

Abstract
Chromohalobacter salexigens, a Gammaproteobacterium belonging to the family Halomonadaceae, shows a broad salinity range for growth. In order to reveal the factors influencing architecture of protein coding genes in C. salexigens, pattern of synonymous codon usage bias has been investigated. Overall codon usage analysis of the microorganism revealed that C and G ending codons are predominantly used in all the genes which are indicative of mutational bias. Multivariate statistical analysis showed that the genes are separated along the first major explanatory axis according to their expression levels and their genomic GC content at the synonymous third positions of the codons. Both NC plot and correspondence analysis on Relative Synonymous Codon Usage (RSCU) indicates that the variation in codon usage among the genes may be due to mutational bias at the DNA level and natural selection acting at the level of mRNA translation. Gene length and the hydrophobicity of the encoded protein also influence the codon usage variation of genes to some extent. A comparison of the relative synonymous codon usage between 10% each of highly and lowly expressed genes determines 23 optimal codons, which are statistically over represented in the former group of genes and may provide useful information for salt-stressed gene prediction and gene-transformation. Furthermore, genes for regulatory functions; mobile and extrachromosomal element functions; and cell envelope are observed to be highly expressed. The study could provide insight into the gene expression response of halophilic bacteria and facilitate establishment of effective strategies to develop salt-tolerant crops of agronomic value.
Keywords: Codon usage pattern, Correspondence analysis, Relative synonymous codon usage, Mutational bias, Halophilic bacteria
Background
The genetic code is the sequence of nucleotides in DNA or RNA that determines specific amino acid sequence in synthesis of proteins. It employs 64 codons, which can be grouped into 20 disjoint families, one family for each of the standard amino acid, and 21st family for translation termination signal. Different codons that encode the same amino acid are called synonymous codons and they usually differ by nucleotide at the third codon position. According to the number of synonymous codons related to each amino acid, thus for a gene using the universal code, there are two amino acids with one codon choice, nine with two, one with three, five with four, and three with six. These represent five synonymous families types (SF), designated as SF types 1, 2, 3, 4 and 6 [1]. The unequal or preferred usage of a particular codon by an amino acid among the SF family is termed as synonymous codon usages (SCU). Specific SCU patterns may be due to mutational bias, bias in G+C content, natural selection etc. However, SCU pattern is non-random and species-specific [2]. It has also been reported that there is significant variation of codon usage bias among different genes within the same organism [3–4].
Microorganisms belonging to diverse genetic lineages of bacteria and archeans are adapted to unusual limits of one or more abiotic factors in environment such as temperature, pressure, pH, radiation, salinity, etc. Salinity is an important deterrent to agriculture in many parts of the world, but investigations on its molecular effects are very few. Moderately halophilic bacteria, which are distributed through wide range of saline environments, constitute a heterogeneous group of microorganisms of different genera. These organisms are characterized by optimum growth at concentrations between 0.5M and 2.5M-NaCl [5]. Most of these have been isolated from either salted food or the Dead Sea, which are specialized hypersaline environments. The molecular basis of microbial resistance to salt stress is not fully understood in relation to regulation of gene expression during salinity, or anaerobic stress and this has little been examined in microorganisms. Understanding of molecular mechanisms involved in the halophilic adaptation will not only provide insight into factors responsible for genomic and proteomic stability under high salt conditions, but also, has importance for potential applications in the field of agriculture.
Chromohalobacter salexigens is a gram-negative aerobic bacterium, which is moderately halophilic in nature. It grows on a wide range of simple carbon compounds at NaCl concentrations between 0.5M and 4M, with an optimum growth at 2.0–2.5M and at an optimum temperature of 37°C [6–9]. So far codon usage bias in C. salexigens has not been investigated in detail, and it is not clear how different genes are expressed under saline environment in this organism. Therefore, it is of interest to understand factors that shape codon usage bias in this organism. Thus in this study, codon usage bias of C. salexigens genes was investigated using codon usage statistic, multivariate statistical technique and correlation analysis. The pattern of codon usage of this organism was studied based on values of codon usage indices and their correlation. The factors responsible for codon usage variation among genes were determined. Moreover, the expressivity level of genes, according to various functions was also determined with a view to understand the highly expressed genes and their optimal codons.
Methodology
The gene sequences (2230) in FASTA format related to various functions in Chromohalobacter salexigens were retrieved from comprehensive microbial resource (http://www.tigr.org/CMR) and given in Table 1 (see supplementary material). In order to minimise sampling errors, sequences with length < 300bp, redundant data and sequences with intermediate termination codons were excluded for this study. Thus the remaining dataset consisting of 2147 gene sequences were used for the analysis. PERL script was developed to merge all the individual gene sequences together for further data processing and analysis.
Codon Usage indices:
In order to investigate the base composition of codons used by these genes, different statistic has been calculated. For each individual gene, the percentage of codons for nucleotides i.e. A, G, T and C at third position, which is represented as A3s, G3s, T3s and C3s respectively were calculated. Apart from this, values of total number of G and C nucleotides in gene i.e. GC content, frequency of codons with G or C at the third positions (GC3s) and GC skewness [(G-C)/(G+C)] were also calculated for each gene.
Measures of codon usage:
In order to investigate the characteristics of synonymous codon usage without the confounding influence of amino acid composition, the Relative Synonymous Codon Usage (RSCU) values among different codons in each gene was calculated. RSCU is defined as the ratio of the observed frequency of a codon to the expected frequency, if all the synonymous codons for those amino acids are used equally [10]. RSCU values greater than 1.0 indicate that the corresponding codons are used more frequently than the expected frequency, whereas, the reverse is true for RSCU value less than 1.0. The effective number of codons of a gene (NC) [1] was also used to quantify the codon usage bias of a gene (Wright 1990). It is calculated by the equation: Nc= 2 + s + [29/ {s2 + (1- s2)}], where, s is the value of GC3s. Nc can take values from 20 to 61, when only one codon, or all synonymous in equal frequencies were used per amino acid respectively. The sequences in which Nc values are <30 are considered as highly expressed, while those with >55 are considered as poorly expressed genes [11–12]. Another measure used for identification of gene expression is Codon Adaptation Index (CAI) is described in supplementary material. CAI value ranges from 0 to 1.0, and a higher value indicates a stronger codon bias and higher expression level [13]. In order to understand the properties of protein coding sequences, hydrophobicity ‘GRAVY’ score [14] and frequency of aromatic amino-acids ‘Aromo’ [15] in the translated gene product were also estimated.
Multivariate Statistical Analysis:
Correspondence analysis (CA) is a data dimension reduction technique which takes multivariate data and combines them into a small number of variables (axes) that explains most of the variation among the original variables [16]. In this study, CA was applied to RSCU values of 59 codons (excluding Met, Trp, and stop codons). Further, correlation analysis (P<0.01) was used for explanation of variation and association of gene feature values with axes scores. Correlation analysis was also applied for the statistics obtained from base composition with Nc value. Chi squared contingency test (P<0.01) was performed to estimate the optimal codons i.e. synonymous codons frequently used in highly expressed genes.
Software implementation:
CodonW [17] was employed for calculating the indices and measures of codon usage and also for CA. SPSS 17.0 and SAS 9.2 were used for statistical analysis.
Discussion
Codon usage analysis:
Table 2 (see supplementary material) depicts the codon usage data and RSCU values for each codon. These values were calculated by summing all the 2147 genes of C. salexigen. It was derived that C3s and G3s accounted for 54.2% and 45.3% respectively, whereas, T3s and A3s accounted for 13.2% and 8.3% respectively. This suggests that, there was much greater preferential stability in the usages of codons with C and G nucleotides at the third position as compared to A and T in the genes of this organism. Moreover, C ending codons were preferred over G ending codons. The average percentage of GC content among genes of this group was 64.9%, which is quite high and average value of GC skewness of the genes was low (0.0079), this indicated the influence of mutational bias.
Further, analysis and the usage of amino acid depict maximum usage of acidic amino acids (Asp, Glu), low proportion of hydrophobic amino acids and a high frequency of amino acids such as Gly and Ser which corroborated the work reported by Lanyi [18] for halophilic protein.
Heterogeneity of codon usage:
In order to study the codon usage variation among genes of this organism, two different indices, namely, Nc and GC3s were used to detect the codon usage variation among the genes [19]. Wright [1] suggested that genes, whose codon choice is constrained only by a G+C mutational bias, will lie on or just below the curve of the predicted value in the Nc plot (a plot of Nc versus GC3s). It is evident from Nc plot (Figure 1), that a few points lie on the expected curve towards GC rich regions, which certainly originates from the extreme compositional constraints. However, considerable number of points with low Nc values lie considerably below the expected curve. This suggests that apart from compositional bias, majority of genes have an additional codon usage bias [2]. Furthermore, most of the genes in the Nc plot fall within a restricted cloud, at a relatively narrow range of GC3s between 0.765 to 0.890 (Figure 2A) with large variation of Nc values ranging between 30 and 45 (Figure 2B). This suggests that translational selection is also responsible for codon bias among the genes. However, the presence of significant negative correlation between GC3s and Nc (r= -0.81, P<0.01) suggests strong influence of compositional constraints on codon usage bias in the genes of this organism.
Figure 1.
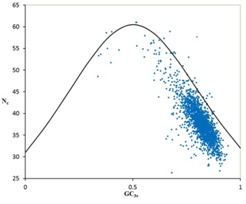
Nc plot of 2147 genes of C. salexigens. The continuous curve between GC3s and Nc under random condon usage.
Figure 2.
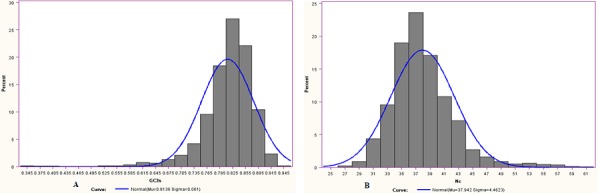
(A) Frequency of GC3s; (B) Frequency of Nc
Relationship between codon bias and gene length:
In order to study the relationship between codon bias and gene length, a plot was drawn between gene length and Nc. From Figure 3A, it is understood that shorter genes have a much wider variance in Nc values, and vice versa for longer genes. The lower Nc values in longer genes may be due to the direct effect of translation time or to the extra energy cost of proofreading associated with longer translating time. A low negative correlation was observed with gene length against Nc (r= -0.15). This reveals that gene length has little influence on codon usage of these genes. However Eyre-Walker [20] has reported that the selection for accuracy in protein translation is likely to be greater in longer genes because the cost of producing a protein is proportional to its length. Correlation between gene length and values of GC3s as well as gene position has also been worked out and similar type of result has been obtained i.e. low correlation (Figure 3B & Figure 3C). These findings suggest that gene length is not playing major role in the case of codon usage bias in this organism.
Figure 3.
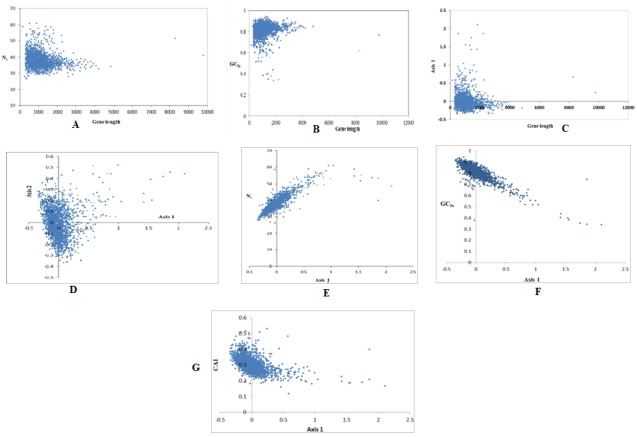
(A) Plot of Nc versus gene length; (B) Plot of GC3s versus gene length; (C) Plot of gene position on axis1 versus gene length; (D) Correspondence analysis of Relative Synonymous Codon Usage values of C. salexigens genes; (E) Scatter plot of gene position on axis 1 and Nc values; (F) Scatter plot of gene position on axis 1 and GC3s values; (G) Scatter plot of gene position on axis 1 and CAI values.
Multivariate Statistical Analysis:
In order to investigate the major possible trends in shaping codon usage variation among the genes, Correspondence Analysis (CA), a method of multivariate statistical analysis, was performed on the data of RSCU values of genes. Axis 1 shows the largest fraction (14.52%) of the variation in the data. Axis 2 describes the second largest trend (6.79%), and so on with each subsequent axis describing a progressively smaller amount of variation. Although, the first principal axis explains a substantial amount of variation of codon usage among the genes in this microorganism, its value is still lower than found in other organisms studied earlier [22]. The low value might be due to the extreme genomic composition of this genome. It is also obvious from Figure 3D that the majority of the points are clustered in an elliptical shape around the origin of axes. This indicates that, these genes have more or less similar codon usage biases. There are very few points that are widely scattered along the positive side of the first major axis, indicating that codon usage biases of these genes are not homogeneous. The first major axis is negatively correlated with G3s and C3s while correlated positively with A3s and T3s Table 3 (see supplementary material). Also, strong positive correlation exists between position of genes along the first axis with Nc (Figure 3E) and high degree of negative correlation with GC3s (Figure 3F). These findings suggest, that highly biased genes, those ending with G and C, are clustered on the negative side, whereas, the codons ending in A and T predominate on the positive side of the first major axis. Further, significant negative correlation is observed with Nc against GC3s (r=-0.81, P<0.01) and GC (r=-0.43, P<0.01), which suggests that highly expressed genes tend to use “C” or “G” at the synonymous positions as compared to lowly expressed genes in this organism. Further study reveals that, C-ending codons are preferred over Gending codons in highly expressed genes. Preference of Cending codons in the highly expressed genes might be related to the translational efficiency of the genes as it has been reported that RNY (R-Purine, N-any nucleotide base, and Ypyrimidine) codons are more advantageous for translation [23]. Thus, compositional mutation bias possibly plays an important role in shaping the genome of this organism.
Effect of gene expression level on synonymous codon usage bias:
CAI has been widely used to estimate the expressivities of genes by many workers and has been considered to be the best measure of gene expressivities [24–26]. In order to assess the effect of expressivities of genes on codon usage biases, codon adaptation index (CAI) values of all the genes of C. salexigens were calculated. In order to investigate, whether, there is a correlation between the codon usage bias and the gene expression level, the correlation coefficients were estimated between CAI values against the positions of genes along the first major axis, nucleotide composition and Nc values. From Table 4 (see supplementary material), it is found that significant negative correlations exist between the gene expression level assessed by CAI value and the positions of genes along axis 1 and Nc values. A significant positive correlation between CAI value and GC3s content is noticed while CAI has negative correlation with GC, though lower negative value. From this analysis, it can be concluded that codon usage bias among genes of C. salexigens is also affected by gene expression level. From the analysis, it can be suggested that genes with higher expression level, exhibiting a greater degree of codon usage bias and distributed at the left side of the first axis, are GC-rich and prefer to the codons with C or G at the synonymous position. A scatter diagram of the position of genes along the first major axis produced by CA on RSCU and their corresponding CAI values is shown (Figure 3G) and it is interesting to note that there is a significant negative correlation between the positions of the genes along the first major axis and their corresponding CAI values (r= -0.49 (P< 0.01), confirming that axis 1 is significantly correlated with the expression level of each gene of C. salexigens. This is a clear indication that gene expression also affects the codon usage variation among the genes in this organism. Correlation analysis of synonymous codon usage bias against hydrophobicity of each protein was also investigated (r=0.22, P< 0.01). The findings indicated that genes, encoding more hydrophobic protein and bias to G/C bases at synonymous third codon positions, showed a stronger codon bias. This was also reported by Liu et al. [27] on synonymous codon usage in maize. Although, the absolute value of this correlation coefficient is low, but it is statistically significant. Subsequently, it has been inferred that the hydrophobicity of the encoded protein played a minor role in affecting codon usage in C. salexigens too. However, no significant correlation has been observed between synonymous codon bias and aromaticity scores.
Translational optimal codons:
The χ2 test (P<0.01) has been applied between top 10% genes (highly expressed) having higher value of the major axis and 10% genes (lowly expressed) having lower value of the axis. Through this analysis, twenty-three codons were determined as the ‘optimal codons’, which were significantly more frequent among the highly expressed genes. From Table 5 (see supplementary material), it was derived that among 23 codons, there are 16 C-ending (69.6%) and 7-G ending codons (30.4%). These optimal codons might be significant to introducing point mutation, and modifying heterologous genes in order to increase the product of specific protein. Ikemura showed that there is a match between these codons and the most abundant tRNAs. In Escherichia coli [28], Drosophila melanogaster [29] and Caenorhabditis elegans [30] highly expressed genes have a strong selective preference for codons with a high concentration for the corresponding acceptor tRNA molecule; the preferred codons are those which are best recognized by the most abundant tRNAs. This trend has been interpreted as the co-adaptation between amino acid composition of protein and tRNA-pools to enhance the translational efficiency. Remarkably, in this study, there is a strong positive correlation (r = 0.84, P <0.01) between the frequency of optimal codons in each gene and respective CAI value. This suggests that translational selection influenced the codon usage of C. salexigens and the optional codons are more frequent in highly expressed genes.
Categorization of genes based on expressivity:
In order to differentiate the genes on the basis of expression level, the scores of axis 1 calculated for CA on RSCU values, were classified into three quantiles representing three different gene expression levels with corresponding cut-off percentages viz., high (75% and above), moderate (between 25% and 75%), and low (below 25%) expressed genes as shown in Table 6 (see supplementary material). The analysis depicted maximum percentage of genes belonging to category of moderately expressed genes. Further analysis revealed that genes related to regulatory functions, mobile and extrachromosomal element functions, cell envelope, protein function and cellular processes are highly expressed. Moderately expressed genes include functions related to DNA metabolism, transcription, signal transduction and protein synthesis, whereas, low expressed genes regulate central intermediary metabolism, purines, pyrimidines, nucleosides and nucleotides; amino acid biosynthesis; energy metabolism; and transport and binding protein (Figure 4). This pattern of expression is expected for this halophilic bacterium.
Figure 4.
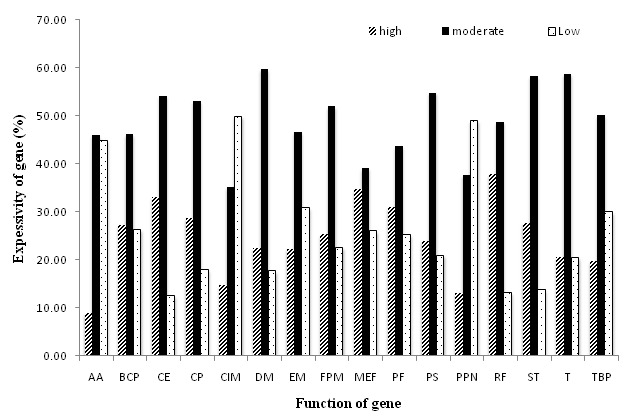
Expression level of functional genes of C. salixigens on the basis of Axis 1 based on RSCU values. AA= Amino acid biosynthesis; BCP=Biosynthesis of cofactor, Prosthetic groups and carriers; CE=Cell envelope; CP=Cellular processes; CIM=Central intermediary metabolism; DM=DNA metabolism; EM=Energy metabolism; FPM=Fatty acid & phospholipid metabolism; MEF=Mobile & extrachromosomal element functions; PF=Protein fate; PS=Protein synthesis; PPN=Purines, pyrimidines, nucleosides & nucleotides; RF=Regulatory functions; ST=Signal transduction; T=Transcription; TBP=Transport & binding protein
Conclusion
It can be inferred that high level of heterogeneity is seen within the genes of various functions in Chromohalobacter salexigens. Though codon usage of C. salexigens is largely determined by compositional constraints, translational selection is also operating in shaping the codon usage variation among the genes. The study revealed that G/C-ending codons are preferred over A/T-ending codons in highly expressed genes. Total number of codons in highly expressed genes is much higher than those in lowly expressed genes. Length of the genes also affects the codon usage bias, while aromaticity and hydrophobicity of the encoded proteins play minor role in shaping codon usage bias. A set of twenty-three codons are identified as the optimal codons. Using χ2 test at P<0.01, it was found that these codons are significantly more frequent among the highly expressed genes. As more genomes of halophilic bacteria with known gene sequences become available at public databases, it will be interesting to see if these effects are universal and whether these bacteria follow a similar trend of codon usage pattern for haloadaptation. The study could be explored to derive unique salt tolerant traits and for prediction of genes responsible for salt stress which could potentially be used in agricultural crops that are almost exclusively glycophytes.
Supplementary material
Acknowledgments
The authors acknowledge the NAIP for financial assistance of this study under the project entitled ‘Establishment of National Agricultural Bioinformatics Grid in ICAR’.
Footnotes
Citation:Sanjukta et al, Bioinformation 8(22): 1087-1095 (2012)
References
- 1.F Wright. Gene. 1990;87:23. [Google Scholar]
- 2.SK Gupta, et al. J Biomol Struc Dyn. 2004;21:527. [Google Scholar]
- 3.M Gouy, C Gautier. Nucleic Acid Res. 1982;10:7055. [Google Scholar]
- 4.T Ikemura. Mol Biol Evol. 1985;2:13. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 5.A Ventosa, et al. Can J Microbiol. 1984;30:1279. [Google Scholar]
- 6.D Canovas, et al. J Bacteriol. 1996;178:7221. doi: 10.1128/jb.178.24.7221-7226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DR Arahal, et al. Int J Syst Evol Microbiol. 2001;51:1443. doi: 10.1099/00207713-51-4-1443. [DOI] [PubMed] [Google Scholar]
- 8.KO Connor, LN Csonka. Appl Environ Microbiol. 2003;69:6334. doi: 10.1128/AEM.69.10.6334-6336.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.C Vargas, et al. Saline Systems. 2008;4:14. doi: 10.1186/1746-1448-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.PM Sharp, WH Li. J Mol Evol. 1986;24:28. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 11.PM Sharp, et al. Nucleic Acid Res. 1986;14:5125. [Google Scholar]
- 12.K Sau, et al. Virus Res. 2005;113:123. [Google Scholar]
- 13.PM Sharp, WH Li. Nucleic Acid Res. 1987;15:1281. [Google Scholar]
- 14.J Kyte, RF Doolittle. J Mol Biol. 1982;157:105. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 15.JR Lobry, C Gautier. Nucleic Acid Res. 1994;22:3174. doi: 10.1093/nar/22.15.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.H Suzuki, et al. DNA Res. 2008;15:357. [Google Scholar]
- 17. http://codonw.sourceforge.net/
- 18.JK Lanyi. Bacteriol Rev. 1974;38:272. doi: 10.1128/br.38.3.272-290.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.K Sahu, et al. J Biochem Mol Biol. 2004;37:487. doi: 10.5483/bmbrep.2004.37.4.487. [DOI] [PubMed] [Google Scholar]
- 20.A Eyre-Walker. Mol Biol Evol. 1996;13:864. doi: 10.1093/oxfordjournals.molbev.a025646. [DOI] [PubMed] [Google Scholar]
- 21.A Pan, et al. Gene. 1998;215:405. [Google Scholar]
- 22.F Alvarez, et al. Mol Biol Evol. 1994;11:790. doi: 10.1093/oxfordjournals.molbev.a040159. [DOI] [PubMed] [Google Scholar]
- 23.JC Shepherd. Proc Natl Acad Sci U S A. 1981;78:1596. [Google Scholar]
- 24.G Gutierrez, et al. Nucleic Acids Res. 1996;24:2525. doi: 10.1093/nar/24.13.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Y Nakamura, S Tabata. Microbiol Comp Genomics. 1997;2:299. doi: 10.1089/omi.1.1997.2.299. [DOI] [PubMed] [Google Scholar]
- 26.ER Tiller, RA Collins. J Mol Evol. 2000;50:249. doi: 10.1007/s002399910029. [DOI] [PubMed] [Google Scholar]
- 27.H Liu, et al. Mol Biol Rep. 2010;37:677. doi: 10.1007/s11033-009-9521-7. [DOI] [PubMed] [Google Scholar]
- 28.T Ikemura. J Mol Biol. 1981;151:389. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 29.EN Moriyama, JR Powell. J Mol Evol. 1997;45:514. doi: 10.1007/pl00006256. [DOI] [PubMed] [Google Scholar]
- 30.L Duret, et al. Trends Genet. 2000;16:287. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.