

Abstract
G protein-coupled receptors (GPCRs) represent a large family of signaling proteins that includes many therapeutic targets; however, progress in identifying new small molecule drugs has been disappointing. The past four years have seen remarkable progress in the structural biology of GPCRs, raising the possibility of applying structure-based approaches to GPCR drug discovery efforts. Of the various structure-based approaches that have been applied to soluble protein targets, such as proteases and kinases, in silico docking is among the most ready applicable to GPCRs. Early studies suggest that GPCR binding pockets are well suited to docking, and docking screens have identified potent and novel compounds for these targets. This review will focus on the current state of in silico docking for GPCRs.
Introduction
GPCRs mediate cellular responses to the majority of hormones and neurotransmitters, and are therefore attractive targets for drug discovery. In the decade following the cloning of the first genes and cDNAs for GPCRs[1, 2], there was great hope that these discoveries would rapidly translate into new and more effective therapeutics. Cloning and later mining the human genome sequence led to the identification of new GPCR subtypes [3] and the establishment of cell lines that could be used for high-throughput screening (HTS) of large compound libraries. The identification of polymorphisms for specific GPCRs suggested the potential for individualized medicines. Unfortunately, the promise of new drugs for new GPCR targets, or safer and more effective drugs for previously identified targets has largely gone unfulfilled [4].
Several reasons may explain the slow pace of drug discovery in the face of more targets and screening modalities. If the advent of the molecular era gave us unprecedented tools and abundant targets, it also disrupted the integrated, tissue-based pharmacology of the classical era of drug discovery [5, 6]; the underlying biology was more complicated than anticipated by the reductionist, molecular view. Many GPCRs signal through multiple pathways, often in a ligand-specific manner. For example, the β2 adrenergic receptor (β2AR) activates specific cellular signaling pathways through Gs, the stimulatory G protein for adenylyl cyclase, and independently through arrestin.
Carvedilol is an inverse agonsit for β2AR activation of Gs, but a partial agonist for activation of arrestin [7]. HTS may not reflect the physiologically relevant signaling pathway [8]. Not only do we need to identify the correct GPCR target and signaling pathway, we must find a drug with the appropriate efficacy profile: agonist, partial agonist, neutral antagonist and inverse agonist. Drugs that satisfy these criteria must then pass through a gauntlet of assays to assess toxicology and pharmacokinetics. For this and other reasons, the cost of drug development has escalated while revenue from new drugs has slipped [9]. Consequently, some pharmaceutical companies are abandoning small molecule development programs in favor of biologics [10] and the cost of the few new drugs that make it to the market will further escalate the cost of healthcare.
In, 2007 we entered the new era of GPCR structural biology. Since the initial crystal structures of the β2AR[11] and the β1AR[12], the number of published GPCRs which have yielded to crystallography has grown to ten and includes the adenosine A2A receptor[13], the D3 dopamine receptor[14], the CXCR4 receptor [15], the histamine H1 receptor, [16], the sphingosine 1 phosphate receptor [17], the M2 and M3 muscarinic receptors [18, 19], and the mu opioid receptor [20], with at least two new structures anticipated in 2012. This is largely attributable to the application of high-throughput methods for lipidic cubic phase (LCP) crystallography [21] and protein engineering with GPCR-T4 lysozyme[11, 22] and thermostabilization[23] methods being generally applicable to structurally diverse GPCRs. Although structural biology is not a panacea for the challenges described above, there is reason to hope that GPCR crystal structures can facilitate drug discovery based on success with soluble protein targets such as kinases and proteases. In this review we will discuss the application of structure-based screens of large compound libraries to GPCR drug discovery.
Structure-based screens for new ligands
Structure-based design has been pivotal in the development of over ten marketed drugs, including recent successes against renin with aliskiren [24] and against hepatitis C virus protease with telapravir [25], and has contributed to the development of multiple others, since the technique came into widespread use in the 1990s. Although this is far fewer than initially promised by advocates of the technique, it is likely larger than the number of drugs whose origins can be traced directly to HTS[6, 26], the dominant technique for new ligand discovery in pharmaceutical research, and has contributed especially to drugs for new targets. Protein structures have contributed in two ways to drug development: guiding the optimization of lead candidates, and enabling the discovery of new chemical series, the latter using molecular docking and related techniques. Whereas structure has arguably had the greater impact on lead optimization in pharmaceutical research, it remains too early to evaluate the impact the new GPCR structures have had on this area, because most of these efforts remain closely held. Conversely, the impact of the new GPCR structures on docking screens has been immediate, with active molecules not only returned with high hit rates, but characterized by substantial novelty, as reflected by the new chemical scaffolds discovered, and potency against each of the four GPCRs targeted thus far in the literature (Table 1, Figure 1). In the case of the new β2-AR ligands, not only were several of the new hits potent, but the most active of them is among the most effective inverse agonist ever found for the receptor, depressing basal activity even more than the standard in the field, ICI-118,551.
Table 1.
Docking hit rates against GPCRs.
Figure 1.

New inverse agonists against the β2AR structure discovered by molecular docking. Top row: four relatively potent ligands that resemble known chemotypes, with Ki values from 9 nM to 2 uM. Bottom row: two novel chemotypes, with Ki values of 1.1 and 3.3 uM.
Structure-based docking begins with a protein structure, either determined experimentally or modeled by homology, a large library of available small organic molecules, and a docking program that fits the two together. The insistence on screening available, typically purchasable, ensures rapid cycles of modeling and experimental testing. For many years the molecular databases and docking programs remained the domain of specialists, but for the last several years databases of over ten million, commercially available small molecules, such as ZINC (zinc.docking.org) [27], have been freely available, as have web-based docking applications (blaster.docking.org) (Box 1), lowering the barriers to entry.
Box 1. Key steps in structure-based docking screens against GPCRs.
Widely used docking programs include AutoDock,[47] ICM,[48] FlexX,[49] Glide,[50] and DOCK.[51, 52] A web-based, automated version of the last is available at http://blaster.docking.org.
A library of small molecules to dock against the GPCR. Over 10 million commercially available molecules, with structures, charges, molecular properties, and links to suppliers, is available for free at http://zinc.docking.org.
Defining the docking site. Optimization of the structure for residue conformations, hydrogen positions, inclusion or exclusion of ordered water molecules. For physics-based programs, calculation of molecular potential functions for the target site.
Control calculations establish to rank known ligands higher than decoy molecules.[53] Such controls are easy to undertake and guide the optimization of parameters for new compounds. Known ligands may often be found from ChEMBL www.ebi.ac.uk/chembl/.
Screen the library against the binding site. We often choose a subset of compounds with “lead-like” properties[54] to dock; these molecules are smaller and often better behaved than the larger “drug-like” molecules, and allow for optimization. Over 3 million are commercially available.
From the top 0.01 to 0.1% of the docking ranked list, pick 25 to 50 to test. This is done by eye, selecting for or against features that the docking scoring function might under- or overweight. This includes complementation of ligand polar groups by the protein, picking diverse chemotypes, balancing between polar and non-polar complementarity.
Careful assays to ensure that any “hits” that emerge are well-behaved, e.g., are reversible, with good dose-response curves, compete with a classical ligand, if available, and do not form colloidal aggregates. [55, 56]
Docking results against the new GPCR structures are given context by comparison to those against soluble proteins. In unbiased screens for new ligands against soluble proteins, docking hit-rates have been about 5 to 10% of the molecules tested, with a hit typically defined as a molecule with micromolar to mid-micromolar affinity. Though two to three logs better than typical HTS hit rates,[28–30] the docking hit rates and the affinity of the hits against soluble proteins are two to three log-orders worse again than those against GPCRs to date. Two factors may contribute to this unusual success rate against GPCRs. First, even supposedly unbiased libraries like ZINC in fact contain a disproportionate number of molecules that resemble GPCR ligands,[31] partly owing to the longstanding interest in these receptors among medicinal chemists. Second, the well-buried GPCR orthosteric sites can almost entirely sequester and complement a small organic molecule, allowing them to recognize small molecules with high ligand efficiency (potency/size). By contrast, for many soluble proteins, there is little certainty that the “right” molecules exist to be found in our libraries,[31, 32] and their binding sites are often larger, flatter and more exposed to solvent than the orthosteric sites of those GPCRs determined to date.
Expanding structural coverage with homology models
As of this writing, we are aware of twelve GPCRs whose structures have been determined; including published and unpublished structures. If this represents a great expansion from only four years ago, it still represents only 4% of the pharmacologically relevant GPCRs, considering that there are approximately 300 non-olfactory GPCRs in the human genome[3]. Even with the current pace of structure determination, for the foreseeable future there are likely to be many fewer structures than good targets for structure-based discovery. For many, the target of particular interest is likely to lack an experimental structure, and there will be a temptation to model a structure based on homology to a structurally determined template. How likely is a model-built structure to lead to a successful screen for new ligands?
This question was taken up in a recent prospective comparison of the dopamine D3 receptor. The community had been challenged to predict D3 dopamine receptor/eticlopride complex, as well as complexes involving the CXCR4 receptor, before their x-ray structure were released [33, 34]. This competition provided an opportunity to prospectively compare docking against a homology model to that against the x-ray structure of the same receptor, once released. As in earlier docking screens against the β2 adrenergic and the A2a adenosine receptors, the screen against the D3 receptor had a high hit rate, 23%, with affinities as good as 200 nM. To our surprise, the docking screen against the homology model, whose predictions were tested before the x-ray structure was released, had hit rates, affinities, and novel structures as good as those against the experimental structure (Table 1)[35]. This suggests that targets that have sequence identities of 35% or greater in the trans-membrane region to GPCRs of known structure may be well enough modeled to allow reliable docking screens, thereby increasing the impact of experimentally determined structures.
Efficacy
State-specific structures
All of the initial GPCR structures captured the receptors in inactive states, and in silico screens against the inactive state structure of the β2AR [32], the A2a adenosine [31, 36], and the D3 dopamine receptor [35] yielded only antagonists and inverse agonists. Whereas retrospective modeling suggested that modest manipulation of the inactive states led to recognition of agonists in docking studies [37, 38], prospective screens—where new molecules were tested—returned no agonists whatsoever. Thus there has been great interest in determining active state structures, but these have been more challenging because, for many GPCRs, agonists alone do not fully stabilize the active state[39].
A common property of many GPCRs is that G protein coupling enhances agonist binding affinity through an allosteric mechanism. For example, the affinity of the β2AR for the agonist isoproterenol is 100 fold greater when coupled to the G protein Gs[40, 41]. As such, stabilization of the cytoplasmic domains by a G protein (or a G protein mimetic binding protein) may be necessary for obtaining the structure of an active-state binding pocket. This was the case for the β2AR, where even a covalent agonist failed to fully stabilize an active conformation[39]. The first active state structure of the β2AR required stabilization with a G protein mimetic camelid single-chain antibody fragment, also known as a nanobody (Nb80)[42]. Like the G protein Gs, Nb80 bound to the β2AR in an agonist dependent manner and stabilized the same high affinity state for the agonist isoproterenol. The structure of the binding pocket of the β2AR-Nb80 complex is indistinguishable from that observed for the recent β2AR-Gs crystal structure[43]. A comparison of active and inactive state binding pockets for the β2AR reveals relatively small changes when individual amino acids are considered; however, the overall volume of the active state binding pocket is reduced relative to the inactive state (Figure 2). It will be fascinating to learn whether docking hits against this activated state will be dominated by agonists as docking against the inactive state was dominated by antagonists. This would suggest that the state of the receptor structure strongly influences the efficacies of the ligands that emerge from the docking screens, with antagonists dominating screens against the inactivated states. Preliminary studies from our labs hint that this may be the case.
Figure 2.

Comparison of the binding pocket of the β2AR in the inactive (carbons in blue) and active (carbons in orange) conformations. The inverse agonist carazolol is shown with carbons in green. The largest difference is observed around Ser207 in transmembrane segment (TM) 5.
As noted above, there is evidence that GPCRs have more than one active state. Several GPCRs couple to more than one G protein and signal through G protein independent pathways in a ligand-specific manner. It is likely that the differences in the orthosteric site responsible for this pathway selective signaling are more subtle than what is observed between inactive and actives states of the β2AR. These small structural differences may not be captured by crystallography. Although this may limit the application of in silico screening, it is also possible that the evolution of initial hits through iterative rounds of modifications to improve selectivity and specificity may produce pathway selective ligands. Finally, it is conceivable that determination of the structure of a GPCR in complex with a non-G-protein partner, such as β-arrestin, may illuminate still other conformations of the receptor that may be directly exploited for pathway-specific ligand discovery.
Concluding remarks
When the field began docking against the new GPCR structures, it was uncertain whether the technique would find potent and novel chemical matter at all. What we have learned over the last three years is that GPCRs are unusually well-suited to docking screens, returning molecules whose affinities and hit-rates are several logs better than we have come to expect in docking against soluble proteins. The campaigns to date have focused on technical features of hit rates, affinities, compound novelty, and the use of models, rather than the discovery of chemical probes and leads as ends in themselves. As the field matures, discovering such chemical matter will become more important. The new GPCR structures open avenues to molecules biased to particular signaling pathways and to molecules specific for particular receptors; such bias and specificity in turn enable the exploration of new biology and new therapies. Exploiting this opportunity will demand not only the collaborations between structural-biologists, computational chemists and molecular pharmacologists, but also animal pharmacologists, medicinal chemists, and signaling biologists. Such close engagement has perhaps been underemphasized in the era of molecular pharmacology, contributing to the lower than expected rate of drugs that exploit the target-based approach. With targets as rich in biology and disease as GPCRs, we can re-imagine a pharmacology that brings to bear the structural tools of the modern era with the integrated approach of the classical period.[5] For structural biology to have the greatest impact drug discovery, it will be necessary to have iterative cycles of docking, verification of ligand-receptor interactions by structure determination, and lead optimization (Figure 3). This not currently practicable for most GPCR targets where the quality of the structures is often dependent on the affinity of the bound ligand, and the time required to acquire structures is often longer than needed for the highly competitive pace of most drug discovery efforts. Nevertheless, given the remarkable progress over the past several years and the flood of new structures, structural biology will likely become an integral part of many new drug discovery efforts [44].
Figure 3.
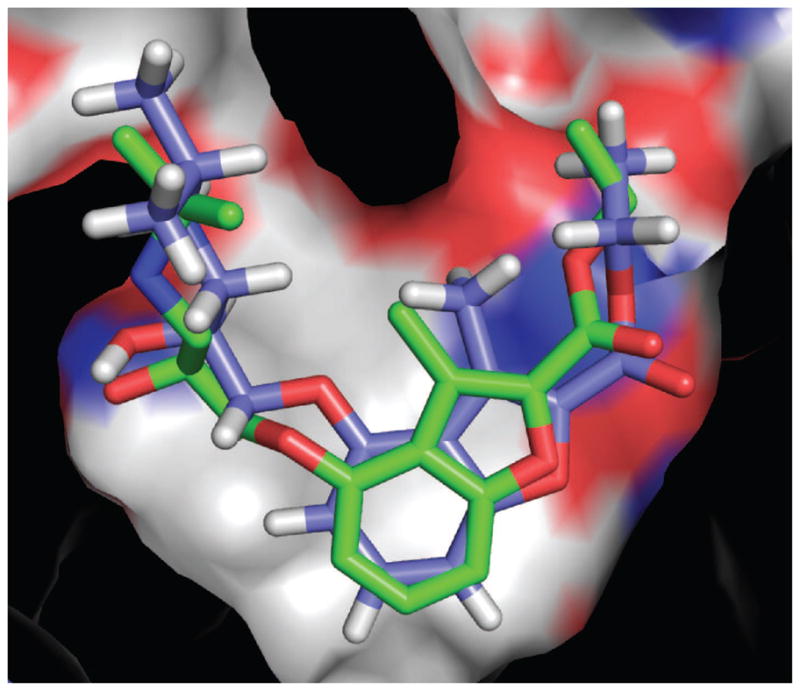
A 9 nM inverse agonist of β2AR discovered by docking: docked orientation (carbons in blue) superposed on the subsequent x-ray crystallographic result (carbons in green).[46]
Acknowledgments
Supported by GM59957 and GM72970 (PI R. Altman) (to BKS) and NS028471 and GM083118 (to BKK). We thank R. Coleman for sequence identity calculations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited: Bibliography
- 1.Dixon RA, et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–79. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 2.Kubo T, et al. Cloning, sequencing and expression of complementary DNA encoding the muscarinic acetylcholine receptor. Nature. 1986;323:411–416. doi: 10.1038/323411a0. [DOI] [PubMed] [Google Scholar]
- 3.Fredriksson R, et al. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 4.Gribbon P, Sewing A. High-throughput drug discovery: what can we expect from HTS? Drug Discov Today. 2005;10:17–22. doi: 10.1016/S1359-6446(04)03275-1. [DOI] [PubMed] [Google Scholar]
- 5.Keiser MJ, et al. The chemical basis of pharmacology. Biochemistry. 2010;49:10267–10276. doi: 10.1021/bi101540g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gleeson MP, et al. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov. 2011;10:197–208. doi: 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajagopal S, et al. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenakin T. Predicting therapeutic value in the lead optimization phase of drug discovery. Nat Rev Drug Discov. 2003;2:429–438. doi: 10.1038/nrd1110. [DOI] [PubMed] [Google Scholar]
- 9.Paul SM, et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 10.Arrowsmith J. A decade of change. Nat Rev Drug Discov. 2012;11:17–18. doi: 10.1038/nrd3630. [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 12.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaakola VP, et al. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science. 2008 doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu B, et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science. 2010 doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanson MA, et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haga K, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruse AC, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manglik A, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012 doi: 10.1038/nature10954. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherezov V, et al. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallogr D Biol Crystallogr. 2004;60:1795–1807. doi: 10.1107/S0907444904019109. [DOI] [PubMed] [Google Scholar]
- 22.Caffrey M. Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annu Rev Biophys. 2009;38:29–51. doi: 10.1146/annurev.biophys.050708.133655. [DOI] [PubMed] [Google Scholar]
- 23.Serrano-Vega MJ, et al. Conformational thermostabilization of the beta 1-adrenergic receptor in a detergent-resistant form. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahuel J, et al. Structure-based drug design: the discovery of novel nonpeptide orally active inhibitors of human renin. Chem Biol. 2000;7:493–504. doi: 10.1016/s1074-5521(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 25.Lin C, et al. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infectious disorders drug targets. 2006;6:3–16. doi: 10.2174/187152606776056706. [DOI] [PubMed] [Google Scholar]
- 26.Macarron R, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 27.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doman TN, et al. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J Med Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 29.Babaoglu K, et al. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J Med Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira RS, et al. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J Med Chem. 2010;53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlsson J, et al. Structure-based discovery of A2A adenosine receptor ligands. J Med Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolb P, et al. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci U S A. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kufareva I, et al. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam AR, et al. Importance of receptor flexibility in binding of cyclam compounds to the chemokine receptor CXCR4. J Chem Inf Model. 2011;51:139–147. doi: 10.1021/ci1003027. [DOI] [PubMed] [Google Scholar]
- 35.Carlsson J, et al. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katritch V, et al. Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. J Med Chem. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds KA, et al. Identifying conformational changes of the beta(2) adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. J Comput Aided Mol Des. 2009;23:273–288. doi: 10.1007/s10822-008-9257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J Med Chem. 2008;51:4978–4985. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- 39.Rosenbaum DM, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whorton MR, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Lean A, et al. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 42.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Congreve M, et al. The use of GPCR structures in drug design. Adv Pharmacol. 2011;62:1–36. doi: 10.1016/B978-0-12-385952-5.00011-7. [DOI] [PubMed] [Google Scholar]
- 45.de Graaf C, et al. Crystal Structure-Based Virtual Screening for Fragment-like Ligands of the Human Histamine H(1) Receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wacker D, et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris GM, et al. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008;Chapter 8(Unit 8):14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez-Recio J, et al. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins. 2003;52:113–117. doi: 10.1002/prot.10383. [DOI] [PubMed] [Google Scholar]
- 49.Kamper A, et al. Fully automated flexible docking of ligands into flexible synthetic receptors using forward and inverse docking strategies. J Chem Inf Model. 2006;46:903–911. doi: 10.1021/ci050467z. [DOI] [PubMed] [Google Scholar]
- 50.Repasky MP, et al. Flexible ligand docking with Glide. Curr Protoc Bioinformatics. 2007;Chapter 8(Unit 8):12. doi: 10.1002/0471250953.bi0812s18. [DOI] [PubMed] [Google Scholar]
- 51.Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 52.Irwin JJ, et al. Automated docking screens: a feasibility study. J Med Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang N, et al. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oprea TI. Current trends in lead discovery: are we looking for the appropriate properties? Mol Divers. 2002;5:199–208. doi: 10.1023/a:1021368007777. [DOI] [PubMed] [Google Scholar]
- 55.McGovern SL, et al. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 56.Coan KE, et al. Promiscuous aggregate-based inhibitors promote enzyme unfolding. J Med Chem. 2009;52:2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]