

Abstract
AdoMet radical enzymes are involved in processes such as cofactor biosynthesis, anaerobic metabolism, and natural product biosynthesis. These enzymes utilize the reductive cleavage of S-adenosylmethionine (AdoMet) to afford L-methionine and a transient 5'-deoxyadenosyl radical, which subsequently generates a substrate radical species. By harnessing radical reactivity, the AdoMet radical enzyme superfamily is responsible for an incredible diversity of chemical transformations. Structural analysis reveals that family members adopt a full or partial Triose-phosphate Isomerase Mutase (TIM) barrel protein fold, containing core motifs responsible for binding a catalytic [4Fe-4S] cluster and AdoMet. Here we evaluate over twenty structures of AdoMet radical enzymes and classify them into two categories: traditional and ThiC-like (named for the structure of 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate synthase (ThiC)). In light of new structural data, we reexamine the traditional structural motifs responsible for binding the [4Fe-4S] cluster and AdoMet, and compare and contrast these motifs with the ThiC case. We also review how structural data combine with biochemical, spectroscopic, and computational data to help us understand key features of this enzyme superfamily, such as the energetics, the triggering, and the molecular mechanisms of AdoMet reductive cleavage.
Keywords: adenosylmethionine, iron-sulfur clusters, adenosylcobalamin, glycyl radical enzymes, SAM radical
1. Introduction
S-adenosylmethionine (SAM, AdoMet) has been referred to as the “poor man s vitamin B12” [1]. Like vitamin B12 (cobalamin), AdoMet is able to catalyze methyl transfer reactions as well as initiate radical-based chemistry. Unlike cobalamin, AdoMet is a relatively simple molecule, less expensive to biosynthesize (Fig. 1A,B). In enzyme systems using coenzyme B12 (adenosylcobalamin, AdoCbl), generation of a radical species involves homolytic cleavage of the C–Co bond connecting the 5' carbon of the deoxyadenosyl ligand and the cobalt of the corrin ring (Fig. 1A). For AdoMet radical enzymes, generation of a 5'-deoxyadenosyl radical species (dAdo•) involves the reductive cleavage of an AdoMet molecule bound to a [4Fe-4S]1+ cluster (Fig. 1B). Flavodoxin is often the source of the requisite electron that reduces the [4Fe-4S]2+ cluster allowing the reductive cleavage reaction [2-6]. Once dAdo• is generated, H-atom abstraction from substrate can occur to initiate a wide range of chemical reactions (Fig. 2). With an initial estimate of 600 unique sequences [7] that recently was expanded to approximately 3000 proteins [8], AdoMet radical proteins constitute a large enzyme superfamily whose chemical diversity is nothing short of enormous.
Figure 1.

Schematic drawing of cofactors involved in the generation of adenosyl radical species. (A) Cleavage of adenosylcobalamin yielding cob(II)alamin and dAdo•. (B) Cleavage of S-adenosyl-L-methionine yielding L-methionine and dAdo•.
Figure 2.

Examples of AdoMet radical enzyme reactions. (A) BioB, (B) PFL-AE, (C) 2,3-LAM, (D) ThiC, (E) RlmN, and (F) HydE.
While AdoCbl-dependent enzymes can be neatly categorized into three main classes according to their reactivity (carbon skeleton mutases, aminomutases, and eliminases), it is impossible to similarly classify the AdoMet radical reactions into a limited set of categories. A small sampling of the known AdoMet radical reactions is shown in Fig. 2. Akin to their AdoCbl cousins, AdoMet radical enzymes can catalyze aminomutase reactions, with lysine 2,3-aminomutase (2,3-LAM) performing a reaction similar to that of AdoCbl-dependent lysine 5,6-aminomutase (Fig. 2C) [9]. A number of the structurally characterized AdoMet radical enzymes are involved in vitamin or cofactor biosynthesis, catalyzing complex rearrangement reactions in some cases. For example, 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate (HMP-P) synthase (ThiC) transforms 5-aminoimidazole ribonucleotide (AIR) into HMP-P as part of thiamine biosynthesis (Fig. 2D) [10]. Other AdoMet radical enzymes like biotin synthase (BioB) and lipoate synthase catalyze radical-based sulfur insertion reactions in the biosynthesis of their respective vitamins (Fig. 2A) [11, 12]. In still other cases, the exact reaction catalyzed by the AdoMet radical enzyme in the biosynthesis of the vitamin or cofactor is either not well established or controversial [13–16]. Examples include MoaA, involved in molybdenum cofactor biosynthesis, and HydE, involved in H-cluster biosynthesis (Fig. 2F). A relatively simple protein-based radical cofactor is the glycyl radical species, found in enzymes such as pyruvate formate-lyase (PFL) and class III ribonucleotide reductase (RNR). These protein-bound glycyl-radical species are formed through the action of AdoMet radical activases (Fig. 2B) [17, 18]. Finally, Figure 2E shows the involvement of AdoMet radical enzymes in modification of ribonucleotides. In the particular case shown, RlmN carries out a radical-based methylation reaction, utilizing both the radical-generation and methylation functions of AdoMet [19]. Insights into this dual use of AdoMet from the recent structural analysis of RlmN are discussed below.
More than twenty structures representing over ten unique AdoMet radical enzymes have been characterized by X-ray crystallography. Structural analysis reveals that these enzymes adopt full or partial Triose-phosphate Isomerase Mutase (TIM) barrels with a (β/α)6-8 architecture, described in detail below [7, 20, 21]. The majority of these enzymes coordinate a unique [4Fe-4S] cluster using a loop from the full or partial TIM barrel comprising a CX3CXφC motif (where φ represents a conserved aromatic residue). Intriguingly, the newly characterized ThiC uses a separate protein domain and a different Cys motif to coordinate its cluster onto a full TIM barrel [22]. These structural motifs are described in detail below.
Regardless of the structural method for coordination of the unique [4Fe-4S] cluster, all AdoMet radical enzymes appear to face the same challenges, that is, how to make the reductive cleavage of AdoMet energetically favorable and how to avoid unwanted cleavage of cluster-bound AdoMet in the absence of substrate. The activation barrier for reductive cleavage of AdoMet is high (32 kcal mol−1), raising the question of how the enzyme catalyzes this reaction at all. Below we discuss studies on 2,3-LAM that suggest that cofactor and substrate binding events decrease the barrier for reductive AdoMet cleavage from 32 kcal mol−1 to 9 kcal mol−1, explaining how AdoMet cleavage can be made energetically feasible [23]. In terms of the molecular mechanism of reductive cleavage, a recent high-resolution structure of HydE and accompanying density functional theory (DFT) calculations provide insight into this process [24]. Additionally, structures of PFL activating enzyme (PFL-AE) suggest one mechanism by which an AdoMet radical enzyme can prevent unwanted cleavage of AdoMet in the absence of substrate [25]. The last few years have provided a wealth of new structural data to help us understand the diverse reactivity of this radical superfamily. This review describes some of the latest findings, providing an overview of the structural motifs in this enzyme family, as well as our current understanding of how AdoMet radical chemistry is harnessed. We suspect that more surprises are in store and that this family will continue to amaze in the years to come.
2. The traditional AdoMet radical structural core
Analysis of the twenty-four AdoMet radical structures listed in Table 1 has provided a picture of the structural requirements for a canonical AdoMet radical core fold. The ThiC structure [22], which represents a departure from the standard fold, is discussed below. Here we will synthesize the structural information obtained since our last review [21] and consider new insights into reactivity provided by these structures.
Table 1.
Structurally characterized AdoMet radical enzymes considered in this review
| Enzyme Name | Role | Main Structural Features | PDB | Organism | Ligands |
|---|---|---|---|---|---|
| Pyruvate Formate Lyase Activase (PFL-AE) | Glycyl radical formation in PFL | partial (β/α)6 TIM barrel [4Fe-4S] cluster |
3C8F, 3CB8 [25] 3CAN (To be published)a |
Escherichia coli Bacteroides vulgatus ATCC 8482 | AdoMet, peptide substrate |
| Lysine 2,3- aminomutase (2,3-LAM) | β-lysyl antibiotic biosynthesis | partial (β/α)6 TIM barrel [4Fe-4S] cluster |
2A5H [36] | Clostridium subterminale | AdoSeMet, PLP, lysine |
| RimO | S-methylthiolation of ribosomal-protein aspartate | partial (β/α)6 TIM barrel 2 x [4Fe-4S] clusters* |
2QGQ [28] | Thermotoga maritima | |
| MiaB | S-methylthiolation of tRNA nucleoside | partial (β/α)6 TIM barrel 2 x [4Fe-4S] clusters |
Homology model [28] | Thermotoga maritima | |
| RlmN | Radical-mediated methylation of ribosomal RNA | partial (β/α)6 TIM barrel [4Fe-4S] cluster |
3RF9, 3RFA [90] | Escherichia coli | AdoMet |
| TYW1 | Wye-base biosynthesis | partial (β/α)6 TIM barrel 2 x [4Fe-4S] cluster* |
2YX0 [37] 2Z2U [38] |
Pyrococcus horikoshii Methanocaldococcus jannaschii | |
| MoaA | Molybdenum cofactor biosynthesis | partial (β/α)6 TIM barrel 2 x [4Fe-4S] clusters |
1TV7, 1TV8 [35] 2FB2, 2FB3 [13] |
Staphylococcus aureus | AdoMet, 5′-GTP, Met, dAdoH |
| FeFe-hydrogenase maturase (HydE) | Cofactor biosynthesis | distorted (β/α)8 TIM barrel [4Fe-4S] cluster [2Fe-2S] cluster |
3CIW, 3CIX [14] 3IIX, 3IIZ[24] |
Thermotoga maritima | AdoMet, AdoHCys, Met, dAdoH, thiocyanate |
| Coproporphyrinogen III oxidase (HemN) | Heme biosynthesis | partial (β/α)6TIM barrel [4Fe-4S] cluster |
1OLT[93] | Escherichia coli | AdoMet |
| Biotin Synthase (BioB) | Biotin synthesis | full (β/α)8 TIM barrel [4Fe-4S] cluster [2Fe-2S] cluster |
1R30 [26] | Escherichia coli | AdoMet, DTB |
| ThiC | Thiamine biosynthesis | full (β/α)8 TIM barrel [4Fe-4S] cluster* |
3EPM, 3EPN, 3EPO [22] |
Caulobacter crescentus | Imidazole ribonucleotide, HMP-P |
Predicted from sequence alignments or spectroscopy
Structure of a deletion construct of PFL-AE, in which the N-terminal segment containing the cluster binding loop is removed.
Abbreviations:
dAdoH, 5′-deoxyadenosine
AdoHCys, adenosyl-L-homocysteine
AdoMet, adenosyl-L-methionine
AdoSeMet, Se-adenosyl-L-selenomethionine
DTB, dethiobiotin
HMP-P, 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate
PLP, pyridoxal phosphate
2.1 Core motifs
The majority of AdoMet radical structures support designation of the core fold as a partial (β/α)6 TIM barrel. The core domain consists of six parallel β strands surrounded on one side by α helices (Fig. 3A). A relatively high level of structural conservation of the core fold is observed (Fig. 3B), with a root mean square deviation (RMSD) of Cα positions falling within a range of 2 - 4 Å. In contrast, sequence conservation between the enzymes is limited, with identity between the enzymes often only 10-20% (Fig. 4). The AdoMet radical active site is located within the lateral opening of the partial (β/α)6 TIM barrel, on the opposite side of the β sheet from the helices (Fig. 3D). This site, which includes a [4Fe-4S] cluster and binding sites for AdoMet and substrate, is formed by residues from the middle and C-termini of the core β strands and loops at the C-terminal face of the barrel. The [4Fe-4S] cluster, which initiates radical chemistry by electron transfer to AdoMet, is coordinated by the CX3CXφC motif from the loop following the first strand of the partial TIM barrel (magenta in Fig. 3B). In addition to “CX3CXφC”, four other AdoMet radical motifs have been identified in the AdoMet radical core fold [21]: “GGE”, “ribose motif”, “GxIxGxxE” and the “β6 structural motif” (Fig. 3C and Fig. 4). All X-ray structures in Table 1 that contain bound AdoMet demonstrate that the basic tertiary structure of these motifs and their function with respect to AdoMet binding are preserved. However, the identity of residues involved has not been retained, and the lack of sequence identity complicates the naming of these motifs. For example, the residues in the “GGE” motif in BioB are AAW, but the structure and function of these residues is equivalent to a classic GGE motif [21, 26]. Thus, as described in more detail below, AdoMet radical enzymes have evolved different sequences to bind AdoMet in similar ways.
Figure 3.

Stereo-depictions of the AdoMet radical core. (A) Traditional AdoMet radical core with loops omitted for clarity. Strands are shown in dark blue, helices in light blue, AdoMet in green carbons, iron in rust, sulfur in yellow, and the CX3CXφC-containing loop in magenta. (B) Overlay of nine AdoMet radical structures, excluding ThiC, with AdoMet, cluster, and the CX3CXφC-containing loop colored as above; strands are in yellow, helices in cyan and loops in black. (C) Example of the conserved AdoMet binding motifs from the structure of PFL-AE. Motifs are colored as follows: magenta, CX3CXφC-containing loop; salmon, “GGE” motif; light pink, ribose motif; tan, “GxIxGxxE” motif; and pale cyan, β6 structural motif. Residues interacting with AdoMet outside of these motifs from β3 are colored gray. (D) An overlay of the radical core domains of five structures reported with bound substrate. The [4Fe-4S] cluster and bound AdoMet are displayed only from PFL-AE for simplification. Substrate carbon atoms are colored as follows: green, PFL-AE; orange, HemN; blue, MoaA; yellow, BioB; and pink, LAM.
Figure 4.

Structure-based sequence alignment of AdoMet radical enzymes highlighting conserved structural motifs. Alignment displays partial or full protein sequences for: PFL-AE (PflA) (residue numbers shown above sequences), HemN (33-331), BioB (23-313), MoaA (1-285), LAM (96-381), TYW1_P. hori (30-342), TYW1_M. Jann (30-311), HydE (35- 335), RlmN (145-384), and RimO (135-430). The ten sequences, including both structurally characterized TYW1 sequences, are aligned with the main secondary structural elements labeled above the alignment and with strand residues in yellow and helical residues in teal. Residues of interest are colored as follows: the CX3CXφC motif cysteines, red; residues that contact AdoMet (or the TYW1 and RimO residues that are expected to contact AdoMet based on an analysis of the structures), green; residues that contact the substrate, blue; residues that contact a cofactor, pink; and residues that contact both AdoMet and the substrate, orange. Residues of the five motifs (see text), boxed in red, are identified as follows: red stars, cysteines of the cluster-binding loop; red pentagon, aromatic of the cluster-binding loop; red circles, the GGE motif; red rectangles, residues that contact the ribose of AdoMet; red triangles, the GxIxGxxE motif; and red squares, the β6 structural motif.
The first and most familiar of the AdoMet radical motifs is CX3CXφC mentioned above (C29-C36 in magenta in Fig. 3C and marked by red stars in Fig. 4). In addition to its role in binding the [4Fe-4S] cluster, CX3CXφC provides hydrophobic interactions to the adenine of AdoMet through its penultimate residue, typically a tyrosine or phenylalanine. The second motif, termed the “GGE motif” for its sequence in PFL-AE and MoaA (G77GE79 in salmon in Fig. 3C and boxed and marked by red circles in Fig. 4), resides at the C-terminal end of strand β2. Interactions between the GGE motif and the amino group of the AdoMet methionine ensure proper ligation of the cluster s unique iron and correct orientation of the AdoMet methionyl moiety. Third, residues originating from β4 often contact the AdoMet ribose hydroxyls, although this ribose motif (light pink in Fig. 3C and marked by red rectangles in Fig. 4) is less well conserved than many of the others. In BioB, residues that contact the AdoMet ribose from β4 have been shown by mutagenesis to be very important, with mutation yielding enzyme that is unable to produce a significant amount of biotin [27]. Interestingly, two mutations of BioB β4 residues, N153S and D155N, support binding and reductive cleavage of AdoMet, but produce an alternate product. This finding, by the Jarrett laboratory, suggests that these residues may help to reposition the dAdo with respect to substrate for H-atom abstraction, thus playing a catalytic role in addition to a binding function [27]. If supported through further studies of other AdoMet radical enzymes, this additional catalytic role of β4 residues would explain the lack of conservation in this motif, since different substrates would require different strategies for dAdo repositioning.
Fourth, the “GxIxGxxE motif”, named for its sequence in BioB (G190-E197 in tan in Fig. 3C and boxed and marked by red triangles in Fig. 4), located on β5 and the following loop, maintains the structure of the AdoMet adenine binding site. The residue at position 2 of the GxIxGxxE motif (V168 in Fig. 3C and the second red triangle in Fig. 4) also supports hydrophobic interactions with the adenine, while the residue at position 1 may play a role in catalysis or cofactor / substrate binding. Finally, the β6 structural motif (L199-H202 in pale cyan in Fig. 3C and boxed and marked by red squares in Fig. 4) provides hydrophobic interactions with the AdoMet adenine. Residues outside of these motifs do occasionally make contacts to AdoMet (see residues boxed in green in Fig. 4); see [21] for a more detailed discussion of the AdoMet binding strategies of this family.
In contrast to AdoMet binding, there is no conservation in the substrate binding sites of AdoMet radical enzymes beyond their location within the lateral opening of the full or partial TIM barrel (Fig. 3D). While we find repeatedly that substrate is bound in the center of the core β strands (Fig. 3D), there is no similarity between these enzymes in terms of sequence motifs used to bind substrate. These substrate binding sites can be formed by residues within the core strands, by residues from elements outside the core fold or by both. Most likely the lack of conservation derives from the large differences in substrates (from small molecules like dethiobiotin to proteins like PFL). In each case, the AdoMet radical enzyme must position its substrates in a very specific orientation within the active site to ensure proper H-atom abstraction, and one binding motif could not possibly accommodate the diversity of all substrates.
2.2. Overall structures of traditional AdoMet radical enzymes
Just like other functional protein domains, the AdoMet radical domain appears at various locations within a given protein chain–near the N-terminus, C-terminus, or center of the chain – as required for correct formation of the active site. As mentioned above, the overall fold of the AdoMet radical core domain is structurally conserved despite low sequence identity, and within this core domain, the largest physical deviations are observed near the putative reductase binding region (see [21]), which could indicate a diversity in the proteins responsible for electron donation. In many cases, the protein responsible for providing the electron is unknown but assumed to be a flavodoxin.
Outside of the canonical core fold, little to no similarity exists between the AdoMet radical enzymes (with the exception of enzymes that belong to the same subfamilies and catalyze similar reactions, such as BioB and lipoate synthase). In light of their widely different reactivities and unrelated substrates, the driving force behind this lack of similarity is easily grasped –each enzyme s structure is highly tailored for its specific reaction.
The tailoring of each enzyme to its substrate and reaction has been accomplished in a variety of ways. Several enzymes (e.g. BioB and HydE) adopt a complete (β/α)8 TIM barrel into which smaller sized substrates, like dethiobiotin, can easily fit (Fig. 5A). 2,3-LAM also binds a small substrate (i.e. lysine), but does not use a complete (β/α)8 TIM barrel. Instead, it sequesters its substrate using both an N-terminal loop and a C-terminal β hairpin comprised of non-core protein elements, which help close the partial TIM barrel core. In addition, an extended loop region from a neighboring molecule of this homotetramer serves to complete the pyridoxal phosphate (PLP) binding site within the bottom of the barrel (Fig. 5F). In other enzymes like HemN and MoaA where the substrates are larger (heme in the case of HemN), the AdoMet radical core is extended to form a longer β sheet by the non-core protein elements, leaving a larger portion of the inner sheet s surface open to solvent. This extension (Fig. 5C) allows access by larger substrate molecules, such as heme, to the enzymes active sites. In the case of particularly large substrates (e.g. glycyl radical enzymes like PFL), the core can also function with minimal tailoring by non-core elements, as observed in PFL-AE. With only 27 residues outside of the core motifs, the crescent-shaped structure of PFL-AE can bind directly to its target enzyme [25]. In a sense, the binding of the target enzyme completes the AdoMet radical fold and creates the active site at the protein-protein interface (Fig. 5D). RimO, which also binds a large substrate (the S12 ribosomal protein), harbors a distinct TRAM domain explicitly used for the purpose of substrate binding (cyan in Fig. 5E) [28, 29]. Taken together, the variability observed amongst these enzymes emphasizes the fascinating modularity and customizability of the AdoMet radical core domain.
Figure 5.
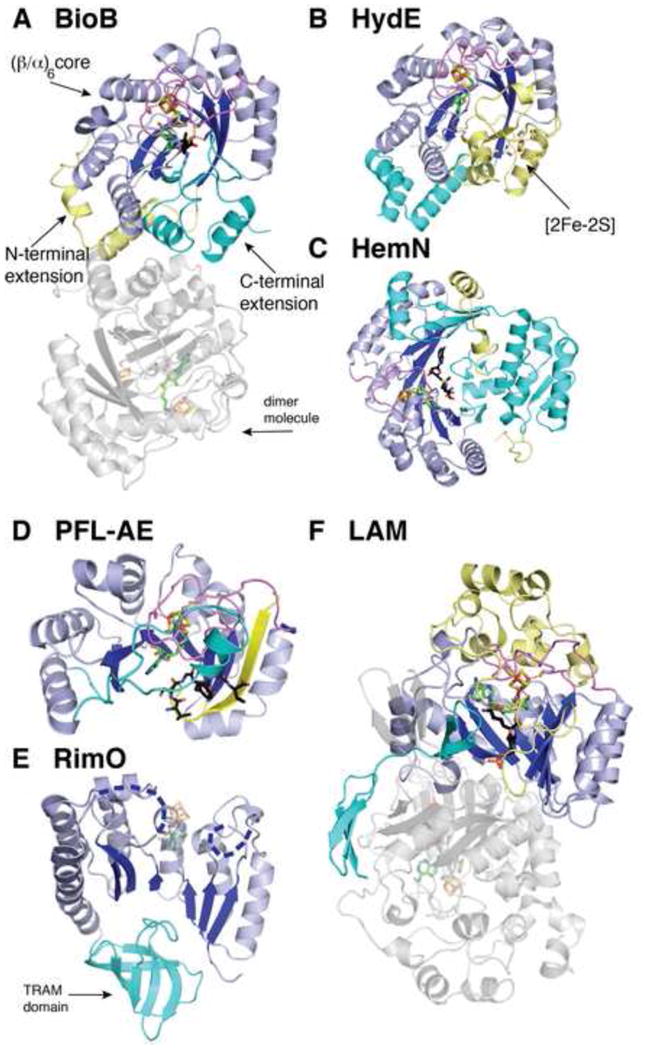
Overall structures of selected AdoMet radical enzymes that contain the traditional AdoMet radical core motifs, colored as described below. (A) BioB with clusters, AdoMet, and dethiobiotin substrate (PDB ID: 1R30). (B) HydE with clusters and AdoMet (PDB ID: 3IIZ). (C) HemN with cluster and AdoMet (PDB ID: 1OLT). (D) PFL-AE with cluster, AdoMet, and peptide substrate (PDB ID: 3CB8). (E) RimO with cluster and AdoMet modeled in transparent sticks based on the structure of PFL-AE, and disordered regions shown as dashed blue lines (PDB ID: 2QGQ). (F) LAM with cluster, AdoMet, and substrate lysine-PLP aldimine (PDB ID: 2A5H). Chain A of each enzyme is colored as follows: yellow, N-terminus; blue, partial (β/α)6 TIM barrel core; and cyan, C-terminal domain. Dimer molecules, if present in the enzyme structure, are colored gray. Only one homodimer of the LAM tetramer is shown for simplicity. Colors are as follows: magenta, the cluster-binding loop; green, AdoMet carbon; black, substrates/ligands carbon; red, oxygen; blue, nitrogen; orange, phosphorous and iron; and yellow, sulfur.
3. ThiC: AdoMet radical chemistry without the AdoMet radical core fold
In 2008, the crystal structure of AdoMet radical enzyme ThiC from Caulobacter crescentus from the Ealick and Begley laboratories revealed substantial departures from the classic AdoMet radical core fold [22]. It is currently unclear as to how many other members of the AdoMet radical superfamily will be ThiC-like. Below we describe what is known about ThiC and how it carries out its AdoMet radical chemistry, and we compare ThiC to traditional family members. This analysis should aid in identifying other ThiC-like AdoMet radical enzymes.
3.1. Discovery of ThiC as a new structural class of AdoMet radical enzyme
The biosynthesis of thiamine in prokaryotes and plants proceeds by the separate synthesis and attachment of thiazole phosphate and thiamine pyrimidine HMP-P (reviewed in [30]). The AdoMet radical enzyme ThiC catalyzes an important step in this process, the rearrangement of AIR to form HMP-P (Fig. 2) [10]. Initial studies of ThiC were carried out with cell-free extract and established the production of HMP-P by this enzyme [10]. Purified ThiC was later shown to be dependent on AdoMet for activity, to harbor an iron-sulfur cluster [31], and to generate a radical [32]. Recent work, in which the AdoMet radical activity of ThiC was demonstrated, enabled further spectroscopic and crystallographic characterization of ThiC, revealing an enzyme that exhibits AdoMet radical activity with a new structural motif [22]. As described above, ThiC lacks the classic CX3CXφC sequence for binding the [4Fe-4S] cluster and lacks the classic AdoMet binding motifs. Instead of using a single loop from a full or partial TIM barrel, ThiC appears to use a separate domain to bind its [4Fe-4S] cluster [22].
3.2. Structure determination of ThiC
Three structures of ThiC (all crystallized aerobically) were solved by selenomethionine single-wavelength anomalous dispersion and molecular replacement to 2.1 Å resolution [22]. The structures comprise residues 5 to 546 and have two chain breaks (residues 99-112 and 223-228 in chain A); similar start points, end points and chain breaks occur in both chains of all three structures. Interestingly, the disordered regions of the enzyme are located adjacent to each other and near the active site (Fig. 6A). Additionally, the C-terminal segment of the enzyme, known to ligate the catalytically essential [4Fe-4S] cluster in ThiC, is one of these disordered regions. Although much is known about the catalytic mechanism of ThiC [33], including that the enzyme is indeed an AdoMet radical enzyme that requires a [4Fe-4S] cluster for activity [31, 32] (Suppl. Fig. 1), all three structures lack the cluster and AdoMet. Here we examine the ThiC structure and discuss its contribution to our understanding of AdoMet radical structure, but a full analysis awaits the availability of the structure in complex with AdoMet and the [4Fe-4S] cluster.
Figure 6.
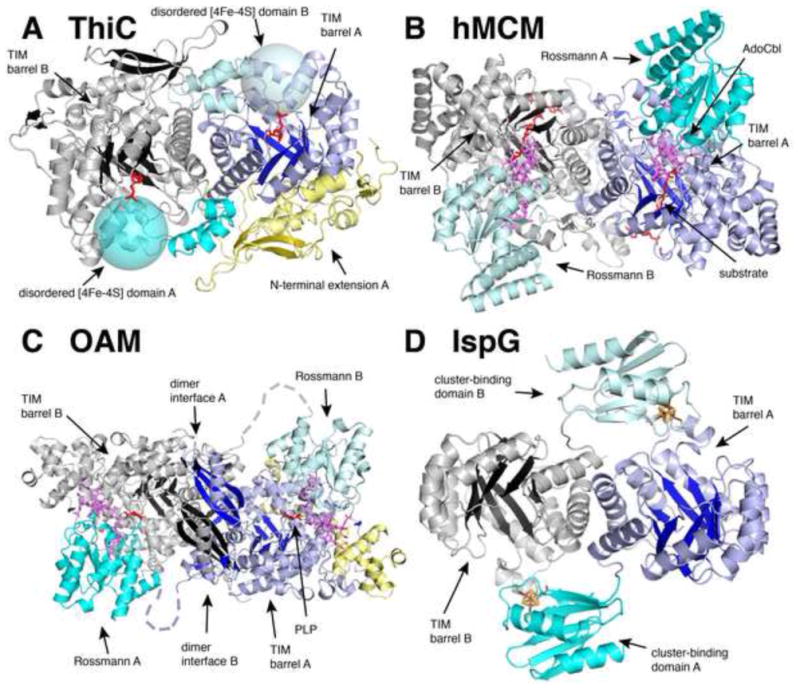
Overall structural comparisons of ThiC with AdoCbl radical enzymes and a [4Fe-4S] cluster-binding protein. (A) ThiC from Caulobacter crescentus (PDB ID: 3EPN) with chain A colored as follows: yellow, N-terminus; dark blue, TIM barrel; and cyan, C-terminal domain. Chain B is colored gray with black β strands, and its C-terminus is colored light cyan to emphasize the domain-swap of the cofactor binding subunit. The expected locations of the ThiC disordered [4Fe-4S] binding domain are represented as large transparent cyan spheres. (B) Human methylmalonyl-CoA mutase (hMCM, PDB ID: 2XIQ) with chain A colored as follows: dark blue, TIM barrel domain; and cyan, AdoCbl-binding Rossmann domain. Chain B is colored gray with black β strands, and its AdoCbl-binding Rossmann domain is light cyan. (C) Ornithine 4,5-aminomutase (OAM) from Clostridium sticklandii (PDB ID: 3KP1) with chain A colored as follows: dark blue, TIM barrel domain; cyan, AdoCbl-binding Rossmann domain; and yellow, accessory protein. Chain B is colored as in chain B of hMCM (B). (D) [4Fe-4S] protein IspG from Aquifex aeolicus (PDB ID: 3NOY) with chain A colored as follows: dark blue, TIM barrel domain; and cyan, cluster-binding domain. Ligands are colored as follows: violet, AdoCbl; red, AIR-analogue and malonyl CoA and PLP; and clusters are colored in rust (iron) and yellow (sulfur). Protein domains are labeled A or B according to which protein chain they belong.
3.3. Structural comparisons with ThiC
With many of the AdoMet radical enzyme structures solved showing partial (β/α)6 TIM barrel protein folds [25, 28, 34-38], ThiC joins the subset of AdoMet radical enzymes that contain full (β/α)8 TIM barrels (see Fig. 5) [14, 26]. In addition, it resembles several AdoCbl-dependent enzymes (Fig. 6B-C), including Clostridium cochlearium glutamate mutase (PDB ID: 1I9C [39], RMSD = 2.9 Å overall, 10% identical to ThiC), human methylmalonyl-CoA mutase (PDB ID: 2XIQ [40], RMSD = 3.0 Å overall, 11% identical to ThiC), lysine 5,6-aminomutase (PDB ID: 1XRS [41], RMSD = 3.1 Å overall, 11% identical to ThiC), and ornithine 4,5-aminomutase (PDB ID: 3KOW [42], RMSD = 3.8 Å overall, 13% identical to ThiC), which all utilize a TIM barrel domain. Finally, there is structural homology to Aquifex aeolicus IspG (PDB ID: 3NOY [43], RMSD = 2.8, 14% identical to ThiC), a protein involved in the non-mevalonate isoprenoid biosynthetic pathway that contains a TIM barrel domain and a separate [4Fe-4S] domain (Fig. 6D); however, this cluster is not involved in AdoMet radical chemistry. ThiC itself is a homodimeric enzyme, with each monomer comprising three domains (Fig. 6A). As with the enzymes listed above, a TIM barrel domain harbors the active site (dark blue in chain A in Fig. 6A), which was identified in the ThiC structure by cocrystallization of the enzyme with either product or substrate analog [22]. N-terminal to this TIM barrel domain is an (α+β) domain unique to ThiC (yellow in chain A of Fig. 6A) that covers the bottom surface (N-terminal face) of the barrel, possibly playing a structural role or helping to seal off the N-terminal side of the barrel. The C-terminal domain (cyan in chain A and light cyan in chain B of Fig. 6A) folds onto the second monomer, extending close to its active site. With the last 76 C-terminal residues missing from the third domain in these structures, including the cluster-ligating CX2CX3φC motif (C561SMC564GPKFC569) (expected locations shown as cyan spheres in Fig. 6A), it is easy to imagine that an intact C-terminal domain will be responsible for radical generation in the neighboring barrel. Thus the ThiC dimer appears to be a domain swapped structure, with the C-terminal [4Fe-4S] cluster domain of one chain binding over the active site of the other chain. Such a domain swapped arrangement is found in AdoCbl-enzyme ornithine 4,5-aminomutase, in which the AdoCbl-binding, Rossmann-like domain of one chain binds over the active site of the TIM barrel of the other chain in the dimer (light cyan Rossmann domain from chain B binds over dark blue TIM barrel of chain A in Fig. 6C) [42]. Domain swapping is also apparent in the structure of IspG, in which the [4Fe-4S] cluster-binding domain of one molecule is closer to the TIM barrel of the dimer molecule (Fig. 5).
Thus, unlike all classic AdoMet radical proteins, dAdo formation in ThiC appears to involve two distinct protein domains, reminiscent of AdoCbl enzymes [22, 44]. In AdoCbl enzymes, substrate binding to the TIM barrel is followed by the appropriate positioning of AdoCbl over the substrate for radical generation (see human methylmalonyl-coenzyme A mutase in Fig. 6B) [40]. For ThiC, substrate binding to the TIM barrel would presumably be followed by the proper positioning of the AdoMet-bound [4Fe-4S] domain over the substrate. Analysis of the conservation of residues in the ThiC TIM barrel shows a broad surface that extends beyond the region likely responsible for catalysis (red in Fig. 7A). These extended areas could delineate where the C-terminal [4Fe-4S] domain and the TIM barrel interact. With no structure of AdoMet bound to ThiC, we can only predict its interactions with the TIM barrel domain based on the solved structures of AdoMet radical and AdoCbl radical enzymes.
Figure 7.
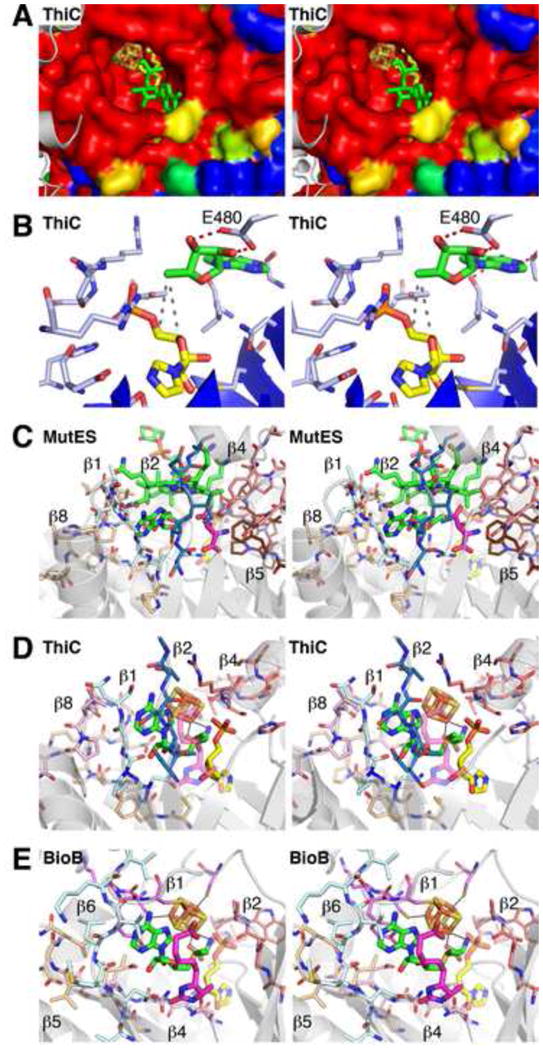
Modeling of AdoMet binding in ThiC based on BioB and glutamate mutase (MutES). Panels shown in stereo. (A) The ThiC active site of chain A displayed as a surface colored by percent sequence conservation with red corresponding to completely conserved residues and blue corresponding to nonconserved residues. Chain B of ThiC is shown as gray cartoon traces, with the last ordered residue (His 546) protruding towards the active site. The bound imidazole ribotide molecule is colored yellow. AdoMet is modeled into the active site by superposition with biotin synthase (PDB ID: 1R30) and colored green. Iron and sulfur are colored rust and yellow, respectively. (B) ThiC residues in the vicinity of the putative dAdo binding site. The dAdo (green) is modeled based on superposition of ThiC with the structure of glutamate mutase (PDB ID: 1I9C). Protein carbon atoms are in blue and substrate carbons in yellow. Dashed red lines connect the ribose of the modeled dAdo and E480 (equivalent to E330 of glutamate mutase); dashed gray lines connect C4' and C5' of the substrate and C5' of the dAdo. (C) Active site of MutES with active site loops labeled and colored as follows: β1 is pale cyan; β2 is dark blue; β4 is salmon; β5 is brown; and β8 is tan. AdoCbl is colored in green carbons and methylaspartate in magenta carbons; the relative position of the ThiC imidazole ribotide is shown in transparent yellow. (D) Active site of ThiC with loops proposed to bind AdoMet, as determined by superposition of ThiC with BioB. Loops of the ThiC structure that resemble AdoMet radical motifs are labeled and colored as follows: β4 and salmon, the GGE motif; β8 and light pink and tan, the β4 and GxIxGxxE motifs; β1 and pale cyan, the β6 motif. The additional β2 loop not present in BioB is colored dark blue. Modeled AdoMet is colored in green carbons and substrate in yellow carbons; the relative position of the BioB dethiobiotin substrate is shown in transparent magenta. (E) Active site of BioB with some of the traditional AdoMet radical enzyme motifs shown as sticks with carbons colored as follows: magenta, cluster-binding loop; salmon, GGE motif following β2; light pink, ribose-binding β4 motif; tan, GxIxGxxE β5 motif; and pale cyan, β6 motif. AdoMet is colored in green carbons and dethiobiotin in magenta carbons; the relative position of the ThiC imidazole ribotide is shown in transparent yellow.
Little is known regarding sequence motifs responsible for the binding of the dAdo group of AdoCbl in the TIM barrel domain of AdoCbl radical enzymes, partly because few structures have been crystallized with intact AdoCbl or both cob(II)alamin and ordered dAdo bound; rather, various structures contain non-catalytic forms of Cbl or an adenosine analog such as adeninylpentylcobalamin, which appears to bind in a non-catalytic orientation compared to the few structures with bound AdoCbl [39–42, 45–49]. One highly identifiable interaction is observed in the isomerases methylmalonyl-coenzyme A mutase, glutamate mutase, and lysine 5,6-aminomutase, in which a conserved glutamate located in the final loop of the barrel interacts with both hydroxyls of the cleaved dAdo ribose. Interestingly, a similar interaction with the ribose of AdoMet is also observed in many AdoMet radical enzymes from a polar residue(s) located after β4 and/or β5, as mentioned above. These protein-ribose interactions can allow the enzyme to tailor generation and control of dAdo• as shown for BioB [27]. An alignment of ThiC with glutamate mutase reveals a glutamate located in the last loop of the TIM barrel (E480) that aligns well with the relevant glutamate in glutamate mutase, suggesting a putative position for dAdo in the ThiC structure (Fig. 7B). The ThiC active site cleft displays an empty surface of the appropriate size and shape for dAdo binding, and this site coincides with the dAdo binding site observed in the glutamate mutase structure (Fig. 7C,D). Modeling of dAdo into the active site by superposition with glutamate mutase puts the C5' atom of dAdo at distances from C4' and C5' of the bound substrate analogue that are consistent with crystallographic distances observed for H-atom abstraction [25, 26, 36, 39, 50] (although the orientation is only correct for C4'). With labeling studies identifying both C4' and C5' as sites of H-atom abstraction during the course of the ThiC reaction [33], the structural model for dAdo binding shown in Fig. 7A,B is likely close to the correct binding site for dAdo in ThiC. An actual structure with AdoMet, or post-homolysis cleavage products dAdo and Met, bound to ThiC in the presence of substrate would make for a fascinating comparison between the AdoMet radical and the AdoCbl radical enzyme superfamilies.
This proposed dAdo binding site in ThiC correlates well with the position of the dAdo moiety of AdoMet in BioB (Fig. 7E). While sequence alignment between ThiC and known AdoMet radical enzymes suggests that ThiC lacks the classical motifs observed in AdoMet binding, which are labeled in Fig. 4 and discussed above, there does appear to be some level of biochemical similarity. Using the structure of E. coli BioB, we can compare the modified sequences observed in the ThiC structure with the canonical AdoMet radical motifs (Fig. 7D,E). First, as is the case with all other structurally characterized AdoMet radical proteins, the ThiC CX2CX3φC cluster binding sequence contains an aromatic residue (φ) known to interact with the adenine of AdoMet. Unfortunately, this loop is disordered in the ThiC structure. Second, a glycine rich sequence following β4 of the ThiC TIM barrel (G330IVSRGGS337) resembles the “GGE” motif located at the end of β2 of BioB (A100AW102, in BioB) (salmon in Fig. 7D,E) [31]. As mentioned above, ThiC residue E480 of (C474YVTPKE480HLGLPDR487) is found in a loop that follows the final strand of the ThiC TIM barrel and superimposes with D155 of BioB, a residue that interacts with the AdoMet ribose hydroxyls and is known to play a role in catalysis (light pink in Fig. 7D,E) [27]. The second half of this loop sequence (L482-R487) of ThiC corresponds to the GxIxGxxE motif located near β5 (G190GIVGLGE197 in BioB), which is known to interact with the adenine and ribose moieties (tan in Fig. 7D,E). Finally, a sequence at the C-terminus of β1 (N219IGN222) of ThiC corresponding to the C-terminus of β6 in BioB (N222MLV225) provides backbone and hydrophobic interactions with the adenine moiety (cyan in Fig. 7D,E). Interestingly, the substrate location in ThiC does not match perfectly with BioB and other AdoMet radical enzymes upon superposition (Fig. 7D,E). The presence of a loop after β2 in ThiC (blue in Fig. 7D) directly clashes with the binding site for dethiobiotin observed in the BioB crystal structure. In ThiC, the substrate binding site (yellow in Fig. 7D) is shifted by roughly 7 Å from the substrate site observed in BioB, but as mentioned above is still predicted to be close enough to the C5' of AdoMet for H-atom abstraction. Further structural analysis of ThiC with AdoMet will be important for confirming these predictions and for a complete analysis of AdoMet structural motifs. Interestingly, the substrate binding location of ThiC is similar to that of the AdoCbl dependent enzymes, such as glutamate mutase (Fig. 7C,D).
Studies of ThiC raise an important question: why utilize a different protein architecture to carry out this particular AdoMet radical reaction? Analysis of the biosynthetic machinery for thiamine pyrophosphate reveals that another protein in the pathway, ThiH, is also an AdoMet radical enzyme. ThiH is a tyrosine lyase [51] and its protein sequence suggests that it adopts the canonical AdoMet radical core fold; thus, thiamine biosynthesis utilizes both types of AdoMet radical folds. Is ThiC a link between AdoCbl and AdoMet radical enzymes? Did it evolve from an AdoCbl enzyme or did AdoCbl enzymes evolve from ThiC? Intriguingly, sequence searching for proteins that may resemble ThiC has been unsuccessful thus far, suggesting that this protein architecture for AdoMet radical chemistry may be limited to ThiC or that our searching algorithms are not sophisticated enough yet.
4. Lessons from 2,3-LAM on energetics of AdoMet cleavage
The common use of a [4Fe-4S] cluster to reduce AdoMet by the AdoMet radical enzyme family, promoting homolysis of the C–S bond and generation of dAdo•, raises very important questions, including how reduction of the [4Fe-4S]2+ to the active 1+ oxidation state is achieved, how the homolysis is regulated, and how this process is made energetically feasible. With an estimated reduction potential of -1800 mV for free AdoMet [23], this reaction seems outside of that which is possible in a biological context. How, then, does an AdoMet radical enzyme achieve cleavage? In terms of understanding the energetics of dAdo• formation by AdoMet radical enzymes, work by the laboratory of Perry Frey on 2,3-LAM is the gold standard [9].
4.1. 2,3-LAM
2,3-LAM is an interesting AdoMet radical enzyme in that its reaction is very reminiscent of an AdoCbl reaction; it catalyzes a 1,2-migration of an amino group much like 5,6-LAM (see [9] for a more comprehensive review on aminomutases). It was originally identified as catalyzing the formation of βlysine from L-lysine and the reverse in Clostridium subterminale strain SB4 [52, 53]. 2,3-LAM activity is important in L-lysine metabolism and the biosynthesis of various antibiotics, and early studies have led to an understanding of the cofactor requirements for 2,3-LAM activity: a [4Fe-4S] cluster, PLP, and AdoMet [52]. Also, along with spore photoproduct lyase [54, 55], 2,3-LAM uses AdoMet catalytically, regenerating the cofactor at the end of each catalytic cycle [56]. The activity of 2,3-LAM is PLP dependent, and in the enzyme resting state the PLP is chemically linked as an aldimine adduct to an internal lysine (Suppl. Fig. 2) [57].
4.2. Structure of 2,3-LAM
The X-ray crystal structure of 2,3-LAM was solved by selenomethionine single-wavelength anomalous dispersion in the laboratory of Dagmar Ringe [36]. Solution studies have indicated the presence of dimers, tetramers and hexamers [58], and 2,3-LAM crystallizes as a dimer of dimers, with each monomer containing a reconstituted [4Fe-4S] cluster, AdoMet, and Michaelis complex mimic lysine-PLP aldimine (Fig. 8A). Oligomerization involves an intimate domain swap between two monomers that is further stabilized by Zn2+ binding. The cofactors bind within the partial (β/α)6 TIM barrel core, similar to other AdoMet radical enzymes, and the C-terminal protein domain constitutes the entire dimer-dimer interface [36] (Fig. 5F). The PLP is housed inside the N-terminal portion of the barrel, which is capped by a helix and β strand from the C-terminal segment of the domain-swapped subunit. Several hydrogen-bonding interactions are observed with the PLP phosphate, phenolic oxygen, and one pyridine nitrogen, as well as with the ε –amino and carboxylate groups of the covalently tethered lysine substrate, which are responsible for positioning L-lysine for hydrogen-abstraction at C3, 4 Å from the expected dAdo• [36]. The protein used for crystallization was expressed in L-Se-methionine media, resulting in a crystal structure with AdoSeMet bound to the [4Fe-4S] cluster. Importantly, the Se atom is 3.2 Å from the unique Fe site of the cluster, positioned to ligate the cluster upon reductive cleavage of AdoSeMet [36]. This structural finding correlates well with X-ray absorption spectroscopy, which revealed a direct ligation of Se to an iron of the cluster in 2,3-LAM activated with AdoSeMet under turnover conditions or complexed with L-Se-methionine in the presence of substrate analogues [59], and similar results are expected for the reductive cleavage of AdoMet.
Figure 8.
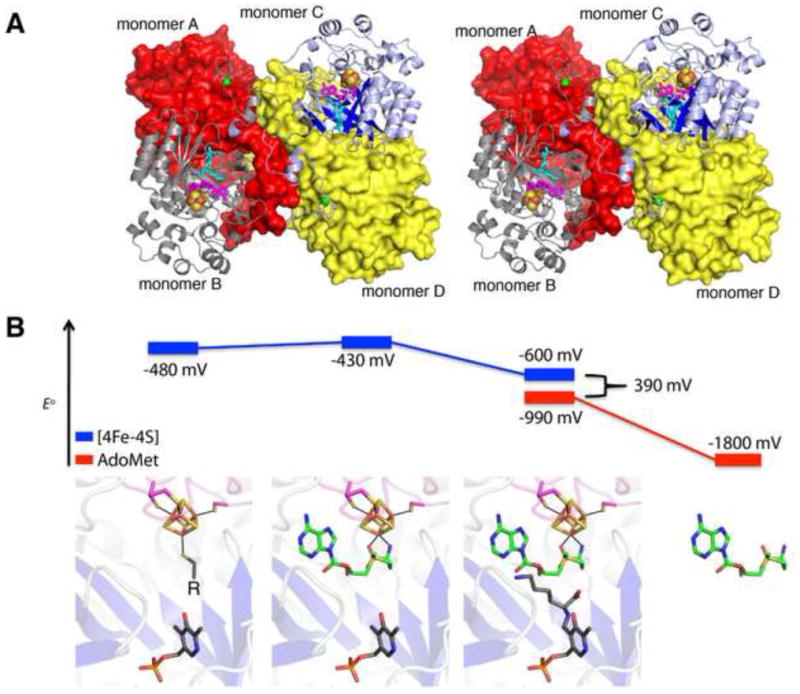
Insight into the energetics of reductive cleavage by 2,3-LAM. (A) Stereo-depiction of the 2,3-LAM tetramer (PDB ID: 2A5H), formed from dimers of monomers A and B, colored red and gray, and monomers C and D, colored blue and yellow, respectively. Monomers B and C are depicted as cartoon traces, while monomers A and D are surface representations. The [4Fe-4S] cluster is depicted as spheres, with sulfur in yellow and iron in rust; AdoMet is colored magenta; and the substrate lysine aldimine is colored cyan. Zinc ions are represented as green spheres. (B) Redox potentials measured for the [4Fe-4S] cluster of 2,3-LAM (blue) and AdoMet (red) under different ligand environments reveal a final difference of approximately 390 mV (~10 kcal) for C–S bond cleavage of AdoMet when all ligands required for the reaction are present [23]. Ligand binding environments are depicted with corresponding models based on the structure of 2,3-LAM (PDB ID: 2A5H). Atoms are colored as follows: red, oxygen; orange, phosphorous and iron; and yellow, sulfur. Ligand carbon atoms are colored as follows: AdoMet, green, and PLP and lysine, gray. All potentials were measured in the presence of LAM in solution except for free AdoMet, which is estimated to be -1800 mV [23]. The unliganded state of the cluster was measured with varying thiol ligands (represented as “R” in the LAM active site) [60]. The measurement for the reduction potential of the cluster in the presence of only AdoMet and PLP, without substrate, revealed a slight increase in reduction potential.
4.3. Redox properties of 2,3-LAM
2,3-LAM is capable of tuning the redox potential of both its [4Fe-4S] cluster and bound AdoMet to facilitate reductive cleavage (Fig. 8B). The redox potential for the “resting” state of the enzyme, i.e. the [4Fe-4S]2+/1+ of 2,3-LAM with various unique iron ligands, including L-cysteine, dithiothreitol, or dihydrolipoate, is approximately -480 mV [60]. Upon AdoMet binding directly to the [4Fe-4S] cluster, its redox potential has been measured to increase to −430 mV, an important step in bringing the redox potential into a regime relevant for physiological reduction to generate the activated [4Fe-4S]1+ state [60]. Consistent with the observation from the X-ray structure that lysine binds in between the PLP and the AdoMet (Fig. 8), Frey and co-workers find that the binding of the lysine substrate in the presence of PLP and AdoMet lowers the cluster reduction potential by 170 mV to a value of −600 mV. While this value is closer to the expected reduction potential of AdoMet, estimated as −1800 mV from solution studies of trimethylsulfoniums and aryl, vinyl, and allyl substituted sulfoniums [23], these values are still too disparate for effective electron transfer. To bring the values closer together, the enzyme functions by altering the reduction potential of AdoMet. The potential of the AdoMet molecule is observed to increase by 810 mV to a value of −990 mV when bound to the enzyme, based on the measured value for S-3,4-anhydroadenosyl-L-methionine [23], bringing the gap in reduction potentials for AdoMet homolysis to 390 mV. Structural comparisons between 2,3-LAM and other family members do not reveal any significant differences in how this enzyme binds AdoMet (see Section 2.1), raising the question of whether all family members will modulate the reduction potential of AdoMet in the same manner. Fully addressing this question will require additional data on the redox behavior of other AdoMet radical enzymes.
Overall, we have learned from 2,3-LAM that cofactor and substrate binding events can decrease the barrier for reductive AdoMet cleavage from 32 kcal mol−1 to 9 kcal mol−1, converting this radical initiation event into a process that is biologically relevant [23].
5. Insights into the mechanism of dAdo formation through the high resolution HydE structure
The molecular mechanism by which the C–S bond of AdoMet is reductively cleaved has been the subject of debate [61]. Before the first X-ray structure was available, selenium X-ray absorption (Se-XAS) spectroscopic studies of 2,3-LAM, PFL-AE and BioB revealed that for intact selenium AdoMet (AdoSeMet), the O/N atoms of the amino acid moiety, and not the Se of AdoSeMet, interact with the unique iron of the [4Fe-4S]1+ cluster [59, 62]. Further electron-nuclear double resonance (ENDOR) studies using PFL-AE by the Broderick and Hoffman laboratories with 2H- or 13C-labeled AdoMet at the methyl position identified an isotropic hyperfine contribution that suggested covalent interactions between the cluster and AdoMet [63]. Calculated distances suggested that the AdoMet sulfonium would be close enough to a sulfur atom of the cluster to mediate electron transfer [62–64]. With the first two X-ray structures of AdoMet radical enzymes confirming the coordination of AdoMet to the [4Fe-4S] cluster [26, 34], the recent determination of the 1.25 Å high resolution structure of AdoMet radical enzyme HydE [24] prompted calculations aimed at a molecular understanding of how AdoMet is reductively cleaved. Below we describe some of the recent studies on HydE.
5.1. HydE
HydE is part of a triad of maturase enzymes (HydE/HydF/HydG) involved in assembling the unusual diiron-containing cofactor (H-cluster) of iron-dependent hydrogenase, although its exact role in this assembly is not fully established [14, 65–68]. The H-cluster of iron-dependent hydrogenase is a remarkable cofactor, containing two cyanide and two carbon-monoxide ligands, which only recently have been discovered to derive from the decomposition of tyrosine through the lyase activity of HydG, another AdoMet-dependent enzyme [69]. The two irons of the H-cluster are bridged by a dithiolate ligand, the exact nature of which has been the source of some controversy (see Fig. 2F), although recent EPR data support the placement of a nitrogen in the middle position [70].
5.2. Structure of HydE
Thermotoga maritima HydE was generated by heterologous expression of the protein in E. coli and crystallographic characterization was carried out using iron single-wavelength anomalous dispersion and molecular replacement by Yvain Nicolet and coworkers [14, 24]. The HydE crystal structures locate the catalytic AdoMet and [4Fe-4S] cluster, and thus the active site [14, 24], at the top of a distorted (β/α)8 TIM barrel, consistent with the structure of the closely-related E. coli biotin synthase (RMSD = 2.5 Å for 292/348 Cα atoms) [26] (Fig. 5B). HydE shares the canonical AdoMet binding motifs described above and shown in Fig. 4. With the exact nature of the substrate still an open question, the large size of the HydE barrel to which the substrate would bind (991 Å3), relative to that in BioB of 717 Å3, suggests a substrate larger than dethiobiotin [14]. HydE binds two distinct iron-sulfur clusters, the [4Fe-4S] cluster that is involved in binding AdoMet and another, identified as a [2Fe-2S] cluster in the X-ray crystal structure, which is sandwiched between helices 7 and 8. This latter cluster, which is refined in the crystal structure with at most 70% occupancy, has been identified as a [4Fe-4S] cluster in solution studies [71]. Mutagenesis studies of the related enzyme from C. acetobutylicum indicate this second cluster is not involved directly in the cofactor biosynthesis [14], and the absence of the binding motif for this second cluster within other HydE proteins provides further evidence for this assertion [72].
5.3. DFT calculations based on high resolution structures of HydE
Structures of HydE in the presence of AdoMet or Ado and Met have been utilized in computational investigations to shed light on the underlying mechanisms behind AdoMet cleavage in AdoMet radical proteins [24]. The structure of HydE complexed with Ado and Met represents a post-homolysis state and shows ligation of the Sδ of methionine to the unique iron in the [4Fe-4S] cluster with an observed distance of 2.7 Å and pseudo-octahedral coordination geometry around the unique iron (Fig. 9). Prior to AdoMet cleavage, the geometry is distorted square pyramidal and distance between the Sδ of AdoMet and the unique iron is 3.3 Å. Using these HydE crystal structures as a starting model for QM/MM calculations, the determined barrier height for AdoMet cleavage is 12.9 kcal mol−1 [24]. This value is remarkably similar to that determined for LAM in the absence of substrate of 13.0 kcal mol−1 [23]. The calculated spin densities in HydE disfavor electron transfer from sulfur S3 of the cluster to the sulfonium of AdoMet [24], a possibility raised by the earlier spectroscopic studies with PFL-AE and BioB [62-64]. Instead, orbital energies of the AdoMet Sδ+ and the unique iron site from HydE were found to match well, which is a prerequisite for electron transfer (Fig. 9). Given the observation that AdoMet binding is similar among AdoMet radical enzymes [21], a common mechanism for AdoMet cleavage is likely. These calculations suggest that the single electron transfer involves passage from the unique iron site to the AdoMet Sδ+ orbital, promoting reductive cleavage and radical generation at the C5' of AdoMet [24]. Alternatively, cleavage can be described as a radical displacement reaction, similar to that observed at sulfoxide centers [73].
Figure 9.
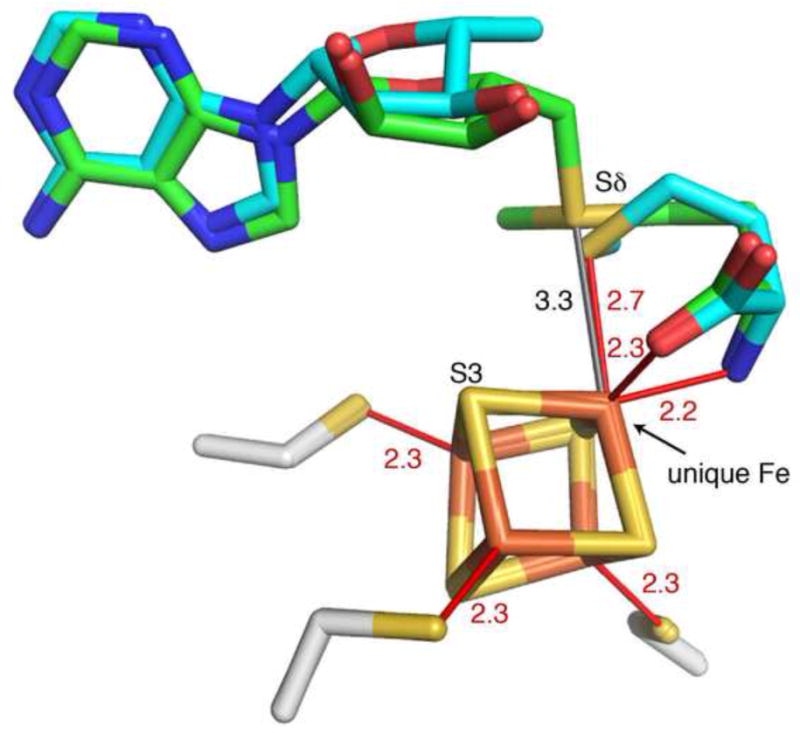
Interaction of [4Fe-4S] cluster (iron in rust, sulfur in yellow) with intact AdoMet (green carbons) and cleavage products, Ado and Met (blue carbons), from HydE structures at 1.62 Å and 1.25 Å resolution, respectively (PDB ID: 3IIZ and 3IIX). Distances from the [4Fe-4S] cluster to interacting atoms of ligands (cleavage product Met and three Cys of CX3CXφC motif) are indicated in red, and the AdoMet-cluster Sδ – Fe distance prior to reductive cleavage is indicated in gray. S3 of the cluster is labeled for comparative purposes (see text). The distance from S3 of the cluster to the sulfonium of AdoMet is 3.7 Å.
6. Mechanism to prevent uncoupling of AdoMet cleavage from catalysis: PFL-AE case study
Despite the fact that reductive cleavage of AdoMet is energetically unfavorable, several AdoMet enzymes (e.g. lipoate synthase, HydE, RNR, and BioB) show uncoupling of AdoMet cleavage from catalysis under certain in vitro reducing reaction conditions [71, 74–77]. If this uncoupling also occurs in vivo, it would be wasteful and detrimental to the organism. We posit that AdoMet radical enzymes must have evolved mechanisms to prevent excessive cleavage in the absence of substrate, albeit imperfect mechanisms in some cases. Here we consider what we have learned from structures of AdoMet-bound PFL-AE, with and without substrate, about preventing AdoMet cleavage in the absence of substrate.
6.1. PFL-AE
Pyruvate formate lyase (PFL) catalyzes the reversible conversion of pyruvate and coenzyme A to formate and acetyl coenzyme A during anaerobic microbial growth [17]. In order to be catalytically functional, PFL requires an activating enzyme known to catalyze the post-translational generation of a radical on Gly734 of PFL [78]. This glycyl radical activating enzyme, PFL-AE, is an AdoMet radical enzyme that utilizes the formation of dAdo• to abstract an H-atom from Gly734, which is located within a specific peptide recognition sequence in a loop in the enzyme active site (Fig. 2B) (Suppl. Fig. 3) [79]. This glycine-loop is buried in the large (β/α)10 barrel [80–82], and, during catalysis, this loop must remain buried both to transfer the protein radical to a conserved active site cysteine and to limit quenching of the Gly734 radical [81, 82]. As an example of the latter, exposure to dioxygen is well known to quench the glycyl radical and cleave the protein backbone [78]. However, for activation the loop must be exposed to react with PFL-AE, indicating the need for PFL to undergo conformational changes. Recent studies with PFL in the Broderick laboratory suggest that this loop samples both open and closed positions in the presence of varying amounts of PFL-AE, with the addition of PFL-AE shifting the equilibrium towards the open/solvent exposed conformation [83]. Once PFL-AE is able to bind substrate, it would be most advantageous to have all cofactors bound and in the correct oxidation state for catalysis. How, then, does the activating enzyme prevent premature cleavage of AdoMet before binding the substrate? While the energetic landscape for reductive AdoMet cleavage is most likely similar to that in 2,3-LAM, crystal structures of PFL-AE help provide a molecular understanding for how AdoMet cleavage is regulated.
6.2. Structure of PFL-AE
Two crystal structures of PFL-AE have been reported by our laboratory [25], one cocrystallized with a [4Fe-4S] cluster, AdoMet, and a 7-residue substrate peptide corresponding to the sequence of PFL surrounding the glycyl radical site (R731VSG734YAV737), which we will refer to here as pept-PFL-AE, and one with only the [4Fe-4S] cluster and AdoMet present (peptide-free PFL-AE). The structures of peptide-free and peptide-bound PFL-AE were solved by multiwavelength iron anomalous dispersion to 2.25 Å and 2.77 Å resolution, respectively [25]. Thus far, PFL-AE is the smallest structural example of an AdoMet radical enzyme; it is only 246 amino acids in length and exists as a monomer in both solution and crystal studies. This structure primarily consists of the AdoMet radical core, with extensions including only two loops located at both the N- and C-termini of the core and an additional βstrand located at the end of the cluster-binding loop (Fig. 5D).
Both crystal structures contain density for replete [4Fe-4S] clusters; however, the active sites of these two enzyme forms are quite different. The pept-PFL-AE structure reveals a well-ordered AdoMet bound to the [4Fe-4S] cluster and residues 2-7 of the bound substrate peptide. The Cα of the substrate glycine residue is positioned 4.1 Å from the C5' of AdoMet, consistent with direct H-atom abstraction of the substrate glycine residue by generated dAdo• (Fig. 10A). In contrast, AdoMet in the peptide-free PFL-AE structure is bound in a non-catalytic conformation with the L-amino acid moiety bound in the expected sulfonium binding site, and the adenosine moiety too disordered to model (Fig. 10B) [25]. Various residues in the active site are observed in different conformations as compared to the pept-PFL-AE structure. In particular, a loop located at the N-terminus before the start of the radical core is found to be in two differing conformations (yellow loop in Fig. 10). In the structure with peptide bound this loop is positioned within the barrel, proximal to the substrate, where it provides various interactions with the peptide. Without peptide, this loop is positioned further away from the active site, leaving the barrel more open. These two binding modes provide an excellent example of how substrate binding can influence AdoMet positioning and therefore reactivity; in the absence of substrate, AdoMet interacts with the cluster as has been reported by Mössbauer and ENDOR spectroscopic studies [63, 84, 85], but in the crystal structure it is not bound appropriately for reductive cleavage of the C–S bond (Fig. 10B) [25]. Once substrate enters, the active site rearranges such that AdoMet is now positioned with its sulfonium approximately 3.2 Å from the unique iron of the cluster, poised for reductive cleavage. Indeed, PFL-AE is not observed to cleave AdoMet in the presence of a reducing agent (flavodoxin, 5-deazariboflavin, or dithionite) when substrate is absent [79, 86].
Figure 10.

Structural changes observed upon substrate binding within PFL-AE (PDB ID: 3CB8 and 3C8F). (A) PFL-AE (strands in blue and helices in gray) bound to a peptide (dark gray) derived from its natural PFL substrate. Six residues of the heptamer peptide (V732SG734YAV737) used in the crystallization are visible in the structure. With peptide bound, Gly734 is 4.1 Å from the 5'-C of AdoMet (green carbons), an appropriate distance for H-atom abstraction (a dashed gray line connects Cα of Gly734 and the 5'-C of AdoMet). The CX3CXφC-containing loop is in magenta and the substrate-binding loop that undergoes a conformational change (see text) is in yellow. (B) PFL-AE in the absence of peptide (colored as above) has a more open structure. The cluster-bound AdoMet is disordered; only the Met moiety of AdoMet (green carbons) can be visualized in the electron density.
The majority of AdoMet radical enzyme structures to date lack snapshots of these enzymes both with and without bound substrate in the presence of AdoMet. As additional structures of AdoMet radical enzymes in various liganded states become available, other family members may display the overall mechanism of PFL-AE: binding AdoMet non-productively in the absence of substrate, which prevents uncoupled AdoMet cleavage and substrate radical generation.
7. Structure of RlmN suggests how one enzyme can harness both methylation and radical generation abilities of AdoMet
Methylation of nucleic acids is common in nature, and while most methylation reactions involve simple SN2 nucleophilic chemistry, methylation of non-nucleophilic positions like the C2 and C8 of adenosine requires a different mechanism. The recent characterization of the enzymes RlmN and Cfr has provided an initial understanding of how these “electrophilic methylations” can occur [19, 87]. The enzymes in question belong to a class of AdoMet-dependent enzymes known as radical SAM methyltransferases (RSMTs), which has recently been reviewed [88]. RlmN, which catalyzes the methylation of C2 at adenosine 2503 of the rRNA 23S subunit, has been found to follow a three-step sequence involving two cysteine residues from outside of the AdoMet radical CX3CXφC motif: (1) methylation of an internal conserved cysteine residue (C355) by an SN2-type mechanism from AdoMet to afford S-methylcysteine (mC355); (2) generation of a mC355 radical and subsequent addition of the methylene group to the C2 position of the purine ring forming a covalent bond between nucleotide and C355; and (3) release of methylated tRNA from this covalent interaction via formation of a disulfide bond between C355 and conserved C118 (Suppl. Fig. 4) [19].
Recent work by the Booker and Rosenzweig laboratories has revealed through biochemical and structural analysis that RlmN uses a single [4Fe-4S] cluster [89] and a single AdoMet binding site [90] to catalyze both the methylation and radical-based chemistry. The uniqueness of this reaction, in which two molecules of AdoMet are expected to bind consecutively to a single [4Fe-4S] cluster to first transfer a methyl group and then initiate radical chemistry, raises the critical question of how only one binding site for AdoMet is able to support these two functions.
7.1. Structures of RlmN
RlmN has been visualized by crystallography both with and without AdoMet [90]. For each, the asymmetric unit contains two monomers, providing two examples of each captured state [90]. The structures confirm that RlmN contains the partial (β/α)6 TIM barrel radical core, with the [4Fe-4S] cluster coordinated by three cysteines within the loop following β1 (Fig. 11A). In addition to the radical core, RlmN contains an N-terminal accessory domain consisting of four α-helices that adopt a variant of the so-called HhH2 fold expected to be involved in nucleic acid binding [90, 91] (Fig. 11A). At the C-terminus, the core is extended by a seventh strand, β7' (cyan in Fig. 11A), followed by a loop (I351DAACGQL358, also in cyan) that contains the conserved C355 and runs through the active site, ending in an interaction with three N-terminal β-strands (β1'-β3', colored yellow in Fig. 11A). The C-terminus of the protein contains an alpha helix observed to interact with a symmetry partner in the crystal [90].
Figure 11.
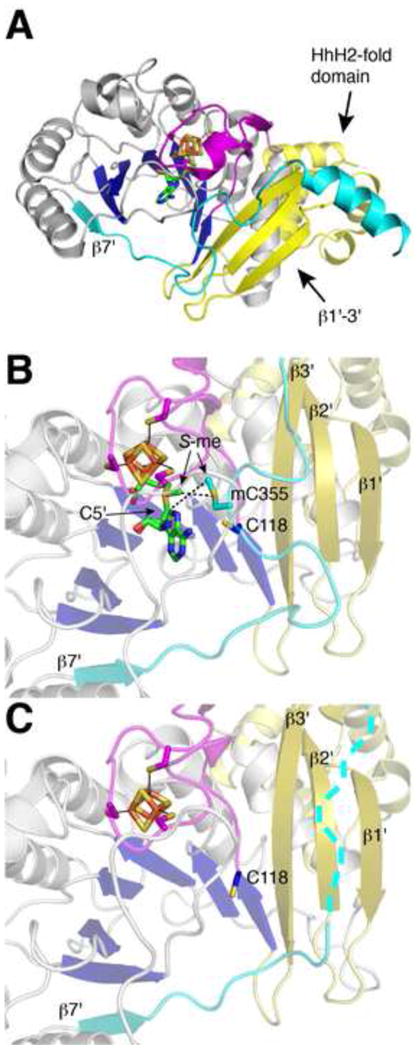
Structural changes observed within crystal structures of RlmN with and without AdoMet (PDB ID: 3RF9 and 3RFA). (A) Structure of the RlmN monomeric subunit. Colors are as follows: yellow, N-terminal domain including the HhH2-like domain; blue and gray, β strand and α helical protein elements of the core domain, respectively; cyan, C-terminal extension containing C355; magenta, cluster-binding loop; green, AdoMet carbons; orange, iron; yellow, sulfur; red, oxygen; and blue, nitrogen. (B) RlmN structure with bound AdoMet and methylated-C355 (mC355). Methyl groups bound to S are labeled S-me. Colors are as in (A). (C) Unliganded RlmN with a disordered mC355, in which the C-terminal extension is disordered and indicated with dashes.
The structure of RlmN complexed with AdoMet reveals electron density for a methylated cysteine residue 355 from within the β7' loop (cyan in Fig. 11B) [90], suggesting that the first step in catalysis (the methylation step) has already occurred. Presumably the AdoMet present in the structure is thus available to catalyze step two upon the binding of nucleotide substrate and reduction of the [4Fe-4S] cluster. With the knowledge that AdoMet has only a single binding site on RlmN to carry out both methylation and H-atom abstraction, this structure can be used to consider the molecular basis for these two catalytic steps. For the SN2-type methyl transfer reaction, this AdoMet-binding site allows for a close interaction (3.6 Å) between the methyl moiety of bound AdoMet and the S of C355, which would facilitate methyl transfer (Fig. 11B). In terms of H-atom abstraction from the methyl of mC355, the C5' of AdoMet is positioned near the methyl of mC355 (6.0 Å). Although 6.0 Å is too far for H-atom abstraction, it is reasonable to expect that substrate binding will bring the already close AdoMet and mC355 even closer. Following the reaction of mC355 radical species with purine substrate, release of product can be accomplished via nucleophilic attack of C118 on C355 (these cysteines are 6.0 Å away in the substrate-free structure) (Fig 11B and Suppl. Fig 4).
Additional differences between RlmN structures with and without AdoMet bound reveal subtle intricacies likely to be important in catalysis. Markedly, the β7' loop, which contains C355, appears rather flexible; it is only observed in one of the two AdoMet-bound monomers, with high B-factor values and poor connectivity for the amino acid backbone in places. In the other monomer, and the structure of RlmN without AdoMet, this entire loop segment is disordered, leaving the carboxyl end of the barrel rather exposed to solvent (Fig. 11C) [90]. Also in the structure without AdoMet, the S of C118 is oriented away from the active site, whereas in the AdoMet bound structure, it now points toward mC355. Although still too far for disulfide formation, these changes reflect the conformational flexibility requisite for the reaction between C118 and C355 as well as for other enzymatic steps.
While the current structures suggest how RlmN becomes primed with a mC355 and bound AdoMet, ready to bind rRNA substrate and reductively cleave AdoMet, key structural information is still missing regarding the changes that will occur upon substrate binding. In order for hydrogen abstraction to occur at the generated mC355, and for C118 to form a disulfide bond with C355 as proposed in the mechanism, promoting loss of the methylated rRNA adenosine, further rearrangement of the active site will be necessary to achieve more favorable distances for this chemistry. Structures with rRNA bound will hopefully aid in determining how RlmN switches from standard methylation chemistry to reductive AdoMet cleavage and hydrogen abstraction.
8. Concluding Remarks
Nature has adapted a variety of methods to catalyze energetically unfavorable reactions. Here, we discuss a broad family of enzymes that utilize a [4Fe-4S] cluster-bound AdoMet molecule to generate a highly reactive oxidant (i.e. dAdo•) for hydrogen abstraction from unactivated C–H bonds. While radical chemistry is powerful, it also comes at a price, and structural analysis of AdoMet radical enzymes such as 2,3-LAM, HydE and PFL-AE help us understand how reactivity is controlled. With the structural data currently in hand, we find that most AdoMet radical enzymes utilize a full or partial TIM barrel fold with CX3CXφC motif to coordinate the [4Fe-4S] cluster; however, ThiC was recently found to represent a new structural class, perhaps providing a link between AdoMet radical and AdoCbl radical enzymes. With most family members using only one function of AdoMet (radical generation), RlmN was recently found to use both methylation and radical generation functions of AdoMet. One can only imagine what further variations of this enzyme architecture and chemistry await discovery. As we write this, a structure of methylornithine synthase has became available [92], demonstrating the rapid rate of structural advancement for this family of enzymes. With each new crystal structure, we are learning how this radical family manipulates AdoMet to catalyze the unexpected.
Supplementary Material
Highlights.
Structural motifs for AdoMet radical enzymes are reexamined
Structures are classified into two categories: traditional and ThiC-like
Recent insights into function through structure are reviewed.
Acknowledgments
The authors thank Michael Funk, Jeremy Setser, Christina Stock, Peter Goldman, and Martin McLaughlin for helpful discussion in preparation of this manuscript. We acknowledge support from the Wellcome Trust [091162/Z/10/Z] (to A.K.C.) and National Science Foundation Grant MCB-0543833 (to C.L.D.). C.L.D is an HHMI investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Anna K. Croft, Email: a.k.croft@bangor.ac.uk.
Catherine L. Drennan, Email: cdrennan@mit.edu.
References
- 1.Frey PA. Lysine 2,3-aminomutase: is adenosylmethionine a poor man's adenosylcobalamin? FASEB J. 1993;7:662–670. doi: 10.1096/fasebj.7.8.8500691. [DOI] [PubMed] [Google Scholar]
- 2.Brazeau BJ, Gort SJ, Jessen HJ, Andrew AJ, Liao HH. Enzymatic activation of lysine 2,3-aminomutase from Porphyromonas gingivalis. App Env Microbiol. 2006;72:6402–6404. doi: 10.1128/AEM.01143-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaschkowski HP, Neuer G, Ludwig-Festl M, Knappe J. Routes of flavodoxin and ferredoxin reduction in Escherichia coli: CoA-acylating pyruvate: Flavodoxin and NADPH: Flavodoxin oxidoreductases participating in the activation of pyruvate formate-lyase. Eur J Biochem. 1982;123:563–569. [PubMed] [Google Scholar]
- 4.Mulliez E, Padovani D, Atta M, Alcouffe C, Fontecave M. Activation of class III ribonucleotide reductase by flavodoxin: A protein radical-driven electron transfer to the iron-sulfur center. Biochemistry. 2001;40:3730–3736. doi: 10.1021/bi001746c. [DOI] [PubMed] [Google Scholar]
- 5.Ifuku O, Koga N, Haze SI, Kishimoto J, Wachi Y. Flavodoxin is required for conversion of dethiobiotin to biotin in Escherichia coli. Eur J Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 6.Sanyal I, Gibson KJ, Flint DH. Escherichia coli biotin synthase: An investigation into the factors required for its activity and its sulfur donor. Arch Biochem Biophys. 1996;326:48–56. doi: 10.1006/abbi.1996.0045. [DOI] [PubMed] [Google Scholar]
- 7.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nuc Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 9.Frey PA, Reed GH. Pyridoxal-5'-phosphate as the catalyst for radical isomerization in reactions of PLP-dependent aminomutases. BBA-Proteins Proteom. 2011;1814:1548–1557. doi: 10.1016/j.bbapap.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Lawhorn BG, Mehl RA, Begley TP. Biosynthesis of the thiamin pyrimidine: the reconstitution of a remarkable rearrangement reaction. Org Biomol Chem. 2004;2:2538–2546. doi: 10.1039/B405429F. [DOI] [PubMed] [Google Scholar]
- 11.Sanyal I, Cohen G, Flint DH. Biotin synthase: purification, characterization as a [2Fe-2S]cluster protein, and in vitro activity of the Escherichia coli bioB gene product. Biochemistry. 1994;33:3625–3631. doi: 10.1021/bi00178a020. [DOI] [PubMed] [Google Scholar]
- 12.Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J Bacteriol. 1993;175:1325–1336. doi: 10.1128/jb.175.5.1325-1336.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hänzelmann P, Schindelin H. Binding of 5 '-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine – enzyme MoaA provides insights into its mechanism. Proc Natl Acad Sci USA. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicolet Y, Rubach JK, Posewitz MC, Amara P, Mathevon C, Atta M, Fontecave M, Fontecilla-Camps JC. X-ray structure of the [FeFe]-hydrogenase maturase HydE from Thermotoga maritima. J Biol Chem. 2008;283:18861–18872. doi: 10.1074/jbc.M801161200. [DOI] [PubMed] [Google Scholar]
- 15.Kanaujia SP, Jeyakanthan J, Nakagawa N, Balasubramaniam S, Shinkai A, Kuramitsu S, Yokoyama S, Sekar K. Structures of apo and GTP-bound molybdenum cofactor biosynthesis protein MoaC from Thermus thermophilus HB8. Acta Crystallogr. 2010;D66:821–833. doi: 10.1107/S0907444910019074. [DOI] [PubMed] [Google Scholar]
- 16.Leimkühler S, Wuebbens MM, Rajagopalan KV. The history of the discovery of the molybdenum cofactor and novel aspects of its biosynthesis in bacteria. Coord Chem Rev. 2011;255:1129–1144. doi: 10.1016/j.ccr.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knappe J, Sawers G. A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol Rev. 1990;75:383–398. doi: 10.1111/j.1574-6968.1990.tb04108.x. [DOI] [PubMed] [Google Scholar]
- 18.Tamarit J, Mulliez E, Meier C, Trautwein A, Fontecave M. The anaerobic ribonucleotide reductase from Escherichia coli. The small protein is an activating enzyme containing a [4Fe-4S]2+ center. J Biol Chem. 1999;274:31291–31296. doi: 10.1074/jbc.274.44.31291. [DOI] [PubMed] [Google Scholar]
- 19.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. A radically different mechanism for S-adenosylmethionine dependent methyltransferases. Science. 2011;332:604–607. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- 20.Nicolet Y, Drennan CL. AdoMet radical proteins--from structure to evolution--alignment of divergent protein sequences reveals strong secondary structure element conservation. Nuc Acids Res. 2004;32:4015–4025. doi: 10.1093/nar/gkh728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vey JL, Drennan CL. Structural insights into radical generation by the radical SAM superfamily. Chem Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE. Reconstitution of ThiC in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nat Chem Biol. 2008;4:758–765. doi: 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang SC, Frey PA. Binding energy in the one-electron reductive cleavage of S-adenosylmethionine in lysine 2,3-aminomutase, a radical SAM enzyme. Biochemistry. 2007;46:12889–12895. doi: 10.1021/bi701745h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolet Y, Amara P, Mouesca JM, Fontecilla-Camps JC. Unexpected electron transfer mechanism upon AdoMet cleavage in radical SAM proteins. Proc Natl Acad Sci USA. 2009;106:14867–14871. doi: 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc Natl Acad Sci USA. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrar CE, Jarrett JT. Protein residues that control the reaction trajectory in S-adenosylmethionine radical enzymes: mutagenesis of asparagine 153 and aspartate 155 in Escherichia coli biotin synthase. Biochemistry. 2009;48:2448–2458. doi: 10.1021/bi8022569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arragain S, Garcia-Serres R, Blondin G, Douki T, Clemancey M, Latour JM, Forouhar F, Neely H, Montelione GT, Hunt JF, Mulliez E, Fontecave M, Atta M. Post-translational modification of ribosomal proteins. J Biol Chem. 2010;285:5792–5801. doi: 10.1074/jbc.M109.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anantharaman V, Koonin EV, Aravind L. TRAM, a predicted RNA-binding domain, common to tRNA uracil methylation and adenine thiolation enzymes. FEMS Microbiol Lett. 2001;197:215–221. doi: 10.1111/j.1574-6968.2001.tb10606.x. [DOI] [PubMed] [Google Scholar]
- 30.Settembre E, Begley TP, Ealick SE. Structural biology of enzymes of the thiamin biosynthesis pathway. Curr Opin Struct Biol. 2003;13:739–747. doi: 10.1016/j.sbi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Gomez NC, Downs DM. ThiC is an [Fe-S] cluster protein that requires AdoMet to generate the 4-amino-5-hydroxymethyl-2-methylpyrimidine moiety in thiamin synthesis. Biochemistry. 2008;47:9054–9056. doi: 10.1021/bi8010253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Gomez NC, Poyner RR, Mansoorabadi SO, Reed GH, Downs DM. Reaction of AdoMet with ThiC generates a backbone free radical. Biochemistry. 2009;48:217–219. doi: 10.1021/bi802154j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterjee A, Hazra AB, Abdelwahed S, Hilmey DG, Begley TP. A "radical dance" in thiamin biosynthesis: mechanistic analysis of the bacterial hydroxymethylpyrimidine phosphate synthase. Angew Chem, Int Ed. 2010;49:8653–8656. doi: 10.1002/anie.201003419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hänzelmann P, Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc Natl Acad Sci USA. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc Natl Acad Sci USA. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goto-Ito S, Ishii R, Ito T, Shibata R, Fusatomi E, Sekine S-i, Bessho Y, Yokoyama S. Structure of an archaeal TYW1, the enzyme catalyzing the second step of wye-base biosynthesis. Acta Crystallogr. 2007;D63:1059–1068. doi: 10.1107/S0907444907040668. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki Y, Noma A, Suzuki T, Senda M, Senda T, Ishitani R, Nureki O. Crystal structure of the radical SAM enzyme catalyzing tricyclic modified base formation in tRNA. J Mol Biol. 2007;372:1204–1214. doi: 10.1016/j.jmb.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 39.Gruber K, Reitzer R, Kratky C. Radical shuttling in a protein: ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12 dependent enzyme glutamate mutase. Angew Chem, Int Ed. 2001;40:3377–3380. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 40.Froese DS, Kochan G, Muniz JRC, Wu X, Gileadi C, Ugochukwu E, Krysztofinska E, Gravel RA, Oppermann U, Yue WW. Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation. J Biol Chem. 2010;285:38204–38213. doi: 10.1074/jbc.M110.177717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL. A locking mechanism preventing radical damage in the absence of substrate, as revealed by the X-ray structure of lysine 5,6-aminomutase. Proc Natl Acad Sci USA. 2004;101:15870–15875. doi: 10.1073/pnas.0407074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolthers KR, Levy C, Scrutton NS, Leys D. Large-scale domain dynamics and adenosylcobalamin reorientation orchestrate radical catalysis in ornithine 4,5-aminomutase. J Biol Chem. 2010;285:13942–13950. doi: 10.1074/jbc.M109.068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee M, Gräwert T, Quitterer F, Rohdich F, Eppinger J, Eisenreich W, Bacher A, Groll M. Biosynthesis of isoprenoids: crystal structure of the [4Fe-4S] cluster protein IspG. J Mol Biol. 2010;404:600–610. doi: 10.1016/j.jmb.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 44.Marsh ENG, Patterson DP, Li L. Adenosyl radical: reagent and catalyst in enzyme reactions. ChemBioChem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsson KM, Logan DT, Nordlund P. Structural basis for adenosylcobalamin activation in AdoCbl-dependent ribonucleotide reductases. ACS Chem Biol. 2010;5:933–942. doi: 10.1021/cb1000845. [DOI] [PubMed] [Google Scholar]
- 46.Mancia F, Evans PR. Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Structure. 1998;6:711–720. doi: 10.1016/s0969-2126(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 47.Mancia F, Keep NH, Nakagawa A, Leadlay PF, McSweeney S, Rasmussen B, Bösecke P, Diat O, Evans PR. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure. 1996;4:339–350. doi: 10.1016/s0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 48.Masuda J, Shibata N, Morimoto Y, Toraya T, Yasuoka N. How a protein generates a catalytic radical from coenzyme B12: X-ray structure of a diol-dehydratase-adeninylpentylcobalamin complex. Structure. 2000;8:775–788. doi: 10.1016/s0969-2126(00)00164-7. [DOI] [PubMed] [Google Scholar]
- 49.Shibata N, Tamagaki H, Hieda N, Akita K, Komori H, Shomura Y, Terawaki S-i, Mori K, Yasuoka N, Higuchi Y, Toraya T. Crystal structures of ethanolamine ammonia-lyase complexed with coenzyme B12 analogs and substrates. J Biol Chem. 2010;285:26484–26493. doi: 10.1074/jbc.M110.125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mancia F, Smith GA, Evans PR. Crystal structure of substrate complexes of methylmalonyl-CoA mutase. Biochemistry. 1999;38:7999–8005. doi: 10.1021/bi9903852. [DOI] [PubMed] [Google Scholar]
- 51.Martinez-Gomez NC, Robers M, Downs DM. Mutational analysis of ThiH, a member of the radical S-adenosylmethionine (AdoMet) protein superfamily. J Biol Chem. 2004;279:40505–40510. doi: 10.1074/jbc.M403985200. [DOI] [PubMed] [Google Scholar]
- 52.Chirpich TP, Zappia V, Costilow RN, Barker HA. Lysine 2,3-aminomutase. Purification and properties of a pyridoxal phosphate and S-adenosylmethionine-activated enzyme. J Biol Chem. 1970;245:1778–1789. [PubMed] [Google Scholar]
- 53.Costilow RN, Rochovansky OM, Barker HA. Isolation and identification of β-lysine as an intermediate in lysine fermentation. J Biol Chem. 1966;241:1573–1580. [PubMed] [Google Scholar]
- 54.Mehl RA, Begley TP. Mechanistic studies on the repair of a novel DNA photolesion: the spore photoproduct. Org Lett. 1999;1:1065–1066. doi: 10.1021/ol9908676. [DOI] [PubMed] [Google Scholar]
- 55.Cheek J, Broderick JB. Direct H atom abstraction from spore photoproduct C-6 initiates DNA repair in the reaction catalyzed by spore photoproduct lyase: evidence for a reversibly generated adenosyl radical intermediate. J Am Chem Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 56.Wu W, Booker S, Lieder KW, Bandarian V, Reed GH, Frey PA. Lysine 2,3-aminomutase and trans-4,5-dehydrolysine: characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism. Biochemistry. 2000;39:9561–9570. doi: 10.1021/bi000658p. [DOI] [PubMed] [Google Scholar]
- 57.Chen D, Frey PA. Identification of lysine 346 as a functionally important residue for pyridoxal 5'-phosphate binding and catalysis in lysine 2,3-aminomutase from Bacillus subtilis. Biochemistry. 2001;40:596–602. doi: 10.1021/bi002265w. [DOI] [PubMed] [Google Scholar]
- 58.Song KB, Frey PA. Molecular properties of lysine-2,3-aminomutase. J Biol Chem. 1991;266:7651–7655. [PubMed] [Google Scholar]
- 59.Cosper NJ, Booker SJ, Ruzicka F, Frey PA, Scott RA. Direct FeS cluster involvement in generation of a radical in lysine 2,3-aminomutase. Biochemistry. 2000;39:15668–15673. doi: 10.1021/bi0022184. [DOI] [PubMed] [Google Scholar]
- 60.Hinckley GT, Frey PA. Cofactor dependence of reduction potentials for [4Fe-4S]2+/1+ in lysine 2,3-aminomutase. Biochemistry. 2006;45:3219–3225. doi: 10.1021/bi0519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fontecave M, Mulliez E, Ollagnier-de-Choudens S. Adenosylmethionine as a source of 5'-deoxyadenosyl radicals. Curr Opin Chem Biol. 2001;5:506–511. doi: 10.1016/s1367-5931(00)00237-4. [DOI] [PubMed] [Google Scholar]
- 62.Cosper MM, Cosper NJ, Hong W, Shokes JE, Broderick WE, Broderick JB, Johnson MK, Scott RA. Structural studies of the interaction of S-adenosylmethionine with the [4Fe-4S] clusters in biotin synthase and pyruvate formate-lyase activating enzyme. Prot Sci. 2003;12:1573–1577. doi: 10.1110/ps.0302203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe 4S]+ cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 64.Buis JM, Broderick JB. Pyruvate formate-lyase activating enzyme: elucidation of a novel mechanism for glycyl radical formation. Arch Biochem Biophys. 2005;433:288–296. doi: 10.1016/j.abb.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 65.Mulder DW, Ortillo DO, Gardenghi DJ, Naumov AV, Ruebush SS, Szilagyi RK, Huynh B, Broderick JB, Peters JW. Activation of HydA(Delta EFG) requires a preformed 4Fe-4S cluster. Biochemistry. 2009;48:6240–6248. doi: 10.1021/bi9000563. [DOI] [PubMed] [Google Scholar]
- 66.Pilet E, Nicolet Y, Mathevon C, Douki T, Fontecilla-Camps JC, Fontecave M. The role of the maturase HydG in FeFe -hydrogenase active site synthesis and assembly. FEBS Lett. 2009;583:506–511. doi: 10.1016/j.febslet.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 67.Posewitz MC, King PW, Smolinski SL, Zhang LP, Seibert M, Ghirardi ML. Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active Fe hydrogenase. J Biol Chem. 2004;279:25711–25720. doi: 10.1074/jbc.M403206200. [DOI] [PubMed] [Google Scholar]
- 68.Mulder DW, Shepard EM, Meuser JE, Joshi N, King PW, Posewitz MC, Broderick JB, Peters JW. Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure. 2011;19:1038–1052. doi: 10.1016/j.str.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 69.Nicolet Y, Martin L, Tron C, Fontecilla-Camps JC. A glycyl free radical as the precursor in the synthesis of carbon monoxide and cyanide by the [FeFe]-hydrogenase maturase HydG. FEBS Lett. 2010;584:4197–4202. doi: 10.1016/j.febslet.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 70.Silakov A, Wenk B, Reijerse E, Lubitz W. 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge. Phys Chem Chem Phys. 2009;11:6592–6599. doi: 10.1039/b905841a. [DOI] [PubMed] [Google Scholar]
- 71.Rubach JK, Brazzolotto X, Gaillard J, Fontecave M. Biochemical characterization of the HydE and HydG iron-only hydrogenase maturation enzymes from Thermatoga maritima. FEBS Lett. 2005;579:5055–5060. doi: 10.1016/j.febslet.2005.07.092. [DOI] [PubMed] [Google Scholar]
- 72.Böck A, King PW, Blokesch M, Posewitz MC. Maturation of hydrogenases. In: Robert KP, editor. Advances in Microbial Physiology. Vol. 51. Academic Press; 2006. pp. 1–225. [DOI] [PubMed] [Google Scholar]
- 73.Kampmeier JA. Regioselectivity in the homolytic cleavage of S-adenosylmethionine. Biochemistry. 2010;49:10770–10772. doi: 10.1021/bi101509u. [DOI] [PubMed] [Google Scholar]
- 74.Ollagnier S, Mulliez E, Schmidt PP, Eliasson R, Gaillard J, Deronzier C, Bergman T, Gräslund A, Reichard P, Fontecave M. Activation of the anaerobic ribonucleotide reductase from Escherichia coli. The essential role of the iron-sulfur center for S-adenosylmethionine reduction. J Biol Chem. 1997;272:24216–24223. doi: 10.1074/jbc.272.39.24216. [DOI] [PubMed] [Google Scholar]
- 75.Ollagnier-de-Choudens S, Sanakis Y, Hewitson KS, Roach P, Münck E, Fontecave M. Reductive cleavage of S-adenosylmethionine by biotin synthase from Escherichia coli. J Biol Chem. 2002;277:13449–13454. doi: 10.1074/jbc.M111324200. [DOI] [PubMed] [Google Scholar]
- 76.Padovani D, Thomas F, Trautwein AX, Mulliez E, Fontecave M. Activation of class III ribonucleotide reductase from E. coli. The electron transfer from the iron-sulfur center to S-adenosylmethionine. Biochemistry. 2001;40:6713–6719. doi: 10.1021/bi002936q. [DOI] [PubMed] [Google Scholar]
- 77.Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- 78.Wagner AFV, Frey M, Neugebauer FA, Schäfer W, Knappe J. The free radical in pyruvate formate-lyase is located on glycine-734. Proc Natl Acad Sci USA. 1992;89:996–1000. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Knappe J, Neugebauer FA, Blaschkowski HP, Gänzler M. Post-translational activation introduces a free radical into pyruvate formate-lyase. Proc Natl Acad Sci USA. 1984;81:1332–1335. doi: 10.1073/pnas.81.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Logan DT, Andersson J, Sjöberg BM, Nordlund P. A gycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science. 1999;283:1499–1504. doi: 10.1126/science.283.5407.1499. [DOI] [PubMed] [Google Scholar]
- 81.Becker A, Fritz-Wolf K, Kabsch W, Knappe J, Schultz S, Wagner AFV. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat Struct Mol Biol. 1999;6:969–975. doi: 10.1038/13341. [DOI] [PubMed] [Google Scholar]
- 82.Leppänen VM, Merckel MC, Ollis DL, Wong KK, Kozarich JW, Goldman A. Pyruvate formate lyase is structurally homologous to type I ribonucleotide reductase. Structure. 1999;7:733–744. doi: 10.1016/S0969-2126(99)80098-7. [DOI] [PubMed] [Google Scholar]
- 83.Peng Y, Veneziano SE, Gillispie GD, Broderick JB. Pyruvate formate-lyase, evidence for an open conformation favored in the presence of its activating enzyme. J Biol Chem. 2010;285:27224–27231. doi: 10.1074/jbc.M109.096875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating enzyme: a mössbauer spectroscopic study. J Am Chem Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 85.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 86.Broderick JB, Duderstadt RE, Fernandez DC, Wojtuszewski K, Henshaw TF, Johnson MK. Pyruvate formate-lyase activating enzyme is an iron-sulfur protein. J Am Chem Soc. 1997;119:7396–7397. [Google Scholar]
- 87.Yan F, LaMarre JM, Röhrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG. RlmN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J Am Chem Soc. 2010;132:3953–3964. doi: 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang Q, van der Donk WA, Liu W. Radical-mediated enzymatic methylation: a tale of two SAMS. Acc Chem Res. 2011 doi: 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grove TL, Radle MI, Krebs C, Booker SJ. Cfr and RlmN contain a single [4Fe-4S] cluster, which directs two distinct reactivities for S-adenosylmethionine: methyl transfer by SN2 displacement and radical generation. J Am Chem Soc. 2011;133:19586–19589. doi: 10.1021/ja207327v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boal AK, Grove TL, McLaughlin MI, Yennawar NH, Booker SJ, Rosenzweig AC. Structural basis for methyl transfer by a radical SAM enzyme. Science. 2011;332:1089–1092. doi: 10.1126/science.1205358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shao X, Grishin NV. Common fold in helix-hairpin-helix proteins. Nuc Acids Res. 2000;28:2643–2650. doi: 10.1093/nar/28.14.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quitterer F, List A, Eisenreich W, Bacher A, Groll M. Crystal structure of methylornithine synthase (PyIB): Insights into the pyrrolysine biosynthesis. Angew Chem, Int Ed. 2012;51:1339–1342. doi: 10.1002/anie.201106765. [DOI] [PubMed] [Google Scholar]
- 93.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.