

Abstract
The recent invention of super-resolution microscopy has brought up much excitement in the biological research community. Here, we will focus on Stochastic Optical Reconstruction Microscopy/Photoactivated Localization Microscopy (STORM/PALM) to discuss the challenges in applying super-resolution microscopy to the study of developmental biology, including tissue imaging, sample preparation artifacts, and image interpretation. We will also summarize new opportunities that super-resolution microscopy could bring to the field of developmental biology.
In the past several years, the emergence of super-resolution microscopy has extended the resolution of fluorescence microscopy by more than an order of magnitude, almost reaching the scale of macromolecules (Hell; Huang et al.; Huang et al.). This new resolving power has immediately attracted the attention from biologists, wondering whether many unanswered mysteries can now be clarified and old “dogma” be revisited. Indeed, the focus of the super-resolution microscopy field has recently shifted from technological advancement to biological applications, with a number of new discoveries already made in cell biology (Kanchanawong et al., 2010; Wu et al.), neurobiology (Beaudoin et al.; Dani et al.; Frost et al.) and microbiology (Wang et al.). What can super-resolution microscopy do for developmental biology then? Are current technologies adequate to perform imaging in the context of a complex organism? What are the challenges and opportunities?
Super-resolution microscopy refers to a collection of new fluorescence microscopy methods that offer spatial resolutions far beyond the classical limit set by the diffraction of light. All of them achieve diffraction-unlimited spatial resolution by modulating close-by fluorescent molecules into different states, thus distinguishing their fluorescence signal. One approach to achieve this distinction is to spatially modulate the illumination light. The best known techniques using this approach are Stimulated Emission Depletion (STED) microscopy (Hell and Wichmann; Klar and Hell) and Structured Illumination Microscopy (SIM) (Gustafsson, 2005). The other approach is based on stochastically switching individual fluorescent molecules between a fluorescent and a dark state, which was independently invented under the names of Stochastic Optical Reconstruction Microscopy (STORM) (Rust et al.), Photoactivated Localization Microscopy (PALM) (Betzig et al.) and Fluorescence Photoactivation Localization Microscopy (FPALM) (Hess et al.). This approach collects a series of fluorescent images, each containing a sparse subset of fluorophores (either fluorescent proteins or organic dyes) activated into the fluorescent state. A super-resolution image is then reconstructed by determining the positions of individual activated fluorophores. Later implementations and improvements of this approach have added in more names such as PALMIRA (Egner et al.), dSTORM (Heilemann et al., 2008) and GSDIM (Folling et al., 2008). Some new developments even avoid the use of photoswitching (Burnette et al., 2011; Sharonov and Hochstrasser, 2006) and single-molecule localization (Dertinger et al., 2009; Mukamel et al., 2012; Zhu et al., 2012). Nevertheless, all these methods share the same fundamental principle, instrumentation, and, in most cases, the analysis procedure. Therefore, here for simplicity, we refer to this single-molecule approach of super-resolution microscopy techniques by the two best known names as STORM/PALM.
The optical configuration of a STORM/PALM microscopy is almost identical to a common total internal reflection fluorescence (TIRF) microscope. This hardware simplicity, its relatively low cost, and the high spatial resolution it can achieve make STORM/PALM particularly popular among labs who would like to join in as either developers or users of super-resolution microscopy techniques. However, despite being relatively easy to set up, obtaining “perfect” STORM/PALM images in real applications is not necessarily an easy task. Here, we will discuss about the challenges and caveats when applying STORM/PALM to the study of developmental biology. We will then provide a brief survey of opportunities in developmental biology where STORM/PALM can make unique contributions.
Although we only discuss about STORM/PALM here, we note that STED microscopy and SIM are both powerful techniques that often see similar challenges and opportunities as STORM/PALM in biological applications.
1. Super-resolution at a depth
STORM/PALM has been extremely successful in producing beautiful images of subcellular structures, including three-dimensional (Huang et al., 2008a; Huang et al., 2008b; Juette et al., 2008), multi-color (Bates et al., 2007; Bossi et al., 2008; Dedecker et al., 2012; Shroff et al., 2007) and live imaging (Hess et al., 2007; Manley et al., 2008; Shroff et al., 2008) of structures ranging from the plasma membrane to inside the nucleus. Nevertheless, except for a few cases, most of these achievements were done in cultured cells. On the contrary, the study of developmental biology routinely requires imaging in the tissue context, sometimes even long term observation in living organisms. Many of these imaging tasks are challenging even for conventional fluorescence microscopy at diffraction-limited resolution. Understandably, super-resolution microscopy in the tissue context could face much more difficulties.
For STORM/PALM, the major challenge in tissue imaging is the requirement for high sensitivity to detect individual fluorophores. The signal from one fluorescent molecule is very weak. Typical photoactivatible fluorescent proteins allow fewer than 1000 photons detected before photobleaching (Lippincott-Schwartz and Patterson, 2009). Organic dyes can be brighter, but the detected photon number is still smaller than 5000 in most cases (Dempsey et al., 2011). With the development of high numerical aperture objectives and single-photon-sensitive cameras, single-molecule imaging has been made relatively straightforward for in vitro systems and in fixed cells. Nevertheless, detecting one fluorescent molecule in a large tissue volume still has two obstacles: signal loss due to tissue scattering and tissue-induced optical aberrations, and the high background from autofluorescence and out-of-focus fluorophores. Although these two issues affect all fluorescence microscopy methods, conventional or super-resolution, STORM/PALM particularly suffers because the resulted loss of signal-to-noise ratio directly translates into worse precision to determine molecule positions, i.e. the optical resolution (Thompson et al., 2002).
For STORM/PALM in cultured cells, a common trick to reduce out-of-focus fluorescence background is to use an illumination scheme similar to TIRF microscopy but with the incident angle slightly smaller than the critical angle (Tokunaga et al., 2008). In this configuration, the excitation light is restricted to a depth within several micrometers from the coverglass, efficiently illuminating the cell but not the mounting media above. The wide use of this scheme has created the misunderstanding that STORM/PALM only works with thin samples under TIRF illumination. Very much on the contrary, the same strategy can be employed to image tissue sections, as long as the structure of interest is within a few micrometers from the section surface. For example, imaging in mouse brain sections has allowed the characterization of the protein organization of synapses, as well as development-associated changes in the density, morphology and receptor composition of synapses (Figure 1A) (Beaudoin et al., 2012; Dani et al., 2010).
Figure 1.
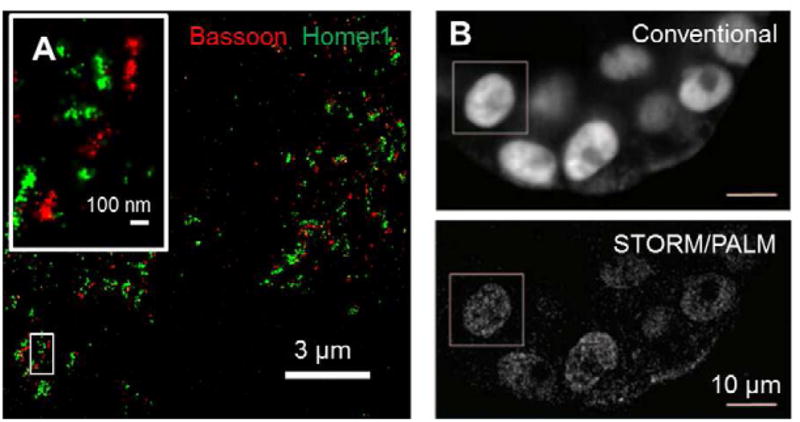
STORM/PALM imaging in tissue samples. (A) synapses in mouse brain cortex, showing immunostained presynaptic scaffolding protein Bassoon and postsynaptic density protein Homer1 imaged in cryostat sections (Beaudoin et al.) (B) Nuclei of human mammary MCF10A cell spheroids. Cells express histone H2B fused to photoactivatible fluorescent protein PAmCherry. Images were recorded at a depth of 100 μm. Panel (B) adapted from (Cella Zanacchi et al.).
To image deeper into the tissue, many fluorescence microscopy techniques have been developed in the past years, including two-photon microscopy (Denk et al., 1990) and selective-plane illumination microscopy (SPIM) (Huisken et al., 2004) for optical sectioning, as well as the use of adaptive optics to correct for tissue-induced aberrations (Girkin et al., 2009). As the field of deep tissue imaging continues to advance rapidly, we have now started to see the application of these techniques in STORM/PALM. Two-photon absorption can restrict fluorophore activation to a thin plane (Vaziri et al., 2008), thus reducing the out-of-focus background and allowing imaging into a depth of several tens of micrometers (York et al., 2011). Using SPIM to confine both activation and excitation, super-resolution images have been acquired at 100 μm depth into mammary cell spheroid (Figure 1B) (Cella Zanacchi et al., 2011). Resolution improvement at this depth and beyond is more limited, though, due to tissue scattering and aberrations. We expect that the use of adaptive optics (Izeddin et al., 2012) and/or new tissue clearing reagents (Hama et al., 2011) could substantially improve the use of super-resolution microscopy in deep tissue imaging. Alternatively, combing optical microscopy with serial physical sectioning could image through a large volume of tissue sample without the complexities in optical sectioning methods (Micheva and Smith, 2007; Nanguneri et al., 2012).
2. Super-resolved artifacts
For STORM/PALM, the precision to determine the position of a fluorescent molecule can routinely reach ~ 10 nm in standard deviation, or ~ 25 nm in full-width at half-maximum, with some of the best numbers smaller than 5 nm in standard deviation (Aquino et al., 2011; Shtengel et al., 2009; Xu et al., 2012). These values are at the same scale as the size of protein molecules. Therefore, they can be smaller than the distance between adjacent fluorophores that label the sample structure. This discrepancy does not prevent us from studying the properties of individual molecules, for example, when trying to observe the trafficking of a low-copy-number membrane protein. However, even by labeling 100% of this membrane protein, the molecule positions measured by STORM/PALM are insufficient to describe the detailed membrane morphology due to the low density (Figure 2A). In other words, the effective resolution in visualizing a “continuous” structure is also limited by the density of fluorescent labels. A simplified description of this relationship is the Nyquist sampling theorem: the distance between adjacent fluorophores needs to be smaller than one half of the feature size (Shroff et al., 2008), whereas a rigorous theory (Fitzgerald et al., 2012) is yet to be established to account for the sample shape and the randomness of probe labeling.
Figure 2.

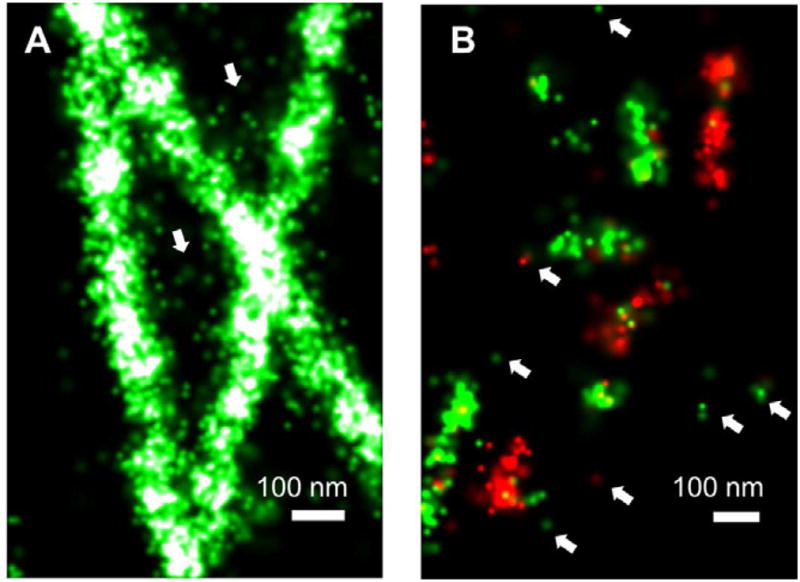
Clustering artifacts in STORM/PALM. (A) 3D super-resolution imaging of Golgi protein Giantin in a B-SC-1 cell reveal the overall morphology of Golgi stacks (left), but the low density of Giantin makes the super-resolution image discontinuous (right). With this low labeling density, judging whether some structures are connected can be difficult (arrows). (B) super-resolution images of immunostained microtubules in Drosophila S2 cells, comparing good fixation (left) and inadequate fixation (right). (C) Zoomed-in view of the boxed region in the microtubule image. Arrows points to clusters generated by individual antibody molecules either nonspecifically bound to the sample or bound to tubulin monomers not incorporated into microtubules. (D) Blinking of fluorescent protein mEos2 cause the non-clustering SrcN15 to appear clustered on the plasma membrane. Panel (D) adapted from (Annibale et al.).
In live sample STORM/PALM, a longer acquisition time for each time step will allow the collection of more single-molecule activation events, thus leading to a higher localization point density and correspondingly a better point-density-limited spatial resolution. Therefore, the spatial resolution requirement limits the temporal resolution of live imaging. Typically, to avoid overlap of single-molecule images, which hinders single-molecule localization, the average density of activated fluorophores is controlled to be no higher than 0.5 per μm2. Under this condition, 2000 camera frames give a 32 nm average point-to-point distance if all points are homogeneously distributed. At a camera frame rate of 60 frames per second, this corresponds to a 33 sec recording time to support a 64 nm spatial resolution. In practice, the effective spatial resolution could be substantially better depending on the shape of the structure. As an example, having all points in a 1 μm2 area concentrated in one clathrin-coated pit (170 nm diameter) would lead to a density-limited resolution of about 10 nm. The temporal resolution can be improved using a faster camera (Jones et al., 2011) or algorithms that can identify single molecules even when their images start to overlap (Holden et al., 2011; Huang et al., 2011; Quan et al., 2011; Zhu et al., 2012).
For fixed sample imaging, on the other hand, the acquisition time can be arbitrarily long in order to sample through all fluorophores. However, inadequate labeling still causes the most commonly observed artifact in super-resolution images: a clustered looking. This problem can be resulted from several possibilities:
imperfect sample fixation and permeabilization. The high spatial resolution of STORM/PALM can easily reveal any disruption of the ultrastructures during sample preparation. For example, immunostained microtubules are widely used to demonstrate the resolution of super-resolution microscopy, but it is highly susceptible to depolymerization during fixation, which creates “broken” looking of microtubules (Figure 2B). Crosslinking and permeabilization may also cause the clustering of membrane proteins. To solve these problems, more electron-microscopy-like sample fixation protocols or live imaging would be necessary.
low labeling efficiency. In the crowded cellular environment, the accessibility to the labeling target by fluorescent probes can restrict the labeling efficiency and hence the ability to resolve small structures. Antigen retrieval sometimes helps in improving antibody accessibility in dense structures such as the postsynaptic density (Beaudoin et al., 2012), albeit with the risk of ultrastructure disruption. Smaller probes, such as nanobodies (Ries et al., 2012), enzymatic tags (Klein et al., 2011; Wombacher et al., 2010), and small molecules (e.g. fluorescently labeled phalloidin for actin (Xu et al., 2012) and a wide varieties of organelle-specific dyes (Shim et al., 2012)), often have higher labeling efficiency compared to full-size antibodies. Labeling efficiency by fluorescent protein fusion to the target protein can be very high, but is still limited by the existence of unlabeled endogenous protein, overexpression artifacts, and the fact that not all fluorescent protein molecules will properly mature and be detected. Therefore, new approaches that can specifically and efficiently label cellular targets with bright fluorophores are much desired.
clustering of the fluorescent probe. In conventional fluorescence microscopy, it is a common practice to “amplify” the fluorescence signal by labeling the antibodies with multiple dyes per antibody molecule, by indirect immunofluorescence staining, and by tyramide signal amplification (TSA) and other peroxidase-based amplification techniques. These methods add many dye molecules to one detected target, thus increasing the fluorescence signal and compensating the low labeling efficiency. However, these practices give no signal increase in STORM/PALM because it always detects individual molecules. In fact, they not only impair the resolution because they increase the effective size of the probe (Huang et al., 2008b; Ries et al., 2012), but also exaggerate the clustering problem because it turns a single probe into many fluorophores clustered together (Figure 2A and B).
Actually, the issue mentioned above exists in all fluorescence microscopy practices. They are usually not a problem for conventional fluorescence microscopy because it does not have the resolution to reveal these artifacts. A sample might appear as “perfect” under a confocal microscopy and even under SIM, but higher resolution methods such as STED microscopy and STORM/PALM could reveal the “clustering” problem. Therefore, higher resolving power always demands more stringent sample preparation, very much resembling the case of electron microscopy.
The three issues mentioned above apply to all super-resolution microscopy techniques. For STORM/PALM in particular, the photophysics of the fluorophores can further exacerbate the clustering problem. Many organic dyes can undergo repetitive switching (Bates et al., 2007; Dempsey et al., 2011; Heilemann et al., 2008). With long acquisition time, multiple points are generated the same fluorophore (Figure 2C). Although most photoactivatible fluorescent proteins can nominally be activated only once, their blinking behavior also create a cluster of localization points (Figure 2D), which might be misinterpreted as clustering of the target protein (Annibale et al., 2011a). Therefore, it is important to develop analysis algorithms that can correctly interpret these “single-molecule clusters” in STORM/PALM super-resolution images (Annibale et al., 2011b).
3. Making sense of super-resolution images
Single-molecule events are by nature quantized and stochastic: there is no such thing as half of a molecule, and reactions at single-molecule level happen by probabilities. These characteristics serve as the basis of STORM/PALM, and also bring in fundamental differences between STORM/PALM and conventional fluorescence microscopy, making it inappropriate to interpret an STORM/PALM image without statistical analysis.
One consequence of imaging at single-molecule level is that noise appears the same as single-molecule signal. For example, the intrinsic noise in a fluorescence image has a low probability to be misidentified as a single-molecule activation event, especially when the background is high and the fluorophore is dim. The resulted false localization point scatters around the super-resolution image and should not be confused with real ones (Figure 3).
Figure 3.
Noise in STORM/PALM. (A) Zoomed-in view of a boxed region in Figure 2B, left panel, showing false localization points (arrows). Particularly, misidentification of two nearby fluorophores activated at the same time creates the false localization points in the region surrounded by three microtubules. (B) Inset of Figure 1A before correction for crosstalk. The crosstalk can be seen as mixed green and red localization points. Arrows point to localization points either from nonspecific antibody binding or from false identification of background noise.
More complicated is the nonspecific binding of fluorescent probes to the sample. Unlike the homogeneous background often detected by conventional fluorescence microscopy, STORM/PALM detects nonspecific probe binding as scattered, individual probe molecules. Unlike the individually scattered false localization points coming from image noise, repetitive activation or blinking of these fluorophores further renders them into a cluster of localization points (Figure 2C).
Another situation for false localization points to appear is the crosstalk in multi-color STORM/PALM. Generally, STORM/PALM has two different approaches for multi-color imaging. One approach is to use fluorescent proteins or dyes with different emission wavelengths (Bossi et al., 2008). The crosstalk can be negligible in this case unless their emission wavelengths are extremely close (Gunewardene et al., 2011). The other approach is to use fluorescent probes (often a pair of organic of dyes) that have the same emission wavelength but can be activated by different wavelengths of light (Bates et al., 2007; Huang et al., 2008a). This approach avoids the necessity to align different emission channels, which can be challenging at nanometer scale, but incurs more crosstalk due to spontaneous activation (Dani et al., 2010). The crosstalk in STORM/PALM appears as points in one color mixed into another color (Figure 3B), which could misinterpreted as colocalization. Unfortunately, the usual approach of crosstalk correction by subtracting pixel values cannot be applied here. Instead, crosstalk subtraction needs to be done for each localization point by analyzing the density of different colors of points surround it (Bates et al., 2007; Dani et al., 2010).
Crosstalk subtraction is one of the examples that demonstrate the challenges in STORM/PALM image analysis. These images usually consist of molecule coordinates instead of pixels, which is incompatible with almost any existing image processing routines. A simple way to adapt a STORM/PALM image to those existing analysis packages is to it into a one-dimensional histograms or a two-dimensional pixelated image based on the number of localization points within each bin or pixel. Calculation of subtraction, correlation and shape fitting can then be performed, as illustrated by the determination of the molecular architecture of focal adhesion complexes and synapses (Dani et al., 2010; Kanchanawong et al., 2010). In both cases, fitting the histogram built from many molecule positions has resolved different domains on the same protein molecule. The binning method has also been used for counting the number of synapses in a brain region (Beaudoin et al., 2012). More recently, methods have been developed to calculate correlation function directly from molecule coordinates, which have enabled quantitative analysis of protein clustering and protein-protein colocalization (Sengupta et al., 2011). We expect that more algorithms and software packages will be developed in the near future to facilitate the interpretation of STORM/PALM images.
4. The Opportunities
The development of an organism consists of three interconnected elements: communication between cells, differentiation and determination of the cell fate, and cell migration and morphogenesis. Understanding the mechanisms behind the development of multicellular organisms requires knowledge covering scales from a molecule to a whole organism. Therefore, a wide variety of biological and physical methods have been demanded for the study of developmental biology. Super-resolution microscopy, with its resolution lies at the scale between molecules and organelles, adds perfectly to this toolbox. It reveals how molecules work inside a cell, thus allowing us to link our structural understandings of biomolecules to their functions in the cellular and tissue context.
Cell communication
During the organism development, a cell constantly exchanges signal with other cells and the outside environment in order to sense as well as establish its developmental context. These signal exchanges are most mediated by receptor molecules on the plasma membrane, especially those in the Notch, Hedgehog, Wnt, TGF(beta) and JAK/STAT pathways (Guruharsha et al., 2012; Krauss, 2008). Adhesion molecules including neuroligin-neurexin, cadherins, and DSCAM also play critical roles in controlling cell recognition and cell morphogenesis (Zipursky and Sanes, 2010). Many of these pathways involve combinatorial use of a relatively small number of signaling molecules among diverse cell types. In addition, the receptors and adhesion molecules are regulated by oligomerization, clustering, anchoring to scaffolding proteins, and partitioning into membrane compartments. Understanding these regulation mechanisms calls for a method to uncover molecular interactions happening at the nanometer to micrometer scale.
The planar geometry of the plasma membrane has made membrane receptors extensively studied targets by STORM/PALM. These studies have been focused on the receptor clustering (Greenfield et al., 2009; Scarselli et al., 2012), participation of receptors in different lipid domains (Hess et al.; Sengupta et al.), and using single-particle tracking to monitor receptor diffusion (Hess et al.; Manley et al.). Many quantitative analysis methods have been developed for these studies. The next challenges would be to probe membrane receptors in the native tissue context.
The three-dimensional and multicolor imaging capability of STORM/PALM also makes it a powerful tool to study the scaffolding and compartmentalizing of signaling molecules. For example, the composition and spatial distribution of neurotransmitter receptors in the postsynaptic density has been characterized by 3D STORM/PALM in the mouse main olfactory bulb and accessory olfactory bulb, revealing the different maturation states of synapses in these two brain regions (Dani et al.). Similar strategies could be applied to other development-related signaling compartment, too, such as the machinery that controls the signaling molecule transportation and localization in the primary cilium.
Cell fate determination
One of the key processes during development is cell differentiation, which is achieved through the integration of external and internal signals by a complicated transcription, translation and epigenetic regulation network. Comparing to the success of microarray and sequencing techniques, imaging has been very much underexplored in studying cell fate determination. This situation can be partially attributed to the fact that much of the information that controls differentiation regulation lies in the DNA and RNA sequence, which perfectly fits microarray and sequencing analysis. Nevertheless, the capability of imaging techniques to probe dynamic processes in individual cells within a heterogeneous population is still uniquely advantageous.
As has been mentioned before, STORM/PALM can routinely achieve ~ 25 nm resolution, which corresponds to just about 70 base pairs for a linear stretch of double-strand DNA. Therefore, super-resolution microscopy might ultimately enable us to map the DNA sequence space to the physical space in the nucleus. This capability could provide the opportunity to examine how gene expression is regulated by chromatin packaging and spatial organization, ranging from nucleosome scale (e.g. heterochromatin formation) to gene level (e.g. allelic exclusion (Yang and Kuroda, 2007)) to whole chromosome scale (e.g. X-inactivation). Most of the chromatin STORM/PALM experiments so far rely on tagging histone proteins (Gunkel et al., 2009; Matsuda et al., 2010; Watanabe et al., 2011; Wombacher et al., 2010), although the development of small molecule probes for DNA and RNA labeling (Benke and Manley, 2012; Flors et al., 2009; Zessin et al., 2012) could further improve the effective spatial resolution.
Another opportunity is to understand how nucleic acids interact with proteins in a cell. STORM/PALM has already demonstrated its potential in understanding how nucleoid-associated proteins help organizing E. coli chromatin (Wang et al., 2011), and in revealing the interaction of centromere-associated histones (Ribeiro et al., 2010) and chromatin-remodeling proteins (Gunkel et al., 2009) with DNA. Sequence specific labeling of DNA could be critical to these applications. In addition to Fluorescence in situ Hybridization (FISH), which has already been used in super-resolution microscopy in a number of cases (Markaki et al., 2012; van de Corput et al., 2012; Weiland et al., 2011), newly developed labeling methods with minimal disruption to the structure of chromosomes will be greatly appreciated.
Cell morphogenesis and migration
Cells exhibit diverse morphologies, ranging from simple flats such as epithelial cells to extremely complicated shapes such as neurons. During development, these different cells sometimes migrate over a long distance to their final locations. Guided by both external signals and internal control networks, cell morphogenesis and migration involve protrusive structures such as filopodia and lamellipodia, cytoskeletons that undergo dynamic reorganization, and mechanical anchors such as focal adhesion and tight junctions. Many of these structures consist of highly organized protein complexes. Dissecting the architecture of these complexes presents a challenge for both conventional fluorescence microscopy (for the lack of spatial resolution) and electron microscopy (for the lack of protein specificity). Therefore, we need a technique that can fill in the gap between molecule level knowledge generated by structural biology and biochemistry and cell to tissue level studies in cell and developmental biology.
Super-resolution microscopy comes in at the perfect position to bridge this gap. Cytoskeleton such as microtubules (Aquino et al., 2011; Bates et al., 2007; Heilemann et al., 2008; Huang et al., 2008b) and actin network (Frost et al., 2010; Izeddin et al., 2011; Xu et al., 2012) has long been the most popular imaging target for super-resolution microscopy study. The morphology of the cell can also be defined with membrane stains (Shim et al., 2012) and membrane-anchored proteins (Aquino et al., 2011; Lakadamyali et al., 2012; Ries et al., 2012). STORM/PALM has revealed the multi-layered molecular architecture of focal adhesions (Kanchanawong et al., 2010) and synaptic terminals (Dani et al.), both demonstrating the capability to resolve different domains on the same protein molecule and thus the orientation of the protein in the complex. What can be even more helpful in the future is to combine super-resolution structural imaging with STORM/PALM with functional imaging such as fluorescence resonance energy transfer (FRET). For example, combining super-resolution microscopy and FRET probes reporting the activity of the Rho family of GTPases could uncover the mechanism of how they regulate cytoskeleton dynamics in a spatial-temporally defined manner (Kamiyama and Chiba).
5. Summary
Super-resolution microscopy is still a young field under rapid development. Considering the immense number of questions awaiting answers in developmental biology, it is impossible for us to give an exhaustive list of what super-resolution microscopy can do now and what new opportunities it will create in the future. As closer collaborations have started to form between research groups on the technology side and those on the biology side, and an ever growing number of biology labs begin to gain access to super-resolution microscopy through home-built instruments, imaging cores and commercial products, we are confident that this new technology will lead to many new discoveries and new insights in the coming years.
Acknowledgments
B.H. receives the Searle Scholarship, the Packard Fellowship for Science Engineering, and the NIH Director’s New Innovator Award (1DP2OD008479-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Identification of clustering artifacts in photoactivated localization microscopy. Nat Methods. 2011a;8:527–528. doi: 10.1038/nmeth.1627. [DOI] [PubMed] [Google Scholar]
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Quantitative photo activated localization microscopy: unraveling the effects of photoblinking. PLoS One. 2011b;6:e22678. doi: 10.1371/journal.pone.0022678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquino D, Schonle A, Geisler C, von Middendorff C, Wurm CA, Okamura Y, Lang T, Hell SW, Egner A. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat Methods. 2011;8:353–359. doi: 10.1038/nmeth.1583. [DOI] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin GM, 3rd, Schofield CM, Nuwal T, Zang K, Ullian EM, Huang B, Reichardt LF. Afadin, a Ras/Rap effector that controls cadherin function, promotes spine and excitatory synapse density in the hippocampus. J Neurosci. 2012;32:99–110. doi: 10.1523/JNEUROSCI.4565-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke A, Manley S. Live-cell dSTORM of cellular DNA based on direct DNA labeling. Chembiochem. 2012;13:298–301. doi: 10.1002/cbic.201100679. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Bossi M, Folling J, Belov VN, Boyarskiy VP, Medda R, Egner A, Eggeling C, Schonle A, Hell SW. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species. Nano Lett. 2008;8:2463–2468. doi: 10.1021/nl801471d. [DOI] [PubMed] [Google Scholar]
- Burnette DT, Sengupta P, Dai YH, Lippincott-Schwartz J, Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci USA. 2011;108:21081–21086. doi: 10.1073/pnas.1117430109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella Zanacchi F, Lavagnino Z, Perrone Donnorso M, Del Bue A, Furia L, Faretta M, Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat Methods. 2011;8:1047–1049. doi: 10.1038/nmeth.1744. [DOI] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedecker P, Mo GC, Dertinger T, Zhang J. Widely accessible method for superresolution fluorescence imaging of living systems. Proc Natl Acad Sci U S A. 2012;109:10909–10914. doi: 10.1073/pnas.1204917109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Dertinger T, Colyer R, Iyer G, Weiss S, Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI) Proc Natl Acad Sci USA. 2009;106:22287–22292. doi: 10.1073/pnas.0907866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner A, Geisler C, Von Middendorff C, Bock H, Wenzel D, Medda R, Andresen M, Stiel AC, Jakobs S, Eggeling C, et al. Fluorescence nanoscopy in whole cells by asynchronous localization of photoswitching emitters. Biophys J. 2007;93:3285–3290. doi: 10.1529/biophysj.107.112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JE, Lu J, Schnitzer MJ. Estimation theoretic measure of resolution for stochastic localization microscopy. Phys Rev Lett. 2012;109:048102. doi: 10.1103/PhysRevLett.109.048102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flors C, Ravarani CN, Dryden DT. Super-resolution imaging of DNA labelled with intercalating dyes. Chemphyschem. 2009;10:2201–2204. doi: 10.1002/cphc.200900384. [DOI] [PubMed] [Google Scholar]
- Folling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- Frost NA, Shroff H, Kong H, Betzig E, Blanpied TA. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 2010;67:86–99. doi: 10.1016/j.neuron.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girkin JM, Poland S, Wright AJ. Adaptive optics for deeper imaging of biological samples. Curr Opin Biotechnol. 2009;20:106–110. doi: 10.1016/j.copbio.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Greenfield D, McEvoy AL, Shroff H, Crooks GE, Wingreen NS, Betzig E, Liphardt J. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 2009;7:e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunewardene MS, Subach FV, Gould TJ, Penoncello GP, Gudheti MV, Verkhusha VV, Hess ST. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101:1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunkel M, Erdel F, Rippe K, Lemmer P, Kaufmann R, Hormann C, Amberger R, Cremer C. Dual color localization microscopy of cellular nanostructures. Biotechnol J. 2009;4:927–938. doi: 10.1002/biot.200900005. [DOI] [PubMed] [Google Scholar]
- Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13:654–666. doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc-Oxford. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, Fukami K, Sakaue-Sawano A, Miyawaki A. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- Hell SW, Wichmann J. Breaking the Diffraction Resolution Limit by Stimulated-Emission - Stimulated-Emission-Depletion Fluorescence Microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ST, Gould TJ, Gudheti MV, Maas SA, Mills KD, Zimmerberg J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc Natl Acad Sci U S A. 2007;104:17370–17375. doi: 10.1073/pnas.0708066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden SJ, Uphoff S, Kapanidis AN. DAOSTORM: an algorithm for high- density super-resolution microscopy. Nat Methods. 2011;8:279–280. doi: 10.1038/nmeth0411-279. [DOI] [PubMed] [Google Scholar]
- Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu Rev Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods. 2008a;5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang WQ, Bates M, Zhuang XW. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008b;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Schwartz SL, Byars JM, Lidke KA. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed Opt Express. 2011;2:1377–1393. doi: 10.1364/BOE.2.001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EH. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Izeddin I, El Beheiry M, Andilla J, Ciepielewski D, Darzacq X, Dahan M. PSF shaping using adaptive optics for three-dimensional single-molecule super-resolution imaging and tracking. Opt Express. 2012;20:4957–4967. doi: 10.1364/OE.20.004957. [DOI] [PubMed] [Google Scholar]
- Izeddin I, Specht CG, Lelek M, Darzacq X, Triller A, Zimmer C, Dahan M. Super-resolution dynamic imaging of dendritic spines using a low-affinity photoconvertible actin probe. PLoS One. 2011;6:e15611. doi: 10.1371/journal.pone.0015611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Shim SH, He J, Zhuang XW. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods. 2011;8:499–505. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, Bewersdorf J. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nature Methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- Kamiyama D, Chiba A. Endogenous activation patterns of Cdc42 GTPase within Drosophila embryos. Science. 2009;324:1338–1340. doi: 10.1126/science.1170615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Opt Lett. 1999;24:954–956. doi: 10.1364/ol.24.000954. [DOI] [PubMed] [Google Scholar]
- Klein T, Loschberger A, Proppert S, Wolter S, van de Linde SV, Sauer M. Live-cell dSTORM with SNAP-tag fusion proteins. Nat Methods. 2011;8:7–9. doi: 10.1038/nmeth0111-7b. [DOI] [PubMed] [Google Scholar]
- Krauss G. Biochemistry of signal transduction and regulation. 4. Willey-VHC; 2008. [Google Scholar]
- Lakadamyali M, Babcock H, Bates M, Zhuang X, Lichtman J. 3D multicolor super-resolution imaging offers improved accuracy in neuron tracing. PLoS One. 2012;7:e30826. doi: 10.1371/journal.pone.0030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Patterson GH. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends in Cell Biology. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Markaki Y, Smeets D, Fiedler S, Schmid VJ, Schermelleh L, Cremer T, Cremer M. The potential of 3D-FISH and super-resolution structured illumination microscopy for studies of 3D nuclear architecture: 3D structured illumination microscopy of defined chromosomal structures visualized by 3D (immuno)-FISH opens new perspectives for studies of nuclear architecture. Bioessays. 2012;34:412–426. doi: 10.1002/bies.201100176. [DOI] [PubMed] [Google Scholar]
- Matsuda A, Shao L, Boulanger J, Kervrann C, Carlton PM, Kner P, Agard D, Sedat JW. Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP- histones. PLoS One. 2010;5:e12768. doi: 10.1371/journal.pone.0012768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel EA, Babcock H, Zhuang X. Statistical deconvolution for superresolution fluorescence microscopy. Biophys J. 2012;102:2391–2400. doi: 10.1016/j.bpj.2012.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanguneri S, Flottmann B, Horstmann H, Heilemann M, Kuner T. Three-dimensional, tomographic super-resolution fluorescence imaging of serially sectioned thick samples. PLoS One. 2012;7:e38098. doi: 10.1371/journal.pone.0038098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TW, Zhu HY, Liu XM, Liu YF, Ding JP, Zeng SQ, Huang ZL. High-density localization of active molecules using Structured Sparse Model and Bayesian Information Criterion. Opt Express. 2011;19:16963–16974. doi: 10.1364/OE.19.016963. [DOI] [PubMed] [Google Scholar]
- Ribeiro SA, Vagnarelli P, Dong Y, Hori T, McEwen BF, Fukagawa T, Flors C, Earnshaw WC. A super-resolution map of the vertebrate kinetochore. Proc Natl Acad Sci U S A. 2010;107:10484–10489. doi: 10.1073/pnas.1002325107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries J, Kaplan C, Platonova E, Eghlidi H, Ewers H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods. 2012;9:582–584. doi: 10.1038/nmeth.1991. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang XW. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli M, Annibale P, Radenovic A. Cell type-specific beta2-adrenergic receptor clusters identified using photoactivated localization microscopy are not lipid raft related, but depend on actin cytoskeleton integrity. J Biol Chem. 2012;287:16768–16780. doi: 10.1074/jbc.M111.329912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci USA. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SH, Xia C, Zhong G, Babcock HP, Vaughan JC, Huang B, Wang X, Xu C, Bi GQ, Zhuang X. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci U S A. 2012;109:13978–13983. doi: 10.1073/pnas.1201882109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, Manley S, Sougrat R, Waterman CM, Kanchanawong P, Davidson MW, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- van de Corput MP, de Boer E, Knoch TA, van Cappellen WA, Quintanilla A, Ferrand L, Grosveld FG. Super-resolution imaging reveals 3D folding dynamics of the beta-globin locus upon gene activation. J Cell Sci. 2012 doi: 10.1242/jcs.108522. [DOI] [PubMed] [Google Scholar]
- Vaziri A, Tang JY, Shroff H, Shank CV. Multilayer three-dimensional super resolution imaging of thick biological samples. Proc Natl Acad Sci USA. 2008;105:20221–20226. doi: 10.1073/pnas.0810636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li GW, Chen C, Xie XS, Zhuang X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science. 2011;333:1445–1449. doi: 10.1126/science.1204697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Punge A, Hollopeter G, Willig KI, Hobson RJ, Davis MW, Hell SW, Jorgensen EM. Protein localization in electron micrographs using fluorescence nanoscopy. Nat Methods. 2011;8:80–84. doi: 10.1038/nmeth.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland Y, Lemmer P, Cremer C. Combining FISH with localisation microscopy: Super-resolution imaging of nuclear genome nanostructures. Chromosome Res. 2011;19:5–23. doi: 10.1007/s10577-010-9171-6. [DOI] [PubMed] [Google Scholar]
- Wombacher R, Heidbreder M, van de Linde S, Sheetz MP, Heilemann M, Cornish VW, Sauer M. Live-cell super-resolution imaging with trimethoprim conjugates. Nat Methods. 2010;7:717–719. doi: 10.1038/nmeth.1489. [DOI] [PubMed] [Google Scholar]
- Wu M, Huang B, Graham M, Raimondi A, Heuser JE, Zhuang XW, De Camilli P. Coupling between clathrin-dependent endocytic budding and F-BAR-dependent tubulation in a cell-free system. Nat Cell Biol. 2010;12:902–908. doi: 10.1038/ncb2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Babcock HP, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods. 2012;9:185–188. doi: 10.1038/nmeth.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PK, Kuroda MI. Noncoding RNAs and intranuclear positioning in monoallelic gene expression. Cell. 2007;128:777–786. doi: 10.1016/j.cell.2007.01.032. [DOI] [PubMed] [Google Scholar]
- York AG, Ghitani A, Vaziri A, Davidson MW, Shroff H. Confined activation and subdiffractive localization enables whole-cell PALM with genetically expressed probes. Nat Methods. 2011;8:327–U373. doi: 10.1038/nmeth.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zessin PJ, Finan K, Heilemann M. Super-resolution fluorescence imaging of chromosomal DNA. J Struct Biol. 2012;177:344–348. doi: 10.1016/j.jsb.2011.12.015. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zhang W, Elnatan D, Huang B. Faster STORM using compressed sensing. Nat Methods. 2012;9:721–723. doi: 10.1038/nmeth.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, Sanes JR. Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell. 2010;143:343–353. doi: 10.1016/j.cell.2010.10.009. [DOI] [PubMed] [Google Scholar]