obtained using only two emission wavelengths, because the value of the GP of the individual components must first be combined with the GP value of the third species, which requires the knowledge of the relative fractional intensity of the third component.
Figure 5.

FRET detection of a RhoA-kRas single chain biosensor, with constitutive membrane localization. (A) Intensity images of COS7 cell transfected with (RBD-Citrine)-1L-(ECFP-RhoA-kRas) before and after LPA stimulation (donor channel). (B) Phasor plot of (RBD-Citrine)-1L-(ECFP-RhoA-kRas) experiment. (C) FRET efficiency calculator of (RBD-Citrine)-1L-(ECFP-RhoA-kRas) experiment (D) Painted FLIM image of this (RBD-Citrine)-1L-(ECFP-RhoA-kRas) experiment. (E) Painted GP images of (RBD-Citrine)-1L-(ECFP-RhoA-kRas) experiment.
Results
Dual chain biosensor FRET detection
The Rac1 dual chain biosensor tested employs a blue-green fluorescent protein CyPet (donor) and a yellow fluorescent protein YPet (acceptor) as the FRET pair. On the donor chain we have the targeted protein Rac1 and on the acceptor chain we have the affinity reagent PBD (binding domain derived from p21-activated kinase1 (PAK1)). The affinity reagent PBD interacts only with the activated state of Rac1, which is induced here by epidermal growth factor (EGF) stimulation. Upon activation, the two fluorescent proteins are brought into an orientation of sufficient spatial proximity to generate FRET. We measure concomitantly the lifetime and intensity image of the Rac1 biosensor in the donor and FRET channel following the experimental set up specified in the Materials and Methods section. This enables a direct comparison of FRET localization by lifetime analysis with FRET localization by intensity analysis on the same data set.
For the FLIM analysis we need only analyze the donor channel and determine the spatial distribution of FRET based on the degree of quenching of the donor lifetime. The precision of the phasor location depends on the number of counts in each pixel and so each measurement was integrated until there was a minimum of 100 counts per pixel (approximately 10 frames). In general we acquired a measurement before EGF stimulation and then twelve measurements after EGF stimulation, each measurement lasting about 10s. FLIM analysis of FRET by the phasor approach requires prior knowledge of the donor phasor in the absence of acceptor. We therefore first measured a COS7 cell transfected with only the donor chain CyPet-Rac1 before and after EGF stimulation (Figure 3A), to locate the phasor of the donor and to check for potential artifacts due to addition of EGF. As seen in the phasor plot of CyPet-Rac1 (Fig. 3B), addition of EGF does not cause a shift in the donor phasor (selected within the cyan cursor) which has an average lifetime of 2.61ns. By use of the reciprocal property of the phasor cursor, we highlight all the pixels in the intensity images of the cell that contain this average lifetime (Fig. 3C). We then measured a COS7 cell transfected with both donor (CyPet-Rac1) and acceptor (YPet-PBD) before and after EGF stimulation (Fig. 3D); the phasor distribution for this experiment is shown in Fig. 3E. We first selected a region in this phasor plot that included only pixels in the background, and marked this as the phasor location for cellular auto-fluorescence (black cursor). A cyan cursor was then placed in the unquenched donor position (2.61ns) and the FRET state of the dual chain biosensor extrapolated by superimposing a theoretical trajectory of all possible donor phasors (defined by equation 9 of the Materials and Methods section) over the experimentally measured distribution. This trajectory is depicted in Fig. 3F and as can be seen the efficiency of the FRET state (where 100% donor is quenched) is determined as 30%. The linear combination of phasor clusters between the donor phasor and FRET state of the biosensor represent the varying contributions of donor fluorescence and quenched donor fluorescence in any one pixel. By moving the phasor cursor along the straight line drawn between these two terminal phasor locations (Fig. 3F), we can calculate the exact fractional contribution of quenched donor fluorescence for each pixel highlighted.
Figure 3.

FRET detection of a Rac1 dual chain biosensor. (A) CyPet-Rac1 before and after EGF stimulation (donor). (B) Phasor plot of CyPet-Rac1 experiment: cluster selected within the cyan cursor corresponds to an average lifetime of 2.61ns. (C) Painted FLIM image of CyPet-Rac1. (D) Intensity images of COS7 cell transfected with both donor (CyPet-Rac1) and acceptor (YPet-PBD) before and after EGF stimulation (donor channel). (E) Phasor plot of CyPet-Rac1 and YPet-PBD experiment. (F) FRET efficiency calculator of CyPet-Rac1 and YPet-PBD experiment (G) Painted FLIM image of this CyPet-Rac1 and YPet-PBD experiment. (H) Average GP ofCyPet-Rac1 and YPet-PBD before and after EGF stimulation. (I) Painted GP images of the CyPet-Rac1 and YPet-PBD experiment.
The spatial distribution of FRET detected by FLIM in the cell over time is represented in Fig. 3G by pseudo coloring the cell images with a palette defined by the series of cursors which extend from the donor lifetime (inside the cyan circle) toward the FRET state. In this colour scheme, pixels highlighted in blue correspond to 0% quenched donor, green to 12% quenched donor, yellow to 24% quenched donor and red to 58% quenched donor. As can be seen in the FLIM images of Fig. 3G before EGF stimulation the entire cell is blue which corresponds to the free state of the biosensor. After addition of EGF the population of donor quenched in the pixels located toward the cell perimeter increases with time (see Fig. S1A for FLIM analysis of this region of interest). This indicates an increase in the number of molecules undergoing FRET in those pixel locations and therefore an increase in Rac1 activity. We next calculated the normalized FRET ratio in each pixel of the images in Fig. 3A using the generalized polarization function and compared the localization of FRET detected by this method to the FRET localization we observed with the FLIM analysis.
The GP function calculates the difference in intensity between the FRET and donor fluorescence intensity images which are normalized to the total fluorescence intensity; mathematically this limits a GP value between +1 and −1. As seen in Fig. 3H before EGF stimulation the average GP value is −0.2 (GP of the free state of this specific biosensor). With time after EGF stimulation the average GP value increases to a maximum of 0.5 (GP of the bound state of the biosensor). The spatial localization of each GP distribution and thus relative level of FRET activity is shown in Fig. 3I. The derived GP images indicate that the perinuclear region of the cell is in an activated state over the duration of the experiment (even before EGF stimulation), and the lower right hand periphery of the cell experiences an incremental increase in FRET with time (see Fig. S1A for GP analysis of this region of interest). This localization of biosensor activity is markedly different from that shown by the FLIM analysis. This is because intensity based analysis is sensitive to large differences in the sub cellular concentration of biosensor components, here causing the low concentration of donor fluorophore in the perinuclear region (Fig. 3D) to be erroneously read as quenching of donor fluorescence due to FRET. Control studies using the inactive (CyPet-T17N-Rac1) and constitutively active (CyPet-Q61L-Rac1) donor chain with the acceptor chain (YPet-PBD) showed only uniform low or high FRET respectively in both FLIM and GP analysis (Fig. S2).
Single chain biosensor FRET detection
Here we measure the FRET activity of two single chain RhoA biosensors (each designed for different sub-cellular localization) using the phasor and GP methods of analysis. We compare the results obtained to dual chain biosensor FRET detection. The first single chain RhoA biosensor tested employs ECFP (donor) and Citrine (acceptor), built into a chain that connects RhoA (the targeted protein) with RBD (a small RhoA binding domain derived from Rhotekin, the affinity reagent), leaving the C terminus free so that the protein is subject to normal regulatory pathways that control localization (i.e. binding of Guanine Nucleotide Dissociation Inhbitors, GDI). The rhotekin fragment binds only to the activated state of RhoA, which is induced here by lysophosphatidic acid (LPA) stimulation. Upon activation, the distance and orientation between the ECFP and Citrine is altered such that the FRET efficiency of the biosensor is enhanced. We measure concomitantly the lifetime and intensity image of the RhoA biosensor in the donor and FRET channels, following the experimental set up specified under the Materials and Methods section and then compare the FRET localization obtained by the two analyses.
Figure 4a, depicts the donor intensity channel of a COS7 cell transfected with the RhoA single chain biosensor before and after LPA stimulation. The phasor distribution of this experiment is depicted in Figure 4b and the unquenched donor phasor position is marked within the cyan circle. As can be seen the phasor distribution of this experiment does not extend from the donor phasor but instead from an origin that is right shifted to a shorter lifetime along the superimposed FRET trajectory depicted in Figure 4c. This corresponds to the low FRET state of the biosensor and is calculated to a have a FRET efficiency of 4% as consequence of the residual FRET interaction inherent to the single chain design. Further shifted to the most quenched lifetime along the FRET trajectory is the high FRET state of this biosensor which is calculated to have an efficiency of 35%. Thus this RhoA single chain design enables a higher FRET efficiency than the coefficient derived by FLIM for the dual chain Rac1 biosensor, but at the expense of sensitivity. The residual FRET signal that is present even when the RhoA is in the off state decreases the dynamic range.
The line that connects the high FRET phasor to the low FRET state gives the fractional population of biosensor active, above the baseline FRET signal in any one pixel. As with the dual chain biosensor analysis we pseudo color the cell images with a palette defined by the series of coloured cursors which extend from the low FRET state (inside the blue circle) toward the high FRET state (red circle). In this colour scheme pixels highlighted in blue correspond to 0% high FRET, green to 22% high FRET, yellow to 38% high FRET and red to 67% high FRET. The pseudo coloured FLIM images show the pixels at the leading edge (top right hand corner) and within the back protrusions of the cell (bottom left hand corner), to show an increase in the population of high FRET state with time after LPA addition (see Fig. S1 B for FLIM analysis of this region of interest). This indicates an incremental increase in the number of molecules undergoing FRET in those pixel locations, which is in agreement spatially with where the literature reports RhoA activity to occur upon cell migration (Machacek and others, 2009). The GP analysis of this experiment (Figure 4e) reveals analogous FRET localization to that obtained with the FLIM analysis. The agreement of the data derived from the two techniques relates to the fact that with a single chain design the two components of the biosensor are ensured to distribute equally throughout the cell.
The second single chain RhoA biosensor tested places the fluorophores in the same positions as that described for the first, but the C terminus is now modified by attachment of a lipid motif from K-Ras in place of the wild-type RhoA lipid modification motif. This modification causes constitutive membrane localization through prevention of RhoA regulation by GDI which regulate reversible membrane localization. Figure 5a depicts the donor intensity channel of a COS7 cell transfected with the RhoA membrane bound single chain biosensor, before and after LPA stimulation. The phasor distribution of this experiment is depicted in Figure 5b, and the unquenched donor phasor position is marked within the cyan circle. The phasor distribution of this experiment extends from the right shifted low FRET state of the biosensor (calculated to a have a FRET efficiency of 7%) toward the high FRET state (calculated to have an efficiency of 39%). However upon closer inspection of the phasor distribution along the line that connects the low FRET to high FRET phasor, we see that the baseline population of high FRET biosensor present is not equal to 0% (blue cursor). In any one pixel we have 9% (green cursor) to 74% (red cursor) active biosensor. When we pseudo color the cell images with the palette defined by the cursors in Fig. 5c, we see that the pixels along a broad section of the cell membrane experience FRET before and after LPA stimulation (Fig. 5D). The GP analysis of this experiment (Fig. 5e) reflects this baseline FRET interaction and active biosensor localization (see Fig. S1 C for FLIM and GP analysis of this region of interest).
Discussion
Detection of FRET by an intensity-based ratiometric method of analysis is advantageous for several reasons: it can be performed on almost every fluorescence microscope and has a good signal to noise ratio, which enables the detection of rapid changes in biochemical concentrations and spatial dynamics. The fundamental problem of ratiometric analysis is that in practice it is very sensitive to the fluorescence artifacts inherent in both a cellular environment and in biosensor designs. Linear un-mixing cannot accurately separate the nonresponsive additional fluorescent species (i.e. which arise from differential photo-bleaching or incomplete protein expression) from the on and off FRET states of the optically active biosensor since these species have the same spectra. For example as demonstrated upon GP analysis of the dual chain Rac1 biosensor the propensity of the donor and acceptor chains to distribute unequally resulted in the perinuclear region of the cell exhibiting a FRET signal that originated from the comparatively low expression level of donor in this zone. Intensity based analysis of a biosensor FRET signal is thus vulnerable to reporting a spatial localization of FRET that is not indicative of the actual biosensor activity, and is unable to determine the exact biochemical concentrations involved in the cellular event being probed.
Given that biosensors are designed to report on such molecular detail, biosensor FRET detection requires a method that is more quantitative, and for this reason fluorescence lifetime imaging (FLIM) is considered advantageous. We show here that FRET detection by the phasor approach to FLIM is able to unequivocally determine the local concentration of the free and bound state of a dual chain biosensor, the low and high FRET species of a single chain biosensor in each pixel of an image, and thus provide a quantitative map of the biosensor activity as a function of time. The robustness of the phasor method is demonstrated by the fact that irrespective of a dual or single chain design, the map of Rac1 and RhoA activity derived is in agreement with the localization reported in the literature upon growth factor stimulation. We also show that the physical properties intrinsic to each biosensor design can be accurately characterized by phasor analysis and thus inform biosensor optimization at the developmental stage. For example the dynamic range of the Rac1 dual chain biosensor is higher than that of the two RhoA single chain designs, which exhibit residual FRET before activation; preventing detection of low level protein activity.
Thus in conclusion we believe that the phasor approach to FLIM is an essential tool not only for the experimental use of FRET biosensors but also for their advancement. It is a reliable method of FRET detection that can accurately determine in vivo the spatiotemporal dynamics of single or dual chain biosensors independent of other sources of fluorescence. We believe that as biosensors become more sophisticated and are multiplexed with other fluorescent molecular tools, the ability of the phasor approach to transform each molecular species into a two dimensional coordinate system which distinguishes independent mixtures from changes in lifetime due to FRET, without having to resolve the decay at each pixel into the individual exponential components, will be imperative.
Supplementary Material
FLIM and GP analysis of biosensor activation in a region undergoing FRET from the experiments presented in Figures 1–3. (A) COS7 cell transfected with the Rac1 DC biosensor, from the experiment described in Fig. 1. The region of interest for analysis is selected with the red cursor. (B) COS7 cell transfected with the RhoA SC biosensor, from the experiment described in Fig. 2. The region of interest for analysis is selected with the green cursor. (C) COS7 cell transfected with the RhoA-kRas SC biosensor, from the experiment described in Fig. 3. The region of interest for analysis is selected with the orange cursor. (D) The percentage of biosensor activation in the selected regions of interest as a function of time (this is indicated by % donor quenching for the dual chain design and % high FRET for the single chain design). (D) Average GP value (which is indicative of biosensor activation) for the selected regions of interest as a function of time.
{kind=link}
Control experiments for detection of FRET from a Rac1 dual chain biosensor. (A) Intensity images of COS7 cell transfected with inactive donor (CyPet-T17N-Rac1) plus acceptor (YPet-PBD) before and after EGF stimulation (donor channel). (B) Phasor plot of CyPet-T17N-Rac1 and YPet-PBD experiment. (C) Painted FLIM image of this CyPet-T17N- Rac1 and YPet-PBD experiment. (D) Intensity images of COS7 cells transfected with constitutively active donor (CyPet-Q61L -Rac1) plus acceptor (YPet-PBD) before and after EGF stimulation (donor channel). (E) Phasor plot of CyPet-T35SQ61L-Rac1 and YPet-PBD experiment. (F) Painted FLIM image of this CyPet-Q61L-Rac1 and YPet-PBD experiment. (G) Painted GP images of the CyPet-T17N-Rac1 and YPet-PBD experiment. (H) Average GP ofCyPet-T17N-Rac1 and YPet-PBD before and after EGF stimulation. (I) Painted GP images of the CyPet-Q61L-Rac1 and YPet-PBD experiment. (H) Average GP ofCyPet-Q61L-Rac1 and YPet-PBD before and after EGF stimulation.
{kind=link}
Acknowledgments
We thank Milka Titin for cultivating and transfecting the cells. This work is supported in part by NIH-P41 P41-RRO3155 (EG, MD), NIH P50-GM076516 (EG, EH), the Cell Migration Consortium grant U54 GM064346 (EG, MD, KH), GM057464 (KH), NIH-T32 GM008719 (CW) and NIH-F30 F30HL094020-02 (CW).
Bibliography
- Bastiaens PIH, Squire A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends in Cell Biology. 1999;9(2):48–52. doi: 10.1016/s0962-8924(98)01410-x. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac Take Center Stage. Cell. 2004;116(2):167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Chandler D, Majumdar Z, Heiss G, Clegg R. Ruby Crystal for Demonstrating Time- and Frequency-Domain Methods of Fluorescence Lifetime Measurements. Journal of Fluorescence. 2006;16(6):793–807. doi: 10.1007/s10895-006-0123-7. [DOI] [PubMed] [Google Scholar]
- Clayton AHA, Hanley QS, Verveer PJ. Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. Journal of Microscopy. 2004;213(1):1–5. doi: 10.1111/j.1365-2818.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- Clegg RM. Fluorescence resonance energy transfer. Current Opinion in Biotechnology. 1995;6(1):103–110. doi: 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- Colyer RA, Lee C, Gratton E. A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microscopy Research and Technique. 2008;71(3):201–213. doi: 10.1002/jemt.20540. [DOI] [PubMed] [Google Scholar]
- Digman MA, Caiolfa VR, Zamai M, Gratton E. The Phasor Approach to Fluorescence Lifetime Imaging Analysis. Biophysical journal. 2008;94(2):L14–L16. doi: 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPilato LM, Zhang J. Fluorescent protein-based biosensors: resolving spatiotemporal dynamics of signaling. Current Opinion in Chemical Biology. 2010;14(1):37–42. doi: 10.1016/j.cbpa.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster T. Delocalized excitation and excitation transfer. In: Sinanoglu O, editor. Modern Quantum Chemistry. Newy York: Academic Press; 1965. pp. 93–137. [Google Scholar]
- Gaits F, Hahn K. Shedding light on cell signaling: interpretation of FRET biosensors. Science’s STKE: signal transduction knowledge environment. 2003;(165) doi: 10.1126/stke.2003.165.pe3. [DOI] [PubMed] [Google Scholar]
- Gratton E, Limkeman M, Lakowicz JR, Maliwal BP, Cherek H, Laczko G. Resolution of mixtures of fluorophores using variable-frequency phase and modulation data. Biophysical Journal. 1984;46(4):479–486. doi: 10.1016/S0006-3495(84)84044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn K, Toutchkine A. Live-cell fluorescent biosensors for activated signaling proteins. Current opinion in cell biology. 2002;14(2):167. doi: 10.1016/s0955-0674(02)00313-7. [DOI] [PubMed] [Google Scholar]
- Hodgson L, Nalbant P, Shen F, Hahn K, William E, Balch CJDaAH. Methods in Enzymology. Academic Press; 2006. Imaging and Photobleach Correction of Mero[hyphen (true graphic)]CBD, Sensor of Endogenous Cdc42 Activation; pp. 140–156. [DOI] [PubMed] [Google Scholar]
- Hodgson L, Pertz O, Hahn KM, Kevin FS. Methods in Cell Biology. Academic Press; 2008. Design and Optimization of Genetically Encoded Fluorescent Biosensors: GTPase Biosensors; pp. 63–81. [DOI] [PubMed] [Google Scholar]
- Hodgson L, Shen F, Hahn K. Biosensors for Characterizing the Dynamics of Rho Family GTPases in Living Cells. John Wiley & Sons, Inc; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson L, Shen F, Hahn K. Biosensors for characterizing the dynamics of rho family GTPases in living cells. In: Bonifacino Juan S, et al., editors. Current protocols in cell biology/editorial board. Chapter. 2010. pp. 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraheem A, Campbell RE. Designs and applications of fluorescent protein-based biosensors. Current Opinion in Chemical Biology. 2010;14(1):30–36. doi: 10.1016/j.cbpa.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Jameson D, Gratton E, Hall R. The Measurement and Analysis of Heterogeneous Emissions by Multifrequency Phase and Modulation Fluorometry. Applied Spectroscopy Reviews. 1984;20(1):55–106. [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac Activation Dynamics Visualized in Living Cells. Science. 2000;290(5490):333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, Laczko G, Cherek H, Gratton E, Limkeman M. Analysis of fluorescence decay kinetics from variable-frequency phase shift and modulation data. Biophysical Journal. 1984;46(4):463–477. doi: 10.1016/S0006-3495(84)84043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR, Masters BR. Principles of Fluorescence Spectroscopy, Third Edition. Journal of Biomedical Optics. 2008;13(2):029901–2. [Google Scholar]
- Llères D, Swift S, Lamond AI. Detecting Protein-Protein Interactions In Vivo with FRET using Multiphoton Fluorescence Lifetime Imaging Microscopy (FLIM) John Wiley & Sons, Inc; 2001. [DOI] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461(7260):99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasassi T, De Stasio G, Ravagnan G, Rusch RM, Gratton E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophysical Journal. 1991;60(1):179–189. doi: 10.1016/S0006-3495(91)82041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy A, Wallrabe H, Chen Y, Barroso M, Correia John J, William Detrich H., III . Methods in Cell Biology. Academic Press; 2008. Chapter 22 Quantitation of Protein-Protein Interactions: Confocal FRET Microscopy; pp. 569–598. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hahn KM. Designing biosensors for Rho family proteins -- deciphering the dynamics of Rho family GTPase activation in living cells. J Cell Sci. 2004;117(8):1313–1318. doi: 10.1242/jcs.01117. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440(7087):1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Suhling K, French PMW, Phillips D. Time-resolved fluorescence microscopy. Photochemical & Photobiological Sciences. 2005;4(1):13–22. doi: 10.1039/b412924p. [DOI] [PubMed] [Google Scholar]
- Tsien RY. THE GREEN FLUORESCENT PROTEIN. Annual Review of Biochemistry. 1998;67(1):509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- van Munster EBGT. Fluorescence lifetime imaging microscopy (FLIM) Adv Biochem Eng Biotechnol. 2005;95:143–75. doi: 10.1007/b102213. [DOI] [PubMed] [Google Scholar]
- Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Current Opinion in Biotechnology. 2005;16(1):19–27. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Yu W, So PT, French T, Gratton E. Fluorescence generalized polarization of cell membranes: a two-photon scanning microscopy approach. Biophysical Journal. 1996;70(2):626–636. doi: 10.1016/S0006-3495(96)79646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FLIM and GP analysis of biosensor activation in a region undergoing FRET from the experiments presented in Figures 1–3. (A) COS7 cell transfected with the Rac1 DC biosensor, from the experiment described in Fig. 1. The region of interest for analysis is selected with the red cursor. (B) COS7 cell transfected with the RhoA SC biosensor, from the experiment described in Fig. 2. The region of interest for analysis is selected with the green cursor. (C) COS7 cell transfected with the RhoA-kRas SC biosensor, from the experiment described in Fig. 3. The region of interest for analysis is selected with the orange cursor. (D) The percentage of biosensor activation in the selected regions of interest as a function of time (this is indicated by % donor quenching for the dual chain design and % high FRET for the single chain design). (D) Average GP value (which is indicative of biosensor activation) for the selected regions of interest as a function of time.
Control experiments for detection of FRET from a Rac1 dual chain biosensor. (A) Intensity images of COS7 cell transfected with inactive donor (CyPet-T17N-Rac1) plus acceptor (YPet-PBD) before and after EGF stimulation (donor channel). (B) Phasor plot of CyPet-T17N-Rac1 and YPet-PBD experiment. (C) Painted FLIM image of this CyPet-T17N- Rac1 and YPet-PBD experiment. (D) Intensity images of COS7 cells transfected with constitutively active donor (CyPet-Q61L -Rac1) plus acceptor (YPet-PBD) before and after EGF stimulation (donor channel). (E) Phasor plot of CyPet-T35SQ61L-Rac1 and YPet-PBD experiment. (F) Painted FLIM image of this CyPet-Q61L-Rac1 and YPet-PBD experiment. (G) Painted GP images of the CyPet-T17N-Rac1 and YPet-PBD experiment. (H) Average GP ofCyPet-T17N-Rac1 and YPet-PBD before and after EGF stimulation. (I) Painted GP images of the CyPet-Q61L-Rac1 and YPet-PBD experiment. (H) Average GP ofCyPet-Q61L-Rac1 and YPet-PBD before and after EGF stimulation.