

Abstract
Latrepirdine (Dimebon; dimebolin) is a neuroactive compound that was associated with enhanced cognition, neuroprotection, and neurogenesis in laboratory animals, and has entered phase II clinical trials for both Alzheimer’s (AD) and Huntington’s diseases (HD). Based on recent indications that latrepirdine protects cells against cytotoxicity associated with expression of aggregatable neurodegeneration-related proteins, including Aβ42 and γ-synuclein, we sought to determine whether latrepirdine offers protection to Saccharomyces cerevisiae (S. cerevisiae). We utilized separate and parallel expression in yeast of several neurodegeneration-related proteins, including α-synuclein, the amyotrophic lateral sclerosis-associated genes TDP43 and FUS, and the HD-associated protein huntingtin with a 103 copy-polyglutamine expansion (HTT gene; htt-103Q). Latrepirdine effects on α-synuclein clearance and toxicity were also measured following treatment of SH-SY5Y cells or chronic treatment of wildtype mice. Latrepirdine only protected yeast against the cytotoxicity associated with α-synuclein, and this appeared to occur via induction of autophagy. We further report that latrepirdine stimulated the degradation of α-synuclein in differentiated SH-SY5Y neurons, and in mouse brain following chronic administration, in parallel with elevation of the levels of markers autophagic activity. Ongoing experiments will determine the utility of latrepirdine to abrogate α-synuclein accumulation in transgenic mouse models of α-synuclein neuropathology. We propose that latrepirdine may represent a novel scaffold for discovery of robust pro-autophagic/anti-neurodegeneration compounds, that might yield clinical benefit for synucleinopathies including PD, Lewy body dementia, REM sleep disorder, and/or multiple system atrophy, following optimization of its pro-autophagic and pro-neurogenic activities.
Introduction
Latrepirdine (Dimebon; dimebolin) is a neuroactive compound with antagonist activity at histaminergic, α-adrenergic, and serotonergic receptors that was associated with enhanced cognition1–4, neuroprotection5, 6, and neurogenesis7 in laboratory animals. Based on its effects on cognition in rodents and its highly favorable safety profile, the compound entered clinical trials for both Alzheimer’s disease (AD)8 and Huntington’s disease (HD)9. Related reports indicate that latrepirdine protects against the cytotoxicity associated with Aβ4210 or γ-synuclein11 by stimulating catabolism of these aggregation-prone, neurodegeneration-related proteins. Here, we sought to determine whether latrepirdine offered protection against the cytotoxicity associated with the accumulation of several neurodegeneration-related proteins including α-synuclein (α-syn), the amyotrophic lateral sclerosis (ALS)-associated genes TDP43 and FUS, and the HD-associated protein huntingtin with a 103 copy-polyglutamine (polyQ) expansion (HTT gene; htt-103Q). We report that latrepirdine improved cell viability in Saccharomyces cerevisiae (S. cerevisiae) expressing α-syn, but offered no protection to yeast strains expressing TDP43, FUS, or htt-103Q.
Parkinson’s disease (PD) and AD are the two most common forms of neurodegenerative diseases. Both PD and AD patients present with progressive cognitive decline resulting from a pathogenic accumulation of insoluble protein aggregates that precedes region-specific synaptic and neuronal loss12. Formation of intracellular Lewy bodies, which consist primarily of α-synuclein (α-syn) and ubiquitin, is associated with the pathogenesis of PD, Lewy body dementia (LBD), REM sleep disorder (REMSD), and multiple system atrophy (MSA)12. Interestingly, 30–50% of AD patients also harbor atypical α-syn pathology (in addition to canonical AD neuropathology) in the cerebral cortex13,14. Indeed, α-syn was originally identified as the non-Aβ component of AD-related senile plaques, and was later determined to represent a primary component of Lewy bodies12,15.
Recent studies suggest that α-syn exists as a stable helically folded tetramer that must undergo destabilization prior to misfolding and formation of the fibrillar aggregates that are observed in synucleinopathies16. We hypothesized that a compound that protects against the cytotoxicity associated with α-syn might provide clinical benefit to patients harboring synucleinopathies, including PD, LBD, REMSD, MSA, and in some instances of AD.
To date, the most promising regulators of α-syn degradation and protection against α-syn-induced cytotoxicity are small molecule enhancers of rapamycin (SMERs; most notably SMER-28). Recently, SMER-28 was shown to induce autophagy, improve cell viability, and promote clearance in cellular models of neurodegenerative disease-related proteins including APP metabolites (among them Aβ17; htt18, and α-syn18). In related studies, we reported that latrepirdine protected against cytotoxicity associated with Aβ42 accumulation in S. cerevisiae10, in mammalian cells, and in the brains of transgenic CRND8 mice19 via induction of mTOR- and Atg5-dependent autophagy. Here, we provide evidence to indicate that latrepirdine: (1) protects against α-syn-related cytotoxicity; (2) enhances autophagy and degradation of α-syn in three different model systems; and (3) when chronically administered, reduces α-syn levels in mouse brain in parallel with the induction of autophagy.
Methods
Preparation and Handling of Latrepirdine
The synthesis and characterization of latrepirdine was described previously20. Briefly, latrepirdine was purchased from SinoChemexper (Shanghai, China) and purity of the compound was determined to be >99% or provided directly by Medivation Inc. For use in vitro, latrepirdine was dissolved directly into culture media to the desired concentration, as described in20. For administration in vivo, latrepirdine was dissolved into 0.9% saline (vehicle) at a final concentration of 3.5 mg/ml (made fresh every 2 days).
Yeast growth curve and spotting assay
Yeast cells were grown at 30°C unless specifically mentioned. Growth of yeast strains for was monitored using Bioscreen (www.bioscreen.fi). Yeast strains were pre-grown in 2% raffinose, diluted to an OD600 of 0.05 and induced with 2% galactose. OD measurements were taken every 1hr. Raw data were averaged among independent experiments and OD600 plotted over time as a growth curve. For spotting assays, yeast cells were grown overnight to mid log phase in raffinose medium. Cultures were then normalized to OD600=5.0, and 10X serially diluted and spotted onto the respective dropout plates containing 2% glucose or galactose. Pictures of plates were taken after 2–3 days of growth.
Assay of non-specific autophagy in yeast
Yeast strain YTS158 (generous gift from Daniel J. Klionsky, University of Michigan) was grown in the absence or presence of different concentrations of latrepirdine (as indicated) to mid-log phase. Cells were then harvested and alkaline phosphatase activity was measured using a spectrophotometric assay as described21.
SH-SY5Y Cell Culture Experiments
SH-SY5Y cells stably transfected with a doxycyline-inducible (“tet-off”) wild-type α-synuclein gene, and control SH-SY5Y cells over-expressing the β-galactosidase (β-gal) gene, (a gift from L. Stefanis, Division of Basic Neurosciences, Biomedical Research Foundation of the Academy of Athens, Athens, Greece)22 were cultured and treated with or without drug. See Supplemental Methods section for detailed methods.
Experimental Animals and Drug Treatment
All mice in this study were maintained on a hybrid C3H/He-C57BL/6 background (referred to here as “wild-type” mice). Male two-month-old littermates received 21 consecutive once-daily intraperitoneal injections of either 3.5 mg/kg latrepirdine (n=4) or 0.9% saline (vehicle; n=4). Following treatment, animals were sacrificed and transcardially perfused with ice-cold PBS (pH 7.4). Brains were dissected and snap-frozen for biochemical analysis. Animals were individually housed and maintained on a 12:12 light:dark cycle (lights on at 7am) with ad libitum access to food and water throughout the course of the entire experiment. All experimental protocols described herein were conducted within NIH guidelines for animal research and were approved by the Institutional Animal Care and Use Committee (IACUC) at Mount Sinai School of Medicine. See Supplemental Methods for detailed methods.
Statistical Analysis
Integrated density of immunoreactive Western blot bands was measured using MultiGauge Software and normalized to % control (vehicle or nTg littermate, where indicated). In all instances, Shapiro-Wilk test for normality of distribution and Levene’s test for homogeneity of variance were utilized for inclusion in parametric tests (p>0.05 for Shapiro-Wilk and Levene’s tests). Independent samples t-tests (parametric design) or Mann-Whitney U tests (non-parametric design) were utilized to determine significant mean differences between two groups. Significance for t-tests and ANOVAs are reported with a p≤0.05 using two-tailed tests with an α-level of 0.05. All statistical analyses were performed using SPSS v18.0 and/or GraphPad Prism 5.
Results
Latrepirdine protected Saccharomyces cerevisiae against α-synuclein toxicity
We employed an S. cerevisiae model systems to investigate whether latrepirdine could protect from α-syn or any of a panel of proteotoxic species associated with neurodegenerative diseases (Table 1). Integration of a single copy of α-syn in S. cerevisiae (1XαSyn) had no appreciable effect on cellular growth23. However, an increase in α-syn gene dosage from one to two copies (2XαSyn) resulted in growth arrest and cell death23. To test the possibility that yeast could be protected from α-syn toxicity by latrepirdine, we monitored the growth of the 2XαSyn strain and the isogenic wild type (W303α) strain in the presence of different concentrations of latrepirdine (Figure 1A–C). Latrepirdine treatment was associated with sustained viability of the 2XαSyn strain, with no effect on growth of the W303α strain. We conclude that latrepirdine treatment was associated with sustained cell viability in the face of α-syn overexpression, which may be related to an effect of the drug on protein degradation.
Table 1.
Yeast strains used in this study.
| Strains | Genotype | Reference |
|---|---|---|
| W303α | MATaade2-1can1-100his3-11,15leu2- 3112, trp1-1, ura3-1 | 31 |
| 2XαSyn | Integration of α-synuclein into HIS3 and TRP1 Loci of W303α (IntTox) | 31 |
| 1XFUS | Integration of FUS/TLS into TRP1 Locus of W303α | 32 |
| 2XTDP43 | Integration of TDP43 into HIS3 and TRP1 Loci of W303α | 33 |
| Htt103Q | Integration of N-terminal huntingtin fragment with stretch of 103 glutamine into HIS3 Locus of W303α | 34 |
| YTS158 | BY4742 with integration of pho8Δ::pho8Δ60(URA3) pho13Δ::Kan | 35 |
| Atg8Δ2XαSyn | 2XαSyn with integration of atg8Δ::LEU2 | This study |
Figure 1. Latrepirdine protects S. cerevisiae from cytotoxicity of α-synuclein, but not TDP-43, FUS, or Htt-103Q.
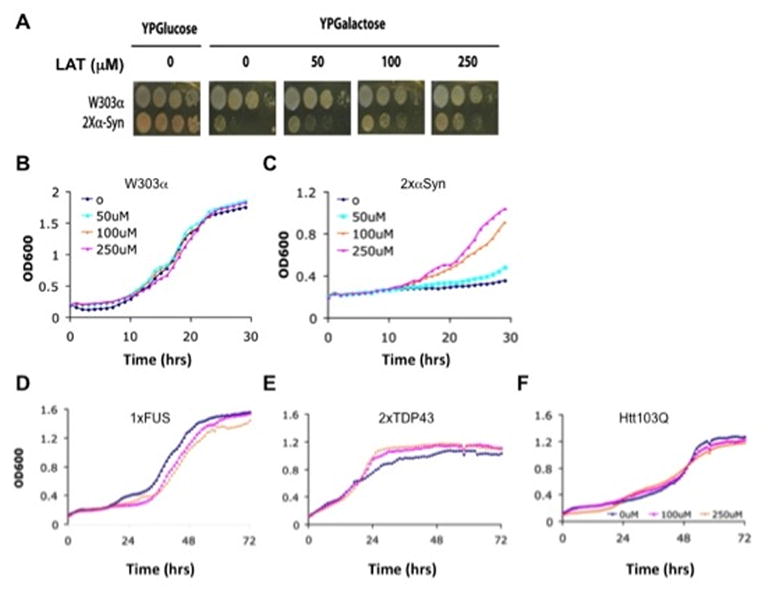
(A) 2XαSyn strain and its isogenic wild type (W303α) strains were grown to mid-log phase in raffinose medium, diluted, and then spotted onto YPGlucose plate (α-syn is “off”) and YPGalactose plates (α-syn “on”) in the absence or presence of latrepirdine (pictures were taken 2 days after growth at 30°C). (B) W303α or (C) 2xαSyn strains were grown in YPGalactose medium (α-syn is “on”) in the absence or presence of latrepirdine (concentration as indicated). (D) 1XFUS, (E) 2XTDP43, and (F) 1XHtt103Q strains were grown in YPGalactose medium (expression is “on”) in the absence or presence of different concentrations of latrepirdine (as indicated). All figures are representative of three or more independent experiments.
To test whether the protection from proteotoxicity by latrepirdine was specific to α-syn, we also studied the effect of latrepirdine on other S. cerevisiae models of proteotoxicity in neurodegenerative diseases, including the ALS-associated genes TDP43 (2XTDP43) and FUS (1XFUS), and the HD-associated htt-103Q. Despite the well-established properties of each of these proteins to form aggregates that are toxic to yeast, latrepirdine offered specific protection from only the proteotoxicity of α-syn (Figure 1A–C); i.e., the drug was unable to afford protection from the proteotoxicity associated with FUS, TDP43, or Htt-103Q (Figure 1D–F). One factor underlying the heterogeneity of proteotoxicity may be attributable to the different intracellular compartments in which the aggregates accumulate. Heterogeneity of proteotoxicity also occurs within the same polypeptide as exemplified by the prion protein (PrP) strain phenomenon, in which some aggregates of PrP are benign and well-tolerated by cells while other aggregates are toxic and pathogenic for transmissible spongiform encephalopathies24. Either or both of these phenomena might explain, at least in part, the differential rescue of yeast from some, but not all toxic aggregates. In related studies, we reported that latrepirdine protects yeast against proteotoxicity associated with Aβ42 accumulation via induction of autophagy10 as well as in mammalian cell lines and in mouse brain19. The specificity of latrepirdine for α-syn shown here, taken together with these two reports, suggests that fibrillar assemblies formed by α-syn or Aβ42 aggregates may be preferentially targeted for degradation by the autophagic pathway following latrepirdine stimulation.
Genetic and pharmacological evidence indicate that latrepirdine-mediated protection of yeast from α-syn toxicity involved autophagy
We sought to determine whether latrepirdine protection of yeast from α-syn toxicity was attributable to activation of autophagy – a conserved pathway regulating the degradation of long-lived proteins, cellular organelles, and protein aggregates. Addition of rapamycin to yeast cultures strongly induced autophagy, even in a nutrient-rich medium25. Using a yeast reporter of non-specific autophagy (in which maturation of the autophagosome activated an alkaline phosphatase; YTS158 strain21), we observed a significant and dose-dependent increase in alkaline phosphatase activity following treatment with latrepirdine, suggesting that latrepirdine activates autophagy (Figure 2A). Since we observed latrepirdine-induced activation of mTOR-dependent autophagy in mammalian cell lines and mouse brain19, we sought to determine whether activation of TOR-dependent autophagy could also protect the 2XαSyn strain from α-syn toxicity. Treatment of yeast with rapamycin (10μM) was also associated with a significant increase in the viability of 2XαSyn cells, further confirming that stimulation of autophagic activity was sufficient to prevent the proteotoxicity associated with α-syn aggregation in the 2XαSyn model (Figure 2B).
Figure 2. Latrepirdine or rapamycin protect against α-synuclein-related cytotoxicity via induction of autophagy.
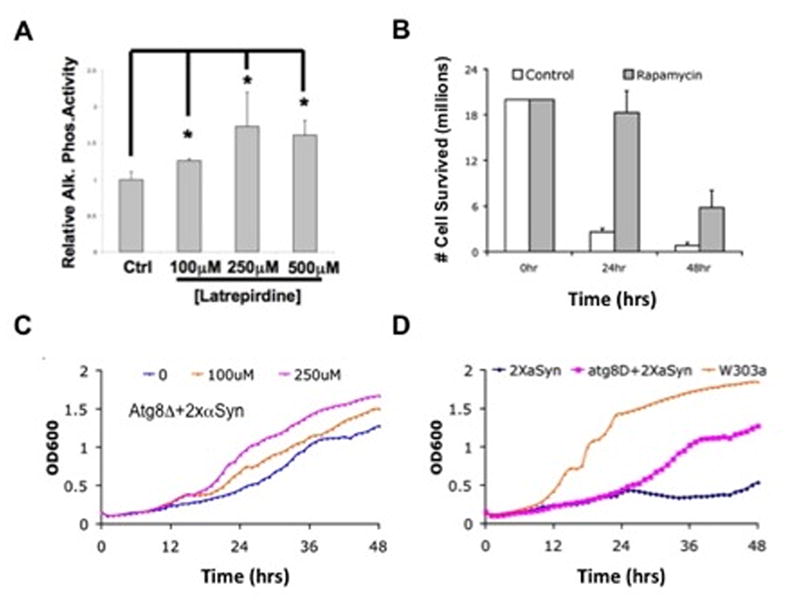
(A) In the YTS158 strain, maturation of the autophagosome results in activation of a functional alkaline phosphatase. Alkaline phosphatase activity was measured from YTS158 stain following growth in synthetic medium to log phase in the absence or presence of the indicated concentrations of latrepirdine (as indicated). (B) 2xαSyn strains were grown in YPGalactose medium (α-syn is “on”) in the absence or presence of rapamycin (concentration as indicated). (C) Atg8Δ+2XαSyn cultures were grown in YPGalactose medium (α-syn is “on”) in the absence or presence of different concentrations of latrepirdine (as indicated) and growth was monitored by measuring OD600. (D) 2XαSyn, its isogenic wild type (W303α), and 2XαSyn with deletion of the ATG8 gene (Atg8Δ+2XαSyn) were grown in YPGalactose (α-syn is “on”) medium at 30oC with shaking. Growth of cells was monitored by OD600 at an interval of 1hr using a Bioscreen machine. All figures are representative of three or more independent experiments, performed in duplicate or triplicate. Graphs are mean ± SEM; *p<0.05.
ATG8 (yeast homolog of LC3) encodes a ubiquitin-like protein required for autophagosome formation. In order to test the hypothesis that latrepirdine-mediated protection from α-syn toxicity involved autophagy, we crossed the 2XαSyn strain with an atg8Δ::LEU2 strain (Atg8Δ2XαSyn; Table 1). Atg8Δ2XαSyn cultures were grown in the absence or presence of 100μM or 250μM latrepirdine, and growth was monitored by OD600 (Figure 2C). Although 100μM latrepirdine was associated with protection from α-syn toxicity in the 2XαSyn strain (Figure 1A–C), this concentration was insufficient to protect the Atg8Δ2XαSyn strain (Figure 2C). However, higher concentrations of latrepirdine (250μM) appeared to offer partial protection from α-syn toxicity in Atg8Δ (Figure 2C). This observation suggests latrepirdine may protect cells against α-syn-related toxicity through at least one pathway that involves the regulation of Atg8-dependent autophagy. In contrast to prior observations18, ablation of Atg8-dependent autophagy resulted in increased cell viability of Atg8Δ2XαSyn after 24 hours of growth, by comparison to 2XαSyn cells, although both exhibited less viability when compared to the isogenic wild-type strain at all time points (Figure 2D). We propose that a compensatory mechanism that is selective for survival of the Atg8Δ2XαSyn strain may be induced after 24 hours of growth, allowing for modest improvement in cell viability. In future studies, we plan to investigate these pathways in more detail in order to understand the mechanisms underlying improved survival after 24 hours in the autophagy-deficient Atg8Δ2XαSyn strain.
Latrepirdine promoted cell viability and protected against α-synuclein toxicity in differentiated SH-SY5Y neurons
We tested latrepirdine altered α-syn aggregation using a stably expressing, inducible tet-off system to overexpress wild-type human α-syn in differentiated SH-SY5Y neurons. In this system, α-syn expression is off in the presence of doxycyline (dox) and switched on in cells deprived of dox. We analyzed the levels of monomeric and oligomeric α-syn in total lysates, Triton X-100 soluble, and insoluble protein fractions. Total levels of α-syn protein were higher than in control cells maintained in the α-syn off (+ dox) state. We observed that treatment with 10 nM latrepirdine by 14 days decreased the levels of all forms of α-syn, including aggregates and soluble monomer in the Triton X-100 soluble, total, and insoluble fractions (Figure 3A). Similar results in Western blots were obtained using the LB509 anti-α-syn antibody (data not shown). However, no significant differences were found in the levels of α-syn mRNA measured by quantitative RT-qPCR at 14 days of culture (Figure 3D), suggesting that latrepirdine regulated α-syn levels via a mechanism that was probably post-translational. This is consistent with a role for protein degradation (e.g., autophagy) in α-syn catabolism.
Figure 3. Latrepirdine stimulates the catabolism of α-synuclein and protects against α-synuclein-related cytotoxicity in differentiated SH-SY5Y neurons.
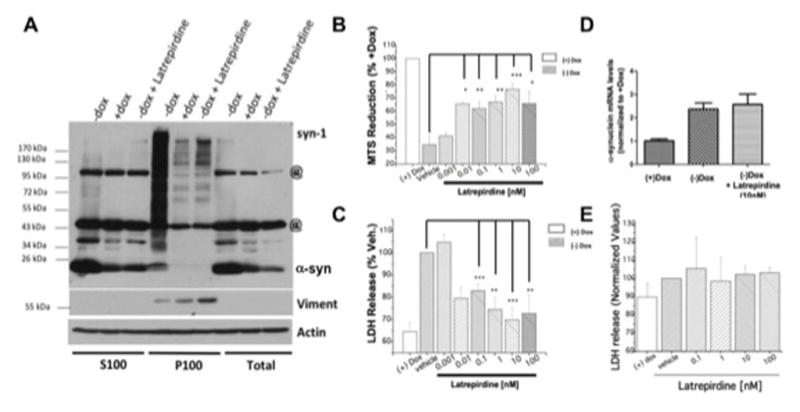
SH-SY5Y cells stably expressing inducible (“tet-off”) human α-syn were differentiated (without dox) using retinoic acid and treated in the absence (Vehicle; without dox) or presence of latrepirdine (without dox) for 14 days. (A) Cells treated for 14 days in the presence of 10nM latrepirdine were lysed and Triton X-100 soluble (S100, left) and insoluble (P100, middle) pellets were obtained by ultracentrifugation (“@” are non-specific bands). Treatment with latrepirdine decreased the levels of all forms of α-syn, including aggregates and soluble monomer in the Triton X-100 soluble (monomer = 52.7% reduction; SEM=10.38; p<0.01), total (monomer = 68.0% reduction; SEM=15.64; p<0.01), and insoluble fractions (aggregated = 74.8% decrease; SEM=9.397; p<0.001). (B) Quantification of LDH released to the culture medium or (C) measurements of tetrazolium MTS reduction in SH-SY5Y cells was used to determine cell viability in the absence of presence of a range of concentratins of latrepirdine (as indicated). (D) α-syn mRNA levels were analyzed using qRT-PCR, normalized to α-tubulin mRNA levels. (E) Inducible SH-SY5Y cells overexpressing β-galactosidase (β-gal) were cultured in the absence of dox in 96-well plates and treated after 24 hours with RA and different concentrations of latrepirdine (as indicated), and LDH release was quantified from the culture medium. All figures are representative of three or more independent experiments. Graphs are mean ± SEM; *p<0.05; **p<0.01; ***p<0.001.
We observed that differentiated SH-SY5Y neurons grown in the absence of dox for 14 days (overexpressing α-syn), exhibited increased necrotic cell death (Figure 3B,C). This conclusion was supported by the observation of a ~36% rise in levels of LDH released into the culture medium in cells over-expressing α-syn as compared to cells maintained in medium containing dox (Figure 3C). In addition, we determined the viability of the remaining non-necrotic cell by measuring the capacity of these cells to metabolize the tetrazolium 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) into a soluble formazan. Reduction in the capacity of MTS metabolism is an indicator of cell injury. We found that latrepirdine treatment decreased cell death of α-syn overexpressing cells as measured by LDH release (Figure 3C) and increased their viability as indicated by their capacity to reduce MTS (Figure 3B). This cytoprotective effect of latrepirdine treatment was observed with concentrations of latrepirdine as low as 0.1 nM. As a control, we also measured cell death and viability in SH-SY5Y differentiated cells overexpressing β-galactosidase in the absence (with or without dox) or presence of latrepirdine. These cells did not display significant changes in cell death or viability (Figure 3E).
Chronic latrepidine treatment was associated with enhanced autophagy and reduction of α-synuclein monomer in the brains of wildtype mice
In a related paper19, we provided evidence that chronic latrepirdine (3.5 mg/kg/day) administration enhanced autophagy, reduced accumulation of Aβ42, and improved behavioral impairment in an AD mouse model. We next investigated whether chronic administration of latrepirdine to wild-type mice for 3 weeks was sufficient to activate autophagy and/or to alter α-syn levels in their brains. Male wild-type mice received 21 once-daily intraperitoneal (i.p.) injections of either latrepirdine (3.5 mg/kg; n=4) or vehicle (0.9% saline; n=4). Following treatment, mice were sacrificed and whole-brain lysates were analyzed for autophagy markers, including LC3 and the polyubiquitin binding protein p62/SQSTM1 (p62), and α-syn levels by Western blot (Figure 4). LC3 is converted from its cytosolic precursor (LC3-I) to its membrane-associated form (LC3-II) by the addition of phosphatidylethanolamine, which allows for the recruitment of LC3-II to the outer membrane of the autophagosome26. When autophagy is induced, autophagosomes accumulate and mature until terminal degradation upon fusion with the lysosome26.
Figure 4. Latrepirdine stimulates α-synuclein catabolism in parallel to markers of enhanced autophagy.
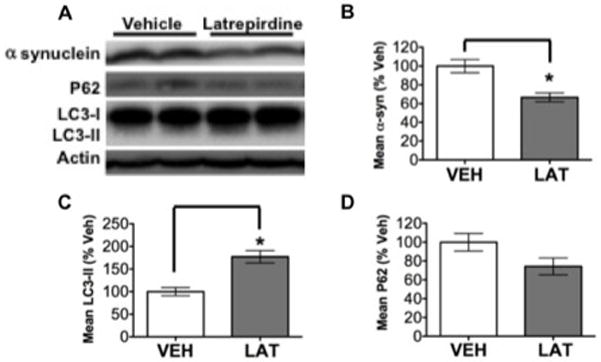
Two-month-old wild-type male mice received 21 consecutive once daily i.p. injections of 3.5mg/kg latrepirdine (n=4; LAT) or equivalent volume of vehicle (0.9% saline; n=4; VEH). (A) Western blot analysis of wild-type mouse brains for protein levels of soluble α-syn, p62, and LC3. (B–D) Quantification of Western blot band integrated density. Graphs are representative littermates; mean ± SEM; *p<0.05.
Following 21 days of latrepirdine treatment, we noted a significant decrease in α-syn monomer in the brains of WT mice, and this was associated with significantly elevated LC3-II accumulation but without accumulation of p62. These results support the hypothesis that latrepirdine stimulates α-syn degradation in vivo in the WT mouse brain, which is likely via induction of autophagy, an observation that dovetails with our recently reports10, 19. Furthermore, the current results suggest that the turnover of α-syn monomer may be determined, at least in part, by autophagic stasis, raising the possibility that enhanced autophagic activity might mitigate the toxicity associated with cellular accumulation of α-syn. Ongoing experiments with latrepirdine in cellular and animal models expressing human α-syn will help to elucidate the specific mechanisms of latrepirdine action on regulation of the autophagic pathway with regard to clearance of pathological α-syn.
Discussion
Protein aggregates are resistant to degradation and therefore inherently longer-lived, allowing for sustained toxicity associated with the buildup of these cytotoxic assemblies27. Based on this principle, a wealth of studies have recently investigated the potential of protein catabolism pathway as therapeutic targets for several neurodegenerative diseases, including AD, HD and PD. Evidence indicates that α-syn is a substrate of autophagy – a conserved catabolic pathway for the degradation of long-lived proteins, cellular organelles, and protein aggregates. Studies of α-syn clearance indicate that α-syn is primarily degraded by the ubiquitin-proteasome system (UPS) under normal conditions in vivo; however, autophagy may be the primary clearance pathway in cases of α-syn overexpression or for the degradation of oligomeric α-syn, which may be too large to fit into the narrow pore of the UPS28. Cellular studies also indicate that α-syn catabolism can be regulated by SMERs, which non-specifically increase Atg5-dependent autophagy18. Another report suggests that conditional deletion of ATG7 results in the accumulation of α-syn aggregates in dopaminergic neurons of mice29.
Herein, we report that latrepirdine, a neuroactive compound with a compelling clinical safety profile, protects S. cerevisiae and differentiated SH-SY5Y neurons against α-syn-induced (or Aβ42-induced10) cytotoxicity. Interestingly, S. cerevisiae expressing TDP43, FUS, or htt-103Q were not protected by latrepirdine in these assays and these findings (specifically regarding htt-103Q) are partially validated by the recent failure of latrepirdine in a phase II trial for HD9. Taken together, we suggest that the latrepirdine-related protection against α-syn cytotoxicity are likely due to promotion of a conserved catabolic pathway (e.g., autophagy). We provide evidence here (Figures 2 and 4), and elsewhere10, 19, that latrepirdine stimulates autophagy, resulting in the degradation of α-syn and increased cell viability yeast, in differentiated SH-SY5Y neurons, and in vivo in mouse brains. Based on these findings, we propose that latrepirdine may represent a potentially viable lead compound that might yield clinical benefit for synucleinopathies following optimization of its pro-autophagic and/or pro-neurogenic activity.
Recently, latrepirdine failed in a US-based phase II replication trial30 of a prior successful Russian phase II trial of mild-to-moderate AD8. We speculate that this may, in part, have occurred due to a lack of understanding of the underlying molecular mechanism(s) of latrepirdine. Given our observations reported herein, any disparity in the contribution of α-syn to the neuropathology in the Russian vs US latrepirdine studies might also explain, at least in part, the inconsistency of the cognitive benefit in the two trials. If this speculation were correct, then one would predict that latrepirdine might be more beneficial in treating synucleinopathies such as PD, LBD, REMSD. and/or MSA by comparison to AD, in which an unpredictable and lower percentage (30–50%) of the clinical population harbors α-syn neuropathology13–15. Since animal models of these diseases are now available, one important direction of this research will be to assess the potential of latrepirdine to improve the neuropathology and/or clinical manifestations of synucleinopathy. These experiments are now underway.
Supplementary Material
Acknowledgments
The work in this manuscript was used in a dissertation by JWS as partial requirement for the fulfillment of the PhD degree. JWS is a trainee in the Integrated Pharmacological Sciences Training Program supported by grant T32GM062754 from the National Institute of General Medical Sciences. MLL was supported by the Deutsche Forschungsgemeinschaft. SG is a member of the Oligomer Research Consortium of the Cure Alzheimer’s Fund.
The authors acknowledge the generous support of the NH&MRC (APP1009295 to RM, GV, SG), McCusker Alzheimer’s Research Foundation (RM, GV); Fidelity Biosciences Research Initiative (SJ, JL, DR, GAP); Cure Alzheimer’s Fund (SG); the US Department of Veterans Affairs (SG); and the NIH (P01AG10491 to SG; P50AG05138 to Mary Sano; P30 NS061777 and S10 RR022415 to RW; R01NS060123 and U54RR022220 to ZY). The authors would also like to thank Rosilyn Kazanjian for her gift in memory of Powel Kazanjian. The authors would like to thank Loren E. Khan and Justine Bonet for technical assistance in animal colony management and Dr. Yun Zhong for technical support.
Footnotes
Supplementary information is available at Molecular Psychiatry’s website.
Potentially Competing Financial Interests: A.P. is Vice President of Preclinical Development for Medivation, Inc. S.G. holds research grant support from Amicus Pharmaceuticals and is a consultant to the Pfizer-Janssen Alzheimer’s Immunotherapy Alliance. G.A.P. is on the scientific advisory boards of Amicus Pharmaceuticals and Neurophage, Inc.
References
- 1.Lermontova NN, Lukoyanov NV, Serkova TP, Lukoyanova EA, Bachurin SO. Dimebon improves learning in animals with experimental Alzheimer’s disease. Bull Exp Biol Med. 2000;129:544–546. doi: 10.1007/BF02434871. [DOI] [PubMed] [Google Scholar]
- 2.Grigorev VV, Dranyi OA, Bachurin SO. Comparative study of action mechanisms of dimebon and memantine on AMPA- and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull Exp Biol Med. 2003;136:474–477. doi: 10.1023/b:bebm.0000017097.75818.14. [DOI] [PubMed] [Google Scholar]
- 3.Giorgetti M, Gibbons JA, Bernales S, et al. Cognition-enhancing properties of Dimebon in a rat novel object recognition task are unlikely to be associated with acetylcholinesterase inhibition or N-methyl-D-aspartate receptor antagonism. J Pharmacol Exp Ther. 2010;333:748–757. doi: 10.1124/jpet.109.164491. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Ferruzzi MG, Varghese M, et al. Preclinical study of dimebon on beta-amyloid-mediated neuropathology in Alzheimer’s disease. Mol Neurodegener. 2011;6:7. doi: 10.1186/1750-1326-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bachurin S, Bukatina E, Lermontova N, et al. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann N Y Acad Sci. 2001;939:425–435. doi: 10.1111/j.1749-6632.2001.tb03654.x. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington’s disease. Mol Neurodegener. 2008;3:15. doi: 10.1186/1750-1326-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pieper AA, Xie S, Capota E, et al. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010;142:39–51. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doody RS, Gavrilova SI, Sano M, et al. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372:207–215. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- 9.Kieburtz K, McDermott MP, Voss TS, et al. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch Neurol. 2010;67:154–160. doi: 10.1001/archneurol.2009.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bharadwaj P, Verdile G, Barr RK, Gupta V, et al. Autophagy promotes the clearance of intracellular Aβ42 in Saccharomyces cerevisiae. submitted manuscript. [Google Scholar]
- 11.Bachurin SO, Shelkovnikova TA, Ustyugov AA, et al. Dimebon Slows Progression of Proteinopathy in gamma-Synuclein Transgenic Mice. Neurotox Res. 2011 doi: 10.1007/s12640-011-9299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein and peptide letters. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 13.Broe M, Shepherd CE, Mann DM, et al. Insoluble alpha-synuclein in Alzheimer’s disease without Lewy body formation. Neurotox Res. 2005;7:69–76. doi: 10.1007/BF03033777. [DOI] [PubMed] [Google Scholar]
- 14.Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–697. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wirths O, Bayer TA. Alpha-synuclein, Abeta and Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:103–108. doi: 10.1016/S0278-5846(02)00339-1. [DOI] [PubMed] [Google Scholar]
- 16.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–1942. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steele JW, Lachenmayer ML, Ju S, Stock A, et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. doi: 10.1038/mp.2012.106. submitted manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steele JW, Kim SH, Cirrito JR, et al. Acute dosing of latrepirdine (Dimebon), a possible Alzheimer therapeutic, elevates extracellular amyloid-beta levels in vitro and in vivo. Mol Neurodegener. 2009;4:51. doi: 10.1186/1750-1326-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klionsky DJ. Monitoring autophagy in yeast: the Pho8Delta60 assay. Methods Mol Biol. 2007;390:363–371. doi: 10.1007/978-1-59745-466-7_24. [DOI] [PubMed] [Google Scholar]
- 22.Vekrellis K, Xilouri M, Emmanouilidou E, Stefanis L. Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J Neurochem. 2009;109:1348–1362. doi: 10.1111/j.1471-4159.2009.06054.x. [DOI] [PubMed] [Google Scholar]
- 23.Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission: an infectious conformation compatible with two highly divergent yeast prion proteins. Cell. 2005;121:49–62. doi: 10.1016/j.cell.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 26.Yue Z, Friedman L, Komatsu M, Tanaka K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta. 2009;1793:1496–1507. doi: 10.1016/j.bbamcr.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knauer MF, Soreghan B, Burdick D, Kosmoski J, Glabe CG. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc Natl Acad Sci U S A. 1992;89:7437–7441. doi: 10.1073/pnas.89.16.7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci. 2011;31:14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman LG, Lachenmayer ML, Wang J, et al. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci. 2012;32:7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller G. Pharmacology. The puzzling rise and fall of a dark-horse Alzheimer’s drug. Science. 2010;327:1309. doi: 10.1126/science.327.5971.1309. [DOI] [PubMed] [Google Scholar]
- 31.Cooper AA, Gitler AD, Cashikar A, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju S, Tardiff DF, Han H, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9:e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- 35.He C, Song H, Yorimitsu T, et al. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol. 2006;175:925–935. doi: 10.1083/jcb.200606084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.