

Abstract
Long-chain polyacetylene alcohols, faulknerynes A–C, were isolated from two specimens of the encrusting sponge, Diplastrella sp., collected from the surface of coral in the Bahamas, along with known compounds diplynes A, C and E. Two CD methods were critically evaluated for their suitability to terminal propargylic glycols and applied to assignment of configurations of faulkneryne A and diplyne C.
Introduction
Long chain polyacetylenic alcohols (PAAs) are characteristic natural products of Haplosclerid and Spirastrellid sponges, including the genera Diplastrella,1 Petrosia, Siphonochalina, Haliclona2 and Cribochalina.3 Among the first to be reported were the highly cytotoxic duryne3 and the HIV reverse-transciptase inhibitor, petrosynol4 from Cribochalina and Petrosia, respectively. Many PAAs are exceedingly cytotoxic towards tumor cell lines (IC50’s ≤ 30 ng/mL), a biological property that has been correlated with the presence of a terminal propargylic alcohol.2 Faulkner and coworkers reported C16 brominated diynes A, B, C (1), E (2) and three related sulfate half-esters from the Philippines sponge Diplastrella sp.1 Fusetani and coworkers described the C23 polynols, callyspongiols A–E, along with related hydrocarbons, from Callyspongia truncata,5 and siphonodiol (3) from Siphonochalina truncata.6,7 In a screen for compounds with antiproliferative activity, extracts of two specimens of the Bahamian sponge, Diplastrella sp., were found to elicit moderate in vitro cytotoxicity against the cultured tumor cell line HCT-116. We describe new brominated long-chain PAAs, faulknerynes A–C (4a–c), from these extracts and assignment of the absolute configuration of 1 and 4a using two microscale methods based on exciton coupling circular dichroism (ECCD). We also establish that 1 obtained from Diplastrella sp. from the Bahamas and the Philipines are both R.1
Results and Discussion
A sample of the thinly-encrusting Diplastrella sp., recovered from small patches, laboriously scraped from coral substrate with a knife, was exhaustively extracted with MeOH. The MeOH extract was progressively adjusted in water content and partitioned against solvents of increasing polarity (hexane, CHCl3 and n-BuOH). The CHCl3-soluble fraction was further purified to give the known diplyne C (1, identified from NMR data1) and new natural products, faulknerynes A–C (4a–c).8 The formula, C16H21BrO2 for 4a was secured from HRMS measurements ([M+Na]+, 347.0633 Δmmu = 1.6). Examination of the 1H NMR spectrum of 4a (Table 1) readily revealed chemical shifts of the two chain termini; a 1,2-disubstituted vinyl bromide (δ 6.01, d, J = 13.5 Hz; 6.17, dt, J 13.5, 7.3 Hz) and a mutually coupled ABX system corresponding to a 1,2-glycol (δ 3.72, dd, J = 11.4, 6.2 Hz, H1a; δ 3.78, dd, J = 11.4, 3.8 Hz, H1b; 4.57, dd, J = 6.2, 3.8, H2). The formula required an additional five degrees of unsaturation which were assigned to a conjugated Z-1,3,5-ene-diyne system, based on a red-shifted UV spectrum [4a; λmax 271 nm] compared to 1 and 3 [3; λmax 228 (ε 15,100)6], the presence of four acetylenic 13C NMR sp signals (δ 71.1 s, 76.5 s, 76.8 s, 80.1 s) and 1H NMR signals ascribed to a Z-1,2-disubstituted double bond, C7-C8 (δ 5.49, d, J = 10.9 Hz; δ 6.11, dt, J = 10.9, 7.6 Hz) conjugated to the diyne. HMBC crosspeaks from H7 to the acetylenic carbon signals of C3-C5 corroborated UV evidence that the Z-double bond was conjugated to the diyne. The remaining signals were assigned to allylic CH2 groups and an unresolved methylene chain. Therefore, 4a is Z-7,8-dehydro-1.
Table 1.
NMR data (CDCl3) for faulkneryne A (4a)
| no. | δC, mult.a | δH, mult. (J in Hz)b | COSYb | HMBC (H→C) |
|---|---|---|---|---|
| 1 | 66.4, CH | 3.72, dd (11.4, 6.2) | H2 | C2, C3 |
| 66.4, CH | 3.78, dd (11.4, 3.8) | H2 | C3 | |
| 2 | 64.0, CH | 4.57, dd (6.2, 3.8) | H1, H7 | C1, C3, C4, C5 |
| 3 | 80.1, C | |||
| 4 | 71.1, C | |||
| 5 | 76.8, Cd | |||
| 6 | 76.5, Cd | |||
| 7 | 107.8, CH | 5.49, d (10.9) | H2, H8, H9 | C3, C4, C5, C8, C9 |
| 8 | 149.5, CH | 6.11, dt (10.9, 7.6) | H7, H9 | C6, C7, C9 |
| 9 | 30.9, CH2 | 2.33, (7.6) | H7, H8, H10 | C6, C7, C8, C11 |
| 10 | 28.7, CH2e | 1.41, m | H9, H11 | C8, C9, C11 |
| 11 | 28.8, CH2e | 1.30, m | H10, H12 | C12 |
| 12 | 28.9, CH2 | 1.31, m | H11, H13 | C11 |
| 13 | 28.6, CH2 | 1.39, m | H12, H14 | C14, C15 |
| 14 | 33.0, CH2 | 2.03, m (7.3) | H13, H15, H16 | C12, C15, C16 |
| 15 | 138.3, CH | 6.17, dt (13.5, 7.3) | H14, H16 | C13, C14, C16 |
| 16 | 104.3, CH | 6.01, d (13.5) | H14, H15 | C14, C15 |
125 MHz.
600 MHz.
Multiplicity assigned from HSQC.
Peaks may be interchanged.
Faulknerynes B (4b) and C (4c) (C16H1779BrO2, m/z 343.0304 [M+Na]+ Δmmu = 0.5), obtained in amounts of 4 μg and 14 μg, respectively, are dehydro-derivatives of 2 and isomeric with each other. Analysis of COSY spectrum and vicinal J values of the vinyl proton signals reveal that the two compounds differ only in the location of the Z-carbon-carbon double bond; in 4b, it is positioned at C-7–C-8 while compound 4c has a 11-Z double bond in conjugation with the terminal ene-yne. Therefore, 4b is C-13,14-di-dehydro faulkneryne A and 4c is the 11-Z- geometrical isomer of 11-E diplyne E (2).
With diplyne C (1) from Diplastrella spp. collected in two different oceans– one from the Pacific, the other from the Atlantic – an opportunity presented itself for configuration analysis by chiroptical comparison, although not without some challenges. The optical rotation of natural 1 was not reported, although a total syntheses of (S)-(+)-1 ([α]D +13.3)9 and unnatural (S)-(+)-diplyne A ([α]D +9.6)10 and chiroptical comparison with levorotatory diplyne A ([α]D −8.7)1 would suggest an R configuration for both diplynes A and C (1). Insufficient amounts of 4a–c were available to record rotations for reliable comparison of [α]D with 1 or 3 so attention was turned to alternative methods. Configurational assignment of secondary alcohols is frequently carried out by NMR using the modified Mosher’s method, 11 however, the limited amount of available sample necessitated deployment of sensitive CD spectroscopy; a method well suited to ‘nanomole-scale’ natural products investigation.12 The stereochemistry of siphonodiol (R)-(−)-(3) was assigned by Fusetani and coworkers using the dibenzoate exciton coupling method; specifically, the observation of a negative bisignate Cotton effect, CE [λ 324 nm (Δε −25.3); 303 (+24.2)] in the exciton coupled CD spectrum of 3a, the derived 4-dimethylaminobenzoate diester (DMB diester).9 From exciton coupling theory,13 the sign of the split CE is predicted from the helicity (twist) of the electronic dipole transition vectors associated with the prominent 1La charge-transfer transition in degenerate benzoate chromophores. These vectors are approximately aligned with the corresponding C-O bond directions; a negative sign for the long-wavelength band of the split CE for 3a indicates a negative helicity of the two C-O vectors and a positive split CE indicates a positive helicity. The strength of the ECCD signal depends upon several other factors and is conveniently reported as the difference between maximum and minimum of the bisignate split CE [e.g. for 3a, A = 24.2 −(−25.3) = 49.5].13
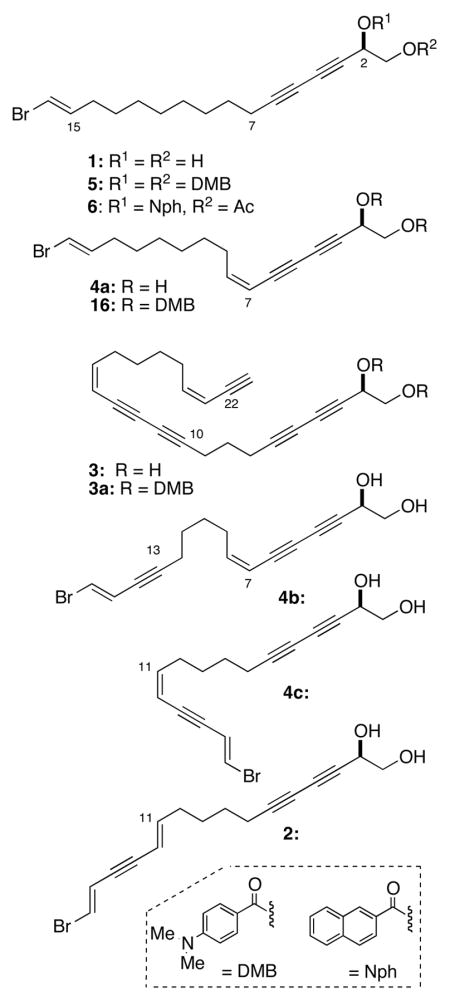
Glycol dibenzoate ECCD is dependent upon the dihedral angle of O-C-C-O around C1-C2. Since acyclic 1,2-glycols have a freely rotating C1-C2 bond, the absolute configurational assignment requires knowledge of the limiting staggered conformers. We were concerned about two implicit assumptions used in the assignment of 3, and consequently, our approach to 1 and 4a; a low barrier to rotation of the C1-C2 bond and similar energies of staggered conformers due to the presence of the relatively sterically unencumbered acetylenic group, and additional exciton coupling from the diyne chromophore with the propargylic DMB chromophore. Dibenzoate esters of 1–4 would be expected to exhibit both types of dichromophoric interactions: yne-benzoate and benzoate-benzoate.14 Consequently, room temperature CD measurements of propargylic benzoates would sample significant proportions of populated staggered conformers a–c, with a and c giving rise opposing signs of ECCDs – and small structural differences may even contribute non-staggered conformers that further influence the magnitude or even the sign of the ECCD. In order to quantify some of these expected differences, we calculated the eneriges of the conformers of propargylic 1,2-glycol dibenzoates (Spartan, PM3) and ordered the lowest lying conformers (Figure 1).
Figure 1.
Conformers of propargylic 1,2-glycol O-dibenzoate analogs of 5. (a)–(c) idealized geometries, helicities and signs of contributions to ECCD (Δε). (d)–(f) Calculated conformers of propargylic 1,2-glycol O-dibenzoate analogs of 5 ranked in relative energy, E (kcal.mol−1), θ, the dihedral angle of the C-O bond vectors, and % Boltzmann populations (Spartan 08, semi-empirical PM3, gas phase) (d) 0, −55.6°, (22.3%) (e) 1.29, −84.1°, (13.3%) (f) 1.67, −55.0° (11.4%).. Only the three best ranked conformations are shown (see text). For ease of calculation, the chain is truncated to a 3,5-diyne.
Perhaps not unexpectedly, the potential energy surface of the model propargylic 1,2-benzoate was found to be fairly shallow. Nevertheless, for the dibenzoates of R 1, the three lowest energy conformers depicted in Figure 1(d)–(e) comprising 47% of the Boltzmann distribution, subtended the same negative helicity corresponding to the idealized geometry (a). The torsional angle, θ, ranged from θ −55° to −84.1° about the C-O vectors of the (C=O)O groups and differed only in minor changes in orientation about the benzoate ester bonds. Surprisingly, the nearest conformers with antiperiplanar dispositions (θ ~ 180°) of the two benzoate groups – and expected to give zero contribution to the ECCD – (c.f. idealized geometry of Figure 1(a)) were higher in energy and ranked fifth and sixth; together, they contribute only 13.2% to the population [E = −0.54 kcal.mol−1 (7.0%); E = −0.27 kcal.mol−1 (6.2%)]. The gauche-gauche conformers (not shown, but cf. Figure 1(b)) which is expected to give ECCD of opposing sign to Figure 1(c) were found to be of even lower ranking and less stable by more than 2.8 kcal.mol−1 than the favored conformers; they represented less than 3% in the conformer population.
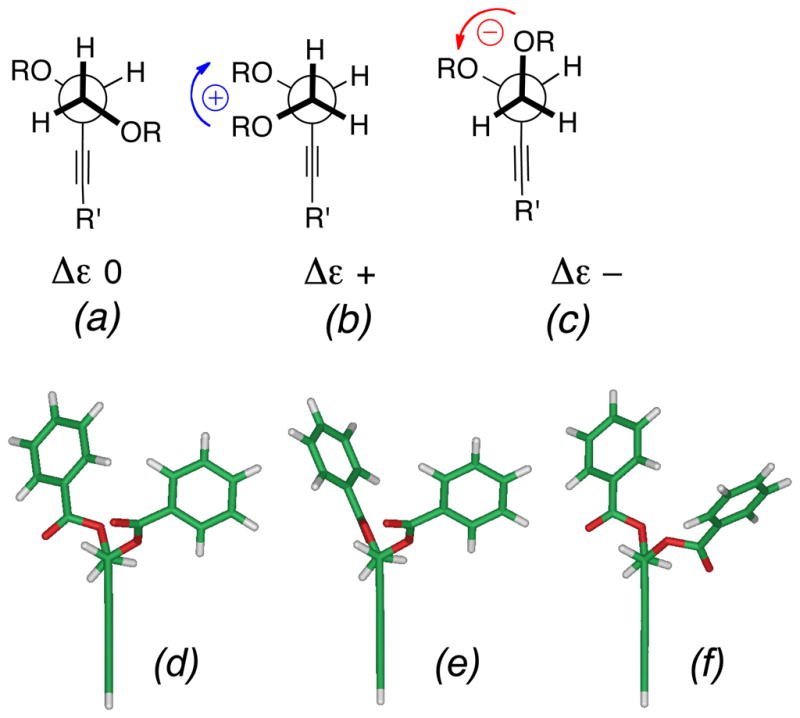
ECCD of arenecarboxylate esters (acylates) of allylic and propargylic alcohols arise from electronic coupling of arene and C-C multiple bond π-π* transitions. Reliable assignments of secondary allylic alcohols have been made from ECCD of the corresponding allylic O-benzoates15 O-naphthoates16, and O-naphthoates of conjugated dienes.17 The model used for interpretation of ECCD in allylic benzoates/naphthoates15 follows from well-understood considerations of allylic strain18 leading to a dominant conformation which is supported by NMR. A large number of examples of chiral allylic benzoates were studied which consistently revealed large vicinal coupling constants (J ~ 9 Hz) for vicinal protons on the adjacent sp2- sp3 carbons that imply the vinyl H is eclipsed by the allylic methine H.15,17 In turn, this sets the torsional angle between the C=C bond and the benzoate C-O vector and the relative helicity of the respective chromophoric transition dipoles. The allylic ECCD method should be extendable to propargylic acylates, however, conformations of the latter are expected to exhibit subtle differences (Figure 2a,b). In both double and triple carbon-carbon bonds the π-π* transition dipoles are polarized along the C-C direction. Unlike the planar C=C bonds in allylic acylates, the radially symmetrical triple bond in a propargylic acylate is essentially a free rotor. A consequence of this, as we showed from molecular mechanics calculations (Spartan, MMFF94, Figure 2c,d), is the dominant conformer places Hα almost syn-coplanar with the conjugated ester Ar(C=O)O group and an angle of ±20° is subtended between the synclinal arenecarboxylate plane and the acetylenic vector. For propargylic esters of (R)-1 (Figure 2c), the respective transition dipoles have a calculated positive helicity (θ = +33.4°), where the dihedral θ is approximated by the dihedral angle, C3-C2-C2′-C-6′ (C2′,C6′ are the ipso and para carbons of the first benzenoid ring of the naphthoate). In turn, this predicts (R)-1 will exhibit a positive Δε in the CD spectrum. Harada has described similar arguments and assignments of alk-1-yn-3-ols by ECCD after a two step derivatization – Sonogashira coupling with a terminal acetylene with an aryl iodide followed by esterification of the secondary alcohol to the corresponding arene-yne benzoate and measurement of CD spectra.19 The sign of the resulting split CE’s conform to the rule, however, the major limitation of the method is its applicability to terminal acetylenes, only.
Figure 2.
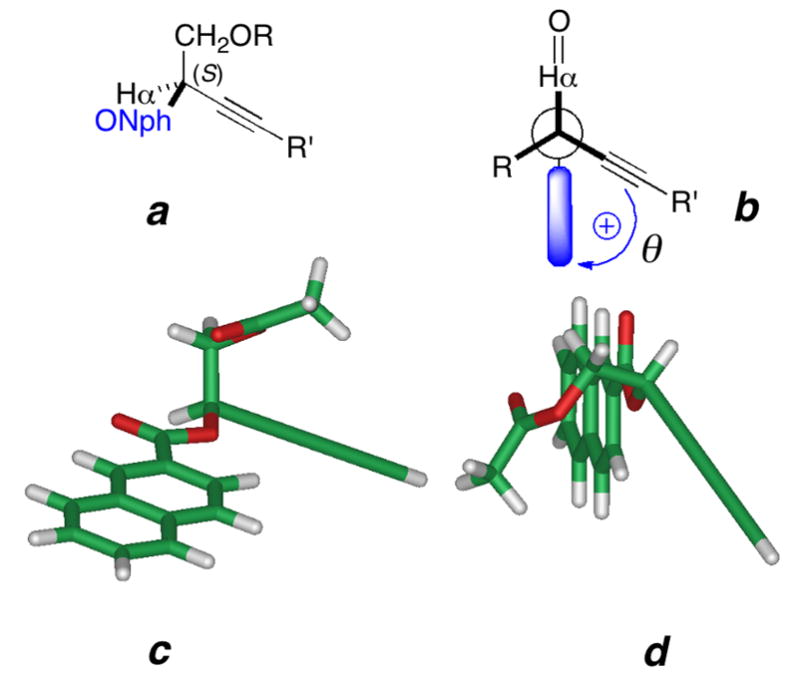
Conformers of propargylic (R)-2-O-(2′-naphthoate) esters of terminal propargylic 1,2-glycols. (a),(b) Idealized geometries (two views) (c.f. 6) and the predicted sign of Δε in the ECCD spectrum. (R = CH2OAc, R′ = alkenyl, alkynyl; Nph = 2′-naphthoyl; shaded bar is 2-naphthyl (C10H11). (c),(d) Two views the energy minimized model (MMFF, Spartan 08) (c.f. 6, however, the chain is truncated to a 3,5-diyne, R′ = ethynyl,) showing a syn-clinal arrangement of the conjugated diyne- and naphthoate plane with a positive helicity (dihedral angle of C-3-C-2-C-2′-C-6′, θ = +33.4°).
An advantage of allylic O-naphthoates over O-benzoates is the very intense Cotton effects (A values of up to ~20017) associated with the 1Bb transition along the skew long axis of the naphthalene ring system, closely parallel with the C-O ester bond, which is better suited for sub-nanomole CD measurements. Unlike vicinal propargylic dibenzoates, interpretation of CD in propargylic naphthoates benefits from the simplicity of only two participant chromophores in exciton coupling.
In order to assess the relative merits of the dibenzoate method and the propargylic O-naphthoate method, we chose to determine the configuration of 1 and 4a using both approaches. Diplyne C (1) was acylated (Scheme 1) (N-(4-dimethylaminobenzoyl)imidazole, DBU, CH3CN, 25 °C) to provide the DMB di-ester 5 (~34 μg)20 which was purified by HPLC. The CD spectrum of 5 (Figure 3a) exhibited a pronounced ECCD [λ 301 nm (Δε +22.4), 326 (−21.5)] of the same sign and similar magnitude observed for the DMB diesters of 3,7 indicating the same C2 configuration. Independently, 1 was converted to a C2-O-(2′-naphthoate) mono-ester 6 as follows (Scheme 1): the primary hydroxyl group was selectively acetylated in the presence of a lipase (Novozyme 435, vinyl acetate, CH3CN) and the free secondary OH group subsequently acylated, as above, to give diester 6 (~24 μg) after HPLC purification. The CD (EtOH) spectrum of 6 (Figure 3b) revealed a positive long wavelength band CE [λ 238 nm (Δε +25.3)]21 that was interpreted as arising from a positive helicity between the diyne and naphthoate chromophores.24 The negative component of the ECCD was observed clearly when the CD spectrum of 6 was recorded in CH3CN [λ 238 nm (Δε +29.7), 188 nm (−50.5)]. Both methods lead to the same answer and we assign the R configuration to 1, 5 and 6.
Scheme 1.

Derivatization of polyacetylenic alcohols to chromophoric derivatives 5, 6 and 16.
Figure 3.

CD spectra (EtOH, 23 °C) of (a) 5 and 16. (b) 6.
In order to validate the outcome of assignment of 1 by the propargylic acyclate ECCD method, CD spectra were obtained of 2-naphthoyl esters (S)-7 and (S)-8 prepared from the known (S)-9 propargylic alcohol22 (note the change in CIP priorities) as shown in Scheme 2. Desilylation of (S)-9 to alcohol (S)-10 was followed by protection as the acetate ester and Sonogashira coupling with E-iododecene in the presence of bis-(triphenylphosphine)palladium (II) dichloride (11) and CuI to give ene-yne acetate ester 13. Exchange of the O-acyl groups (acetyl for 2-naphthoyl) through the intermediary alcohol 14 was achieved in two steps (NH3, MeOH; 2-naphthoic acid, EDC, DMAP, 76 % over two steps) to provide en-yne model ester (S)-7.23 In parallel, (S)-10 was acylated to the naphthoate ester 15, which was desilylated (TBAF, 86%) to give the propargylic O-napthoate 15a and transformed into the diyne naphthoate (S)-8 (CuCl, NH2OH·HCl, n-BuNH2, 1-bromoheptyne).
Scheme 2.

Preparation of en-yne and di-yne propargylic 2-naphthoate esters (S)-7 and (S)-8. Note: CIP priorities of 7–15 differ from those of 1–4.
The CD spectra of (S)-7 and (S)-8 (CH3CN) showed strong bisignate CEs [(S)-7: λ 188 nm (Δε −37.8). (S)-8: 225 (−24.6), 242 (+35.3)] (Figures 4 and 5). Note that the the secondary alcohol (S)-14, corresponding to (S)-7 and lacking a dichromophoric exciton coupling, shows essentially only baseline CD (Figure 3). Because the π-π* transition of the conjugated di-yne occurs at lower wavelenths [λ 214 nm (ε 37,300), Figure 5],1 the short-wavelength component of the exciton couplet of (S)-8 [λ 188 nm (Δε −37.8)] is at the edge of instrument detection limits, however, in (S)-7 this high energy component reveals itself readily [λ 225 nm (Δε −24.6)] due to the presence of a red-shifted extended en-yne chromophore.24 Nevertheless, as noted by Nakanishi and co-workers,15 only the sign of the long-wavelength component is necessary and sufficient to assign allylic benzoates which also applies for propargylic naphthoates due to the stronger oscillator strength of the 1Bb transition. Even acyclic non-conjugated propargylic O-naphthoates should be amenable to chiroptical analysis, unlike the corresponding O-benzoates that show only weak or non-detectable ECCD from the shorter wavelength acetylene chromophore (< λ 190 nm),19. This is demonstrated in the CD spectrum of 15a (Figure 6) which displays a weaker, yet still prominent positive component [λ 240 nm (Δε +4.6)] of the biphasic Cotton effect.
Figure 4.

CD spectra, 25 °C (a) (S)-7 in CH3CN, (b) (S)-7 in MeOH, and (c) (S)-14 in hexane.
Figure 5.

CD spectra for (S)-8 in (a) CH3CN, (b) MeOH (λ < 200 nm, obscured by solvent end-absorption), and (c) hexane at 25°C.
Figure 6.

CD spectrum of (S)-15a (CH3CN, 25 °C).
Since both the dibenzoate and the propargylic naphthoate ECCD methods lead to the same configurational assignment in 1, it can be concluded that both can be reliably used for stereo-assignments of other long-chain propargylic alcohols under appropriate conditions, although the onestep dibenzoate method is attractive for its operational simplicity.
A simple extension of the glycol benzoate method was applied to faulkneryne A (4a). Conversion of 4a, by the aforementioned procedure, gave the di-DMB ester 16 which showed an CD spectrum (EtOH, Figure 3a) with a negative split CE [λ 302 nm (Δε +17.4), 327 (−9.0)], similar to that of 5. As with 5, it is fortuitous that both the short-wavelength component of the dibenzoate ECCD of 16 and the superposed ene-diyne benzoate ECCD interaction (c.f. 6, Figure 3b) are of the same sign (positive) 25 and reinforce at ~λ 302 nm. 26 Therefore, the configurations of 4a and 16, are revealed to be R. Assuming congeneric 4b,c are of the same configuration as 4a, it would appear the biosynthesis of propargylic diols 1–4 from sponges across two different oceans conserves the C2 stereochemistry.27
Diplyne C (1) exhibited cytotoxicity against cultured human colon tumor cells (HCT-116; LD50 3.6 μg/mL), however, insufficient amounts of 4a–c were available for biological evaluation.
Conclusions
The complete structures of new long-chain polyacetylenic diols, faulknerynes A–C (4a–c) were elucidated with the aid of microcryoprobe NMR spectroscopy. The configurations of 4a and the known compound diplyne C (1) were assigned the R configuration by two different microscale ECCD methods that converged upon the same answer. The ECCD spectrum of propargylic en-yne naphthoates are nicely suited for observation of both halves of the exciton couplets for microscale configurational analysis of polyacetylenic alcohols without the necessity for additional synthetic elaboration of the acetylenic chromophore.
Experimental Section
General Experimental Procedures
Animal Material
The sponge Diplastrella sp., Family Spirastrellidae (Ridley & Dendy 1886) (08-05-026) was collected on 31 May 2008 at Sweetings Cay, Bahamas (26° 34.080′ N, 77° 53.407′ W) at a depth of −19 m using scuba, and kept in EtOH at −20 °C until extraction. The tissue was pale pink to red in life, thinly encrusting on coral in small patches. Microscopic spicule analysis (SEM) showed tylostyles with rounded heads, and microscleres with a preponderance of diplasters over spirasters as is typical for Diplastrella.28 A second sponge, Diplastrella sp. (08-07-038), was collected in the same vicinity at similar depths. Voucher samples for all animal material are archived at UCSD.
Extraction and Isolation
The EtOH extract of the sponge Diplastrella sp. (08-05-026, wet sponge vol. ~2–5 mL) was concentrated, and redissolved in MeOH. The MeOH extract was sequentially extracted with solvents of increasing polarity with adjustment of the H2O content at each stage and removal of solvent from the organic layer: hexane, 0% H2O (5.2 mg), CHCl3, 40% H2O (12.4 mg) The aqueous phase was concentrated under reduced pressure to remove MeOH then extracted with n-BuOH (15.6 mg); the latter was shown to contain diplyne E.1 Finally, the aqueous phase was concentrated under reduced pressure (106.9 mg). The CHCl3–soluble phase was separated by flash chromatography (SiO2, 1–100% MeOH-CHCl3 step gradient) to give a diyne-containing fraction (2.1 mg) which was further separated by RP HPLC (C18 5 μ, 10 × 250 mm, CH3CN-H2O, 2 mL/min) to give diplyne C1 (1, 500 μg) and faulkneryne A (4a, 400 μg).
The EtOH extract of a second specimen of Diplastrella sp. (08-07-038; wet sponge vol. ~2–5 mL) was concentrated, and redissolved in MeOH. The MeOH extract was sequentially extracted with solvents of increasing polarity with adjustment of the H2O content at each stage and removal of solvent from the organic layer: hexane, 0% H2O (9.8 mg), CHCl3, 40% H2O (19.5 mg) The aqueous phase was concentrated under reduced pressure to remove MeOH then partitioned against with n-BuOH which was concentrated to give the n-BuOH-soluble fraction (13.3 mg). Finally, the aqueous phase was concentrated under reduced pressure (73.9 mg). The CHCl3–soluble phase was separated by RP HPLC (C18 5 μ, 10 × 250 mm, CH3CN-H2O) to give faulknerynes A–C (4a, 1 mg), (4b, 4.7 μg29), (4c, 14.1 μg29), diplyne A1 (3 mg) and diplyne E (2, 100 μg).1
Faulkneryne A (4a): colorless glass; UV (MeOH) λmax 271 nm. 1H NMR, 13C NMR, see Table 1. HRMS m/z 347.0633 [M+Na]+ calcd for C16H2179BrNaO2 347.0623.
Faulkneryne B (4b): colorless glass: UV (MeOH) λmax 273 nm 1H NMR, see Table S1, Supporting Information. HREIMS: The compound failed to give pseudo-molecular ions..
Faulkneryne C (4c): colorless glass: UV (MeOH) λmax 272 nm. 1H NMR, see Table S1, Supporting Information. HRMS m/z 343.0304 [M+Na]+ calcd for C16H1779BrNaO2 343.0309
Preparation of 1,2-O-bis-(4′-Dimethylaminobenzoyl)diplyne C (5)
A solution of 1 (100 μg, 0.305 μmol) in CH3CN (100 μL) was treated with N-(4-dimethylaminobenzoyl)imidazole (197 μg, 0.917μmol) and DBU (140μg, 0.917 μmol) and the mixture allowed to stir for 6 h, then concentrated to give a crude product, which was purified by normal phase HPLC (17:83 EtOAc/hexane) to give pure 5. UV-vis (EtOH), λmax 313 nm. CD (EtOH) λ 301 nm (Δε +22.4), 326 (−21.5), 1H NMR (600 MHz, CDCl3) 7.91 (d, 2H, J = 8.8 Hz), 7.88 (d, 2H, J = 8.8 Hz), 6.62 (d, 2H, J = 8.8 Hz), 6.60 (d, 2H, J = 8.8 Hz), 6.15 (dt, 1H, J = 13.9, 7.3 Hz), 5.99 (d, 1H, J = 13.7 Hz), 5.96 (m, 1H), 4.56 (m, 2H), 3.03 (s, 6H), 3.02 (s, 6H), 2.25 (t, 2H, J = 7.1 Hz), 2.01 (q, 2H, J = 7.4 Hz), HRMS m/z 621.2330 [M+H]+ calcd for C34H4279BrN2O4 621.2328.
Preparation of (R)-1-O-Acetyl-(2′-O-naphthoyl)diplyne C (6)
A solution of 1 (100 μg, 0.305 μmol) in CH3CN (100 μL) was treated with Novozyme 435 (7 mg) and vinyl acetate (263 μg, 3.05 μmol, 10 equiv), and the mixture stirred at 40–45 °C. After 2 h the reaction was quenched, the enzyme filtered off, and the filtrate was concentrated under reduced pressure to give the crude product. The latter material was redissolved in anhydrous CH3CN (100 μL) and treated with N-(2′-naphthoyl)imidazole30 (137 μg, 0.61 μmol) and DBU (93 μg, 0.61μmol) and allowed to stir at rt for 6 h. The reaction mixture was concentrated under reduced pressure to give the crude product, which were purified by normal phase HPLC (9:91 EtOAc in hexane) to give pure (R)-6. CD (EtOH) λ 238 nm (Δε +25.3, see Figure 2b. CD (CH3CN) λ 238 nm (Δε +29.7), 188 nm (−50.5). 1H NMR (600 MHz) 8.63 (s, 1H), 8.05 (d, 1H, J = 8.5 Hz), 7.97 (d, 1H, J = 8.1 Hz), 7.90 (d, 1H, J = 8.6 Hz), 7.89 (d, 1H, J = 8.1 Hz), 7.62 (m, 1H), 7.56 (m, 1H), 6.15 (dt, 1H, J = 13.8, 7.3 Hz), 5.99 (d, 1H, J = 13.5 Hz), 5.95 (dd, 1H, J = 7.2, 4.1 Hz), 4.47 (dd, 1H, J = 11.8, 4.0 Hz), 4.44 (dd, 1H, J = 11.8, 7.4 Hz), 2.27 (t, 2H, J = 7.0 Hz), 2.08 (s, 3H), 2.02 (q, 2H, J = 7.2 Hz). HRMS m/z 545.1298 [M+Na]+ calcd for C29H3179BrNaO4 545.1303.
(S)-1-Cyclohexylprop-2-ynyl acetate (12)
(S)-9 (52.4 mg, 0.21 mmol) was dissolved in THF (2mL), and TBAF (0.25 mL, 1M in THF) was added dropwise at 0°C. The mixture was stirred at 0 °C for 30 min and the solvent was removed under reduced pressure to give a crude product which was immediately subject to acetylation (Ac2O, 30 μL) in anhydrous pyridine (500μL) at 25 °C for 48 hrs. The volatiles were removed and the mixture separated (silica cartridge, 3:7 hexane-CH2Cl2) to provide (−)-12 (14.1 mg, 38%). [α]24 D −66.6 (c 0.011, CHCl3) [lit. [α]24D −65.5 (c 1.5, CHCl3)].22 MS and NMR data were identical with literature values.
En-yne (13)
A mixture of 1-iododecene (43 mg, 0.12 mmol), (Ph3P)2PdCl2 (5.5 mg, 7.8 μmol), CuI (6.0 mg, 0.032 mmol) and Et3N (2.5 mL) were stirred at rt for 30 min. This solution was then added dropwise to a solution of alkyne 12 in Et3N (1 mL) and the mixture reaction stirred at rt for 1.5 hrs. The volatiles were removed and the residue separated by flash chromatography (silica cartridge, 3:97 EtOAc-hexanes) to yield acetoxy en-yne 13 as an oil (21.5 mg, 79%). [α]24D −88.0 (c 0.004, CHCl3); IR (KBr) vmax 2925, 2854, 1743, 1451, 1369, 1229, 1017, 977, 955 cm−1; 1H NMR (400 MHz, CDCl3) 0.86 (t, J = 7.2 Hz, 3H), 1.04–1.36 (m, 21H), 1.61–1.84 (m, 6H), 2.06 (s, 3H), 2.06 (dq, J = 1.6, 7.2 Hz, 5H), 5.30 (dd, J = 2.0, 6.4 Hz, 1H), 5.45 (dq, J = 2.0, 15.6 Hz, 1H), 6.15 (dt, J = 7.2, 15.6 Hz, 1H); 13C NMR (100MHz, CDCl3) δ 14.1 (CH3), 21.1 (CH3), 22.7 (CH2), 25.7 (CH2), 25.8 (CH2), 26.2 (CH2), 28.1 (CH2), 28.6 (CH2), 29.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 31.9 (CH2), 33.1 (CH2), 42.0 (CH), 68.9 (CH), 74.0 (C), 83.9 (C), 84.7 (C), 108.6 (CH), 146.0 (CH), 170.2 (C). HREIMS m/z 346.2869 [M]+, calcd for C23H38O2 346.2866.
En-yn-ol (14)
A solution of 13 in MeOH (100 μL) was treated with anhydrous NH3 (2M in MeOH, 300 μL), stirred overnight and then concentrated under a stream of N2. The crude mixture was separated by chromatography (silica cartridge, 95:5 hexanes-EtOAc) to yield alcohol 14 as an oil (4.8 mg, 91%). [α]24 D +5.3 (c 0.02, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.8 Hz, 3H), 1.02–1.38 (m, 21H), 1.49–1.86 (m, 7H), 2.07 (dq, J = 1.2, 7.2 Hz, 5H), 4.22 (m, 1H), 5.47 (dq, J = 1.6, 15.6 Hz, 1H), 6.13 (dt, J = 7.2, 15.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH3), 25.9 (CH2), 26.3 (CH2), 28.2 (CH2), 28.5 (CH2), 28.6, 29.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 31.9 (CH2), 33.1 (CH2), 44.3 (CH), 67.7 (CH), 84.5 (C), 87.5 (C), 108.8 (CH), 145.4 (CH). The configuration of the carbinol center in 14 was confirmed by the modified Moshers’s method.11 (R)-MTPA ester of compound 14. Compound 14 (0.8 mg, 2.6 μmol) was dissolved DCM/pyridine (1:1 200 μL). (S)-MTPA-Cl (5 μL, 0.27 mmol) was added and the mixture stirred for 1.5 hrs. The volatiles were removed and the crude product separated by chromatography (silica, 95:5 hexanes-EtOAc) to yield the (R)-MTPA ester of compound 14 (1.3 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.8 Hz, 3H), 1.05–1.36 (m, 21H), 1.63–1.74 (m, 5H),1.81 (brd, J=12.4 Hz, 1H), 2.08 (dq, J = 1.2, 6.8 Hz, 2H), 3.54 (s, 3H), 5.42 (d, J = 5.6 Hz, 1H), 5.43 (dq, J = 1.6, 15.6 Hz, 1H), 6.12 (dt, J = 6.8, 15.6 Hz, 1H). (S)-MTPA ester of compound 14. The same procedure applied to compound 14 (1.0 mg, 3.3 μmol) with (S)-MTPA-Cl (5 μL, 0.27 mmol) to yield the corresponding (S)-MTPA ester (1.4 mg, 99%) yield. 1HNMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.8 Hz, 3H), 0.99–1.38 (m, 21H), 1.60–1.71 (m, 5H),1.71 (m, 1H), 2.09 (dq, J = 1.6, 7.2 Hz, 2H), 3.57 (d, J = 1.2 Hz, 3H), 5.42 (d, J = 5.6 Hz, 1H), 5.46 (dq, J = 1.6, 16.0 Hz, 1H), 6.15 (dt, J = 6.8, 16.0 Hz, 1H).
Naphthoate ester (S)-7
A solution of enyn-ol 14 (2 mg, 6.57 μmol) in CH2Cl2 (200 μL) was treated with 2-naphthoic acid (2.8 mg, 16.4 μmol), EDC (3.0 mg, 19.7 μmol), and a small crystal of DMAP. The resulting mixture was stirred for 48 hrs, concentrated and separated by flash chromatography (SiO2) to afford naphthoate ester (S)-7 (2.4 mg, 82%). [α]24 D +20.2 (c 0.94, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.61 (bs, 1H), 8.07 (dd, 8.4, 1.2 Hz, 1H), 7.95 (d, 8.0 Hz, 1H), 7.86 (d, 8.4 Hz, 2H), 7.55 (m, 2H), 6.17 (dt, 16.0, 7.2 Hz, 1H), 5.62 (dd, 5.6, 1.2 Hz, 1H), 5.49 (dq, 16.0, 0.8 Hz, 1H), 2.07 (dq, 6.8, 1.6 Hz, 2H), 1.66–1.98 (m, 4H), 1.22 (m, 20H); HREIMS m/z 458.3176 [M+] calcd for C32H42O2, 458.3179.
Naphthoate ester (15)
Alkynol (S)-9 (10.4 mg, 0.041 mmol) was converted into naphthoate ester 15 (13.5 mg, 82%) using the procedure described above. [α]24D +12.6 (c 1.93, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.61 (bs, 1H), 8.07 (dd, J = 8.4, 1.2 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.55 (m, 2H), 5.56 (d, J = 6.0 Hz, 1H), 1.67–1.99 (m, 6H), 1.13–1.32 (m, 4H), 0.98 (t, J = 8.0 Hz, 9H), 0.59 (q, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 165.7 (C), 135.5 (C), 132.4(C), 131.2 (CH), 129.4 (CH), 128.3 (CH), 128.1 (CH), 127.7 (CH), 127.4 (C), 126.6 (CH), 125.4 (CH), 103.0 (C), 88.6 (C), 69.3 (CH), 42.1 (CH), 28.7 (CH2), 28.1 (CH2), 26.3 (CH2), 25.8 (CH2), 25.7 (CH2), 7.8 (CH3), 4.2 (CH2); HREIMS m/z 406.2330 [M+] calcd for C26H34O2Si, 406.2323.
Diyne Naphthoate Ester (S)-(8)
Naphthoate ester 15 (13.5 mg, 0.033 mmol) was dissolved in THF (1 mL) and the solution cooled to 0 °C. TBAF (1M in THF, 33.2 μL) was added dropwise and the mixture stirred for 10 min before removal of the volatiles. The residue was purified by flash chromatography (SiO2, 9:1 hexanes-ether) to afford essentially pure propargyl O-naphthoate 15a (8.1 mg, 86%). [α]23D −12.8 (c 1.19, CHCl3); FTIR (KBr) vmax 3298, 2929, 2853, 1719, 1281, 1225, 1195, 1129, 1088, 973, 777, 761 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.61 (bs, 1H), 8.07 (dd, J = 8.4, 1.2 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.55 (m, 2H), 5.51 (dd, J = 6.0, 2.4 Hz, 1H), 2.48 (d, J = 2.4 Hz, 1H), 1.68–2.00 (m, 6H), 1.18–1.32 (m, 4H); 13C NMR (100 MHz, CDCl3) δ165.7(C), 135.6 (C), 132.4 (C), 131.3 (CH), 129.4 (CH), 128.4 (CH), 128.2 (CH), 127.8 (CH), 127.1 (C), 126.7 (CH), 125.3 (CH), 80.3 (C), 74.3 (CH), 68.6 (CH), 41.8 (CH), 28.5 (CH2), 28.2 (CH2), 26.2 (CH2), 25.8 (CH2), 25.7 (CH2).
A mixture of 15a (7.8 mg, 27 μmol), CuCl (0.5 mg, 5.3 μmol), NH2OH·HCl (2.8 mg, 40 μmol) was suspended in MeOH (150 μL) at 0 °C under N2 and treated dropwise with neat n-butylamine (250 μL). The mixture was stirred for 10 min and treated dropwise with a solution of 1-bromoheptyne (4.6 mg, 26.7 μmol) in MeOH (100 μL). The mixture was stirred at 0 °C for 1 hr then poured into ice water (2 mL) before acidification with 5% H2SO4 and extraction with ether (3 × 4mL). The combined ether extracts were washed with brine, dried with anhydrous MgSO4, and separated by flash chromatography (silica cartridge, 8:1, hexanes-diethyl ether) to give diyne (S)-8 as an oil (8.2 mg, 80%). [α]23 D +58.8 (c 1.24, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.60 (bs, 1H), 8.05 (dd, J = 8.4, 1.2 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.55 (m, 2H), 5.55 (d, J = 6 Hz, 1H), 2.25 (t, J = 6.8 Hz, 2H), 1.67–1.99 (m, 6H), 1.50 (p, J = 7.6 Hz, 2H), 1.17–1.37 (m, 4H), 0.87 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.6(C), 135.6 (C), 132.4 (C), 131.3 (CH), 129.4 (CH), 128.4 (CH), 128.2 (CH), 127.8 (CH), 127.1 (C), 126.7 (CH), 125.3 (CH), 81.8 (C), 72.0 (C), 71.2 (C), 69.2 (CH), 64.5 (C), 42.2 (CH), 31.0 (CH2), 28.6 (CH2), 28.3 (CH2), 27.8 (CH2), 26.1 (CH2), 25.8 (CH2), 25.7 (CH2), 22.1 (CH2), 19.2 (CH2), 13.9 (CH3). HRESIMS m/z 386.2238 [M]+ calcd for C37H30O2, 386.2240.
Preparation of (R)-1,2-O-bis-(4′-Dimethylaminobenzoyl)faulkneryne (16)
Using the procedure described above for 1, faulkneryne A (3, 100 μg, 0.307 μmol) was converted into the corresponding DMB diester (R)-16 (21 μg). UV-vis (EtOH), λmax 312 nm. CD (EtOH) λ 302 nm (Δε +17.4), 327 (−9.0), 1H NMR (600 MHz, CDCl3) 7.92 (d, 2H, J = 8.8 Hz), 7.89 (d, 2H, J = 9.0 Hz), 6.63 (d, 2H, J = 9.6 Hz), 6.60 (d, 2H, J = 9.4 Hz), 6.15 (dt, 1H, J = 13.3, 7.5 Hz), 6.09 (dt, 1H, J = 10.8, 7.7 Hz), 6.02 (m, 1H), 5.98 (m, 2H), 5.48 (m, 1H), 4.60 (m, 1H), 2.35(m, 2H), 2.03(m, 2H). HRMS m/z 619.2141 [M+H]+ calcd for C34H3979BrN2O4, 619.2172.
Molecular Modeling
An analog of compound 5 (truncated to the 3,5-diyne) was mimimized using molecular mechanics (MMFF94, Spartan 08, gas phase). Energies were calculated (semi-empirical, PM3) and Monte Carlo conformational searching applied to obtain the lowest 25 models ranging in energy from −1.06 to 25 kcal.mol−1. For the three most stable conformers, see Figure 1(d)–(f). An analog of dibenzoate ester 6 (truncated to the 3,5-diyne) was minimized using semi-empirical methods (PM3, Spartan 08) and the 25 lowest energy conformers determined by Monte Carlo methods. See Figure 2.
Cytotoxicity Assay
Cytotoxicity of 1 against cultured human colon tumor cells (HCT-116) incubated under 5% CO2 in the presence and absence of compound, followed by colorimetric measurement of growth inhibition by the MTS method using a microplate reader as decribed elsewhere.2 Diplyne C (1) exhibited an LC50 of 3.6 μg/mL (etoposide was used as a positive control).
Supplementary Material
Acknowledgments
We thank C. K. Skepper for assistance sponge collection and preparation of S-(+)-9. We’re grateful to S. Zea and and M-K. Harper (University of Utah) for helpful discussions on sponge taxonomy, to J. Siegel (University of Zürich) for stereochemical advice, J. R. Pawlik (University of North Carolina, Wilmington), and the captain and crew of the RV Seward Johnson for logistical support during collecting expeditions. The NSF Biological Oceanography Program (OCE-0095724, 0550468 to J.R.P).) is acknowledged for ship time. The 500 MHz NMR spectrometers were purchased with a grant from the NSF (CRIF, CHE0741968). This work was supported by grants from NIH (CA122256 and AI039987 to T. F. M.), and a Ruth L. Kirschstein National Research Service Award NIH/NCI (T32 CA009523 to B. I. M).
Footnotes
Supporting Information Available. 1H, 13C NMR and 2D NMR spectra of 3, and 1H, 13C NMR of 4 and all synthetic compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References and Notes
- 1.Lerch ML, Harper MK, Faulkner DJ. J Nat Prod. 2003;66:667–670. doi: 10.1021/np020544+. [DOI] [PubMed] [Google Scholar]
- 2.Zhou GX, Molinski TF. Mar Drugs. 2003;1:46–53. [Google Scholar]
- 3.Wright AE, McConnell OJ, Kohmoto S, Lui MS, Thompson W, Snader KM. Tetrahedron Lett. 1987;28:1377–1379. doi: 10.1021/np50050a064. [DOI] [PubMed] [Google Scholar]
- 4.Isaacs S, Kashman Y, Loya S, Hizi A, Loya Y. Tetrahedron. 1993;49:10435–10438. [Google Scholar]
- 5.Tsukamoto S, Kato H, Hirota H, Fusetani N. J Nat Prod. 1997;60:126–130. doi: 10.1021/np50113a027. [DOI] [PubMed] [Google Scholar]
- 6.Tada H, Yasuda F. Chem Lett. 1984:779–780. [Google Scholar]
- 7.Fusetani N, Sugano M, Matsunaga S, Hashimoto K. Tetrahedron Lett. 1987;28:4311–4312. [Google Scholar]
- 8.The compounds are named in honor of the late D. John Faulkner (1940–2002) a pioneer in marine natural products who described diplynes A-E (Reference 1), related polyacetylenic alcohols, and hundreds of other compounds. Andersen RJ, Ireland CM, Molinski TF, Bewley CB. J Nat Prod. 2004;67:1239–1251. doi: 10.1021/np049912v.
- 9.Gung BW, Gibeau C, Jones A. Tetrahedron: Asymmetry. 2005;16:3107–3114. [Google Scholar]
- 10.Gung BW, Gibeau C, Jones A. Tetrahedron: Asymmetry. 2004;15:3973–3977. [Google Scholar]
- 11.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 12.(a) Molinski TF. Curr Opin Drug Discov Devel. 2009;12:197–206. [PubMed] [Google Scholar]; (b) Molinski TF. Curr Opin Biotechnol. 2010;21:819–826. doi: 10.1016/j.copbio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harada N, Nakanishi K. Circular Dichroic Spectroscopy: Exciton Coupling in Organic Stereochemistry. University Science Books; Mill Valley, CA: 1983. p. 162. [Google Scholar]
- 14.Presumably, for this reason, DMB diesters were chosen for the dibenzoate assignment of 2 to move the bisignate split CE to longer wavelengths (λ ~310 nm) and diminish potential interference from di-yne–benzoate interactions (Reference 7).
- 15.Gonnella NC, Nakanishi K, Martin VS, Sharpless KB. J Am Chem Soc. 1982;104:3775–3776. [Google Scholar]
- 16.Molinski TF, Brzezinski LJ, Leahy JW. Tetrahedron: Asymmetry. 2002;13:1013–1016. [Google Scholar]
- 17.Schneider C, Schreier P, Humpf HU. Chirality. 1997;9:563–567. [Google Scholar]
- 18.Johnson F. Chem Rev. 1968;68:375–413. [Google Scholar]
- 19.Naito J, Yamamoto Y, Akagi M, Sekiguchi S, Watanabe M, Harada N. Monatsh Chem. 2005;136:411–445. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.