

Abstract
Farnesyl pyrophosphate synthase (FPS; EC 2.5.1.10) is a key enzyme in isoprenoid biosynthetic pathway and provides precursors for the biosynthesis of various pharmaceutically important metabolites. It catalyzes head to tail condensation of two isopentenyl pyrophosphate molecules with dimethylallyl pyrophosphate to form C15 compound farnesyl pyrophosphate. Recent studies have confirmed FPS as a molecular target of bisphosphonates for drug development against bone diseases as well as pathogens. Although large numbers of FPSs from different sources are known, very few protein structures have been reported till date. In the present study, FPS gene from medicinal plant Bacopa monniera (BmFPS) was characterized by comparative modeling and docking. Multiple sequence alignment showed two highly conserved aspartate rich motifs FARM and SARM (DDXXD). The 3-D model of BmFPS was generated based on structurally resolved FPS crystal information of Gallus gallus. The generated models were validated by various bioinformatics tools and the final model contained only α-helices and coils. Further, docking studies of modeled BmFPS with substrates and inhibitors were performed to understand the protein ligand interactions. The two Asp residues from FARM (Asp100 and Asp104) as well as Asp171, Lys197 and Lys262 were found to be important for catalytic activity. Interaction of nitrogen containing bisphosphonates (risedronate, alendronate, zoledronate and pamidronate) with modeled BmFPS showed competitive inhibition; where, apart from Asp (100, 104 and 171), Thr175 played an important role. The results presented here could be useful for designing of mutants for isoprenoid biosynthetic pathway engineering well as more effective drugs against osteoporosis and human pathogens.
Abbreviations
IPP - Isopentenyl Pyrophosphate, DMAPP - Dimethylallyl Pyrophosphate, GPP - Geranyl Pyrophosphate, FPP - FPPFarnesyl Pyrophosphate, DOPE - Discrete Optimized Protein Energy, BmFPS - Bacopa monniera Farnesyl Pyrophosphate Synthase, RMSD - Root Mean square Deviation, OPLS-AA - Optimized Potentials for Liquid Simulations- All Atom, FARM - First Aspartate Rich Motif, SARM - Second Aspartate Rich Motif.
Keywords: Bacopa monniera, Bisphosphonates, Comparative modeling and docking, Farnesyl pyrophosphate synthase
Background
Farnesyl pyrophosphate synthase (FPS; EC 2.5.1.10) is an important chain elongation enzyme of the terpenoid biosynthetic pathway and belongs to the E-family of the prenyltransferases [1]. It catalyzes the condensation of hydrocarbon moieties of DMAPP (C5) and GPP (C10) to IPP (C5) and furthermore to produce FPP (C15) [2]. Formation of FPP is a branch step responsible for directing carbon flow towards sesquiterpenes, triterpenes, steroids and farnesylated proteins in the isoprenoid pathway. FPS gene has been characterized from several plants [3–6] and human [7]. In plants a number of reports have shown the physiological role of FPS in secondary metabolite biosynthesis. Transgenic plants with overexpression of FPS gene showed that FPS plays a regulatory role in sesquiterpenes biosynthesis [8]. However, in other report it was suggested that FPS supplied the carbon flow into phytosterol and carotenoids biosynthesis in tobacco overexpressing yeast FPS [9]. Three dimensional structure detemination of proteins is a foundation for many biological aspects. Bioinformatics and computational biology provide an important contribution in protein structure prediction. Biophysical techniques like X-ray diffraction and NMR always remain choices for structure determination of biomolecules for researchers. However, due to technical intricacy, the numbers of proteins are modeled by computational methods to interpret the biological functions. Crystal structures of FPS from various species including G. gallus [10], humans [11], Trypanosoma cruzi [12] and Plasmodium vivax [13] have been solved. Some related protein structures were also investigated in plants such as Arabibopsis polyprenyl pyrophosphate synthase [14] and tobacco 5-epi-aristolochene synthase [15], but little structural information about plant FPSs is available.
T. cruzi FPS has been characterized by homology modeling and molecular dynamics studies and it has been proposed as a model for the active site dependent design of novel inhibitors for the treatment of Chagas' disease [16]. In other report, Leishmania donovani and Leishmania major, FPSs have been characterized for development of potential anti leishmanial drug through homology modeling and docking studies [17]. Bisphosphonates are class of drugs that prevent the loss of bone mass and, used to treat osteoporosis and similar diseases. Nitrogen containing bisphosphonates inhibits the growth of various disease causative agents [18]. The bisphosphonates mainly bind to the DMAPP binding site and competitively inhibit the binding of DMAPP [17].
B. monniera is an important medicinal herb in Ayurvedic medicine, used as a “memory booster”. The medicinal property of this plant is mainly attributed to triterpene saponins bacosides. Bacosides are synthesized via isoprenoid pathway and FPS plays a key regulatory role. In this study, we modeled three dimensional structure of BmFPS based on comparative modeling approach to establish a basis for its biological role and interaction properties. Molecular interactions with substrates and inhibitors were also studied by docking simulations, which could certainly provide mechanistic insight in development of mutants for better understanding of reaction mechanisms. These structural studies will give an authoritative approach to study catalysis mechanism of BmFPS and its potential use for pathway engineering in plants for altered medicinal value and to develop the drugs not only for bone related disease but also to inhibit the growth of different disease causative agents.
Methodology
Sequence analysis, alignment, and secondary structure prediction:
Farnesyl pyrophosphate synthase protein sequence of B. monniera, BmFPS (NCBI GenBank Ac. No.GU385740) was retrieved from NCBI GenBank database (http://www.ncbi.nlm.nih.gov). BLAST algorithm against protein data bank (PDB) was used to carry out the sequence homology searches. The sequence and 3D structure of template proteins were extracted from the PDB database [19]. Multiple alignments of the amino acid sequences were carried out with the Clustal W 1.8 program (http://www.ebi.ac.uk/clustalw/). Conserved domains in BmFPS were detected using Conserved Domain Database search tool (CDD) on NCBI server (http://www.ncbi.nlm.nih.gov/structure/cdd/wrpsb.cgi). The PSIPRED V.3 programme was used to predict secondary structure of BmFPS amino acid residues [20].
Comparative modeling of BmFPS:
Comparative modeling was used to build the three dimensional model of BmFPS using template amino acid sequences of closest homologues for which X-ray crystal structures are available. An X-ray crystal structure of G. gallus FPS (PDB code: 1UBY) was used as a template [10]. BmFPS homology models were generated using software Modeller 9v10 [21]. The coordinates of two Mg2+ atoms in the models were obtained from template and positioned in target protein. Out of 50 models generated, the model with the lowest DOPE scores was taken as the final model. The protein model was imported to maestro window and energy was minimized using the protein preparation wizard by applying OPLS_2005 force field (Schrödinger, Inc.). Minimizations were performed until the average root mean square deviation (RMSD) of the non-hydrogen atoms reached 0.3 Å.
Model evaluation:
Stereochemical analyses of the BmFPS homology model was carried out using Ramachandran plot obtained from PROCHECK [22], overall quality factor by ERRAT analysis [23], Verify3D [24] and ProSAII programs [25]. Superimposition and calculation of the (RMSD) between model and template, was made by Chimera software [26] using the Carbon alpha fitting method. Diagrammatic representations of the structures were generated using PyMOL 1.3 software [27].
Docking studies of BmFPS model with substrates and inhibitors:
The structure data files (SDF) of the substrates and inhibitors (ligands) were obtained from the PubChem database. Protein– ligand docking simulations were conducted using Glide module of Schrödinger, Inc. [28–30]. After ensuring that protein and ligands are in correct orientations for docking, the receptorgrid files were generated using grid-receptor generation program. To alleviate the potential for non-polar parts of the receptor, we balanced van der Waals radii of receptors by 1.00 with partial atomic charge 0.25. A grid box dimension was generated at the centroid of the active site. The ligands were docked with the modeled BmFPS active site using the “xtra precision” Glide algorithm. The OPLS-AA force field was used for the refinement of docking results including torsional and rigid-body movements of the ligand. The final energy estimation was done with Glide score (GScore) and a best orientation was taken as the output for a particular ligand. The interactions of the ligands with protein were visualized and the figures were formed using PyMOL 1.3.
Results
BmFPS showed significant sequence identity with four prospective templates for comparative modeling that is, G. gallus (PDB ID-1UBY, identity- 47%), Human (4DEM, 45%), T. cruzi (1YHK, 38%) and P. vivax (3MAV, 36%). Multiple sequence alignment with templates showed highly conserved two aspartate rich motifs (DDXXD) called FARM (100DDIMD104 in BmFPS) and SARM (239DDYLD243 in BmFPS), and residues important for catalytic activity (Figure 1A Green and Red highlighted). FARM is highly conserved and has been designated as chain length determination (CLD) region [1]. In most FPSs, the 4th and 5th aromatic amino acids before FARM motif are involved in the product chain length specificity of the enzyme [3,31–33]. In case of BmFPS, Tyr95 and Phe96 residues are supposed to be involved in chain length specificity.
Figure 1.

A) ClustalW alignment of BmFPS (ADV03080.1) with templates Gallus gallus FPS (PDB ID-1UBY), Human FPS (4DEM), Trypanosoma cruzi (1YHK) and Plasmodium vivax (3MAV). The FARM and SARM (DDXXD) motifs are highlighted in green color and boxed in rectangle. Single conserved residues are highlighted in green, and those in red are important catalytic residue. Yellow highlighted residues (Tyr-Phe in BmFPS) play an important role in determining the product specificity; B) NCBI conserved domain database search (CDD) showing different motifs involved in catalytic activity.
The crystal structure of G. gallus (PDB ID- 1UBY, resolution 2.4Å) with maximum sequence identity was considered as a best hit (E-value 4e-105) and used as a template to generate 3D model of BmFPS. CDD search at NCBI showed specific hits with Trans_IPPS_HT and superfamily of Isoprenoid_Biocyn_C1 (Figure 1B). The secondary structure of BmFPS predicted by PSIPRED server showed only 14 α-helices and 15 coils without any β-sheets (Supplementary Figure S1).
BmFPS model structure and analysis:
Total 50 models of BmFPS were generated with template 1UBY and their discrete optimized protein energy (DOPE) was calculated. The model No.14 (BmFPS.B999900014.pdb) having lowest DOPE score (-44021.3125) was considered as the best model of BmFPS. The RMSD value of alpha carbon (Cα) of the BmFPS model was calculated by superimposition with templates on Chimera and it was found to be 0.953 Å. The overall conformation of the model was very similar to the template. The final 3D BmFPS model and its superimposition with template are shown in (Figure 2 A & B).
Figure 2.
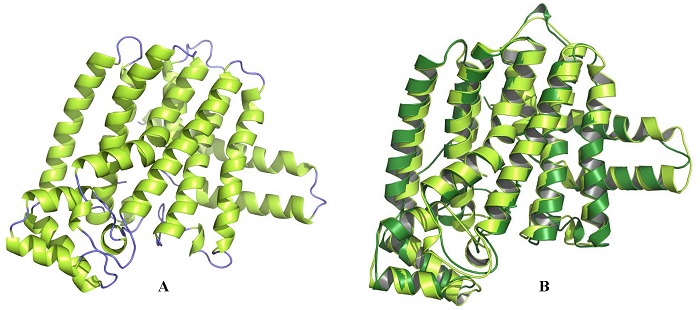
A) Ribbon diagram of BmFPS homology model showing α-helices and coils; B) Superimposition of modeled BmFPS (lemon yellow) and template G. gallus FPS (PDB:1UBY, green color).
PROCHECK analysis of the modeled BmFPS protein showed that 92.1 % of the residues are located in the most favored region, 7.3 % in additional allowed region and 0.6 % in generously allowed region of the Ramachandran plot (Figure 3A; Table 1 (see supplementary material). The overall quality factor of modeled BmFPS in ERRAT analysis was 88.235, expressed as the percentage of the protein for which the calculated error value falls below the 95 % rejection limit (Supplementary Figure S2) and the Verify 3D score was 90 %. Z-plot analysis (ProSA) of the modeled protein measures compatibility between its sequence and structure. Z score value obtained for BmFPS model (−10.16) indicated its location within the space of protein related to X-ray (dark black point Figure 3B). This value was quite comparable with template 1UBY (−10.72) suggested that the obtained model was reliable. Finally, the resultant energy minimized BmFPS model satisfying evaluation criteria's was further used for docking analysis with ligands.
Figure 3.
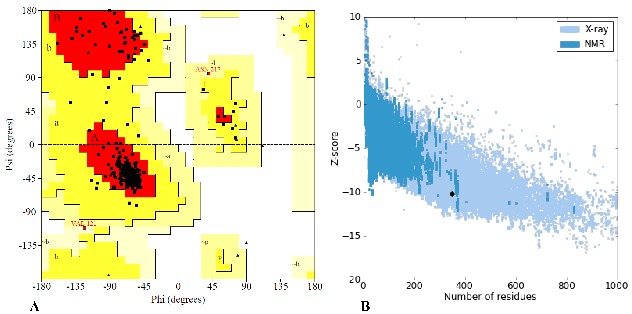
A) Ramachandran plot (PROCHECK) showing the dihedral angles Psi and Phi of amino acid residues, in which residues lie in most favoured regions are in red curves (ABL) and additional allowed regions are [a,b,l,p] in dark yellow curves; B) Z plot analysis (ProSA) of modeled BmFPS. The dark black spot represents the Z-score of the BmFPS model and is located within the space of protein related to X-ray.
Molecular interactions of ligands with modeled BmFPS:
Obtained BmFPS model was docked with substrates IPP, DMAPP and GPP; and with inhibitors, N-containing bisphosphonates (NBPs; risedronate, alendronate zoledronate and pamidronate). When IPP was docked with BmFPS model, Asp (100, 104) of FARM and Asp (171, 257, 260) (Figure 4A) showed interactions; whereas in case of DMAPP, apart from Asp (100, 104, 171), Arg109 and Lys197 (Figure 4B) play an important role in catalysis. When GPP is docked with modeled BmFPS, the amino acid residues involved were Asp (100, 104, and 171), Arg109 and Lys (197, 262) (Figure 4C). The docking interactions of the substrates and modeled BmFPS imply that the DMAPP and GPP have more negative binding energy of - 90.976 kJ/mol and -98.420 kJ/mol respectively, while IPP shows binding energy of -74.978. The Glide score represents docking energy, and therefore a low Glide score indicates strong binding. DMAPP shows lowest Glide score (-5.509) than GPP (- 5.390) and IPP (-2.764), suggested that DMAPP and GPP have strong affinity with BmFPS over IPP Table 2 (see supplementary material). Docking simulations of BmFPS with substrates are quite similar as that of previously observed for other homology models [16, 17] and the structure of other FPSs [10, 34]. Furthermore, docking conformations displayed key interactions of Lys197 and Lys262 residues with substrates, different from catalytic aspartate motif. Recently, Fisher et al. [35] showed that the loss of FPS activity and cell death of Saccharomyces cerevisiae was observed when mutations were subjected at position Lys197 and Lys254. Thus, apart from Asp (100, 104 and 171), Lys197 and Lys262 residues play pivotal role in catalysis mechanism of BmFPS.
Figure 4.
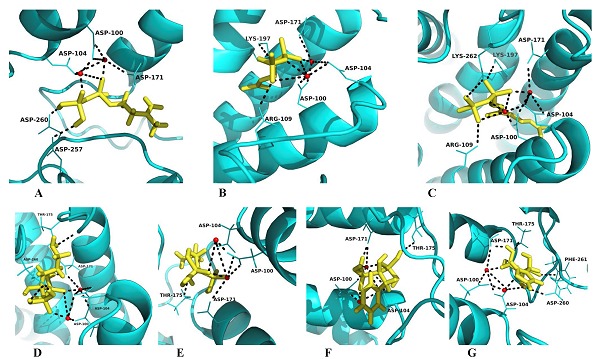
Molecular interactions of modeled BmFPS with ligands. (A) IPP, (B) DMAPP, (C) GPP, (D) Alendronate, (E) Pamindronate, (F) Risedronate and (G) Zolendronate. Ligands are shown in yellow, whereas red coloured spheres denotes Mg2+ molecules. Dotted black line represents H-bonding.
Docking of modeled BmFPS with NBPs showed similar interacting residues as with IPP, DMAPP and GPP (Figure 4D-G). Thr175 and Asp260 residues form electrostatic interactions with alendronate and zoledronate. Furthermore, Phe261 was found to be involved in binding of zoledronate Table 2 (see supplementary material). Gabelli et al. [12] showed that, when risedronate and alendronate bind with FPS enzyme, it undergoes conformational change and usually DMAPP is replaced. This conformational change in FPS structure leads to death of the parasite. On the basis of Glide scores and Glide ligand efficiency, we can conclude that all four nitrogen containing bisphosphonates (NBPs) having almost same affinity (slightly higher for pamindronate) for BmFPS.
Conclusion
FPS is an important regulatory enzyme in isoprenoid biosynthesis pathway which directs carbon flow towards sesquiterpene and triterpenes. Drugs like bisphosphonates are used to treat bone diseases and act as a competitive inhibitor of FPS, and cause the death of different disease causative parasites. In the present study, homology model of BmFPS was generated using G. gallus FPS as a reference structural homologue. Docking analysis with substrates and inhibitors (NBPs) illustrated important catalytic residues, and results were corroborated by experimental results from other sources. Asp100 and Asp104 residues of FARM, and also Lys197 and Lys262 are found to be important for catalysis of BmFPS. Further biochemical and in vivo investigations of in silico interpretations will provide an authoritative approach for novel drugs design and development; ultimately an application in medicinal chemistry for human welfare and society.
Supplementary material
Acknowledgments
Authors thank National Chemical Laboratory for providing all the necessary facilities for completion of the work. This work was supported by grant from Council for Scientific and Industrial Research (CSIR), New Delhi, India
Footnotes
Citation:Vishwakarma et al, Bioinformation 8(22): 1075-1081 (2012)
References
- 1.MK Dhar, et al. N Biotechnol. 2012 [Google Scholar]
- 2.HV Thulasiram, CD Poulter. J Am Chem Soc. 2006;128:15829. [Google Scholar]
- 3.N Cunillera, et al. J Biol Chem. 1996;271:7774. doi: 10.1074/jbc.271.13.7774. [DOI] [PubMed] [Google Scholar]
- 4.J Gaffe, et al. Plant Physiol. 2000;123:1351. doi: 10.1104/pp.123.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.M Closa, et al. Plant J. 2010;63:512. [Google Scholar]
- 6.P Gupta, et al. Plant Growth Regul. 2011;65:93. [Google Scholar]
- 7.DJ Wilkin, et al. J Biol Chem. 1990;265:4607. [PubMed] [Google Scholar]
- 8.Chen, et al. Plant Sci. 2000;155:179. doi: 10.1016/s0168-9452(00)00217-x. [DOI] [PubMed] [Google Scholar]
- 9.S Daudonnet, et al. Mol Breed. 1997;3:137. [Google Scholar]
- 10.LC Tarshis, et al. Proc Natl Acad Sci U S A. 1996;93:15018. [Google Scholar]
- 11.YS Lin, et al. J Med Chem. 2012;55:3201. doi: 10.1021/jm201657x. [DOI] [PubMed] [Google Scholar]
- 12.SB Gabelli, et al. Proteins. 2006;62:80. [Google Scholar]
- 13.JD Artz, et al. J Biol Chem. 2011;286:3315. [Google Scholar]
- 14.FL Hsieh, et al. Plant Physiol. 2011;155:1079. doi: 10.1104/pp.110.168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.JP Noel, et al. ACS Chem Biol. 2010;5:377. [Google Scholar]
- 16.L Sigman, et al. J Mol Graph Model. 2006;25:345. doi: 10.1016/j.jmgm.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 17.P Mukherjee, et al. J Chem Inf Model. 2008;48:1026. doi: 10.1021/ci700355z. [DOI] [PubMed] [Google Scholar]
- 18.V Yardley, et al. Antimicrob Agents Chemother. 2002;46:929. doi: 10.1128/AAC.46.3.929-931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.HM Berman, et al. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LJ McGuffin, et al. Bioinformatics. 2000;16:404. [Google Scholar]
- 21.A Sali, TL Blundell. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 22.RA Laskowski, et al. J Biomol NMR. 1996;8:477. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 23.C Colovos, TO Yeates. Protein Sci. 1993;2:1511. [Google Scholar]
- 24.R Luthy, et al. Nature. 1992;356:83. [Google Scholar]
- 25.M Wiederstein, MJ Sippl. Nucleic Acids Res. 2007;25:407. [Google Scholar]
- 26.EF Pettersen, et al. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27. http://www.pymol.org.
- 28.RA Friesner, et al. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 29.TA Halgren, et al. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 30.EM Krovat, et al. Curr Comp Aided Drug Des. 2005;1:93. [Google Scholar]
- 31.S Ohnuma, et al. J Biol Chem. 1996;271:18831. doi: 10.1074/jbc.271.31.18831. [DOI] [PubMed] [Google Scholar]
- 32.S Ohnuma, et al. J Biol Chem. 1996b;271:10087. doi: 10.1074/jbc.271.17.10087. [DOI] [PubMed] [Google Scholar]
- 33.S Ohnuma, et al. J Biol Chem. 1996c;271:30748. doi: 10.1074/jbc.271.48.30748. [DOI] [PubMed] [Google Scholar]
- 34.LC Tarshis, et al. Biochemistry. 1994;33:10871. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 35.MJ Fischer, et al. Protein J. 2011;30:334. doi: 10.1007/s10930-011-9336-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.