

Abstract
Although an extensive body of scientific and patent literature exists describing the development of HIV-1 integrase (IN) inhibitors, Merck’s raltegravir and Gilead’s elvitegravir remain the only IN inhibitors FDA-approved for the treatment of AIDS. The emergence of raltegravir-resistant strains of HIV-1 containing mutated forms of IN underlies the need for continued efforts to enhance the efficacy of IN inhibitors against resistant mutants. We have previously described bicyclic 6,7-dihydroxyoxoisoindolin-1-ones that show good IN inhibitory potency. This report describes the effects of introducing substituents into the 4- and 5- positions of the parent 6,7-dihydroxyoxoisoindolin-1-one platform. We have developed several sulfonamide-containing analogs that enhance potency in cell-based HIV assays by more than two orders-of-magnitude and we describe several compounds that are more potent than raltegravir against the clinically relevant Y143R IN mutant.
Keywords: HIV-1 integrase, Inhibit, Raltegravir, Sulfonamide
Integrase (IN) is an HIV-1 encoded polynucleotidyl transferase that inserts viral cDNA into the host genome through a process involving two sequential enzymatic steps, termed 3′-processing (3′-P) and strand transfer (ST).1 Integration is required for viral replication and IN is a validated antiviral target for the treatment of AIDS.2,3 There is an extensive body of scientific and patent literature describing the development of IN inhibitors. Reviews 4,5 Raltegravir (1) is the first IN inhibitor FDA-approved for the treatment of HIV/AIDS (Fig. 1).6 Raltegravir binds at the interface of the macromolecular complex formed by IN and the viral DNA substrate, blocking the active site and preventing the insertion of the viral DNA into the host genome.Review 7 Because it more potently blocks the second enzymatic step, it is referred to as an IN strand transfer inhibitor, or INSTI. All anti-HIV drugs select for resistant strains of the virus and raltegravir is no exception. Because there are clinical strains of HIV-1 that exhibit reduced sensitivity to raltegravir,8 “second-generation” inhibitors are being developed to treat patients failing raltegravir-based treatment.9 However, in in vitro experiments exposure to such compounds selected IN mutants showing reduced susceptability,10,11 suggesting that additional work needs to be done to understand the underlying mechanisms of resistance, and to develop new compounds that will be more effective against resistant strains.
Figure 1.

Structures of IN inhibitors discussed in the text.
Raltegravir shares with other well-described INSTIs, the ability to chelate two catalytic divalent metal ions (Mn2+ or Mg2+) through a triad arrangement of heteroatoms (Fig. 1). We have previously described structurally simple bicyclic 6,7-dihydroxyoxoisoindolin-1-one-based IN inhibitors (2) that show good potency and strand transfer selectivity in vitro in the presence of Mg2+ cofactor.12 Our earlier reports focused primarily on the arylamide “right side” of the molecules.13,14 This paper describes the effects of substitutions at the two free aryl positions on the “left side” of the molecule (Fig. 1). In designing new analogs we were guided by the fact that Merck’s bicylic second-generation inhibitor MK-0536 (3)15,16 has carboxamido and isopropyl substituents at what would be equivalent to positions 4 and 5 of our 1-oxoisoindoline ring system (Fig. 1). We applied a variation of this theme to the dihydroxyoxoisoindoline nucleus by placing several different alkyl groups at the 5-position (4) and employing sulfonamido rather than carboxamido functionality at the 4-position (5, Fig. 1).
Synthesis of derivatives of compound 4 modified at the 5-position employed methodologies similar to those used to prepare the unsubstituted congeners.12–14 Substituents that would ultimately occupy the 5-position of the 6,7-dihydroxyoxoisoindolines final products were derived from appropriately substituted methyl dimethoxybenzyl ethers (6, Scheme 1). Methyl-substituted 6b was obtained from vanillin using 4-hydroxy-3-methoxy-5-methylbenzaldehyde17 as an intermediate (see Supporting Information Scheme SI1). The n-butyl-substituted compound (6c) was obtained serendipitously in an attempt to prepare the isopropyl congener (6d). The synthesis started from 5-bromovanillin and there was an unintended introduction of the n-butyl group when n-butyl lithium was used for metalation of the aryl bromide of intermediate 1-bromo-2,3-dimethoxy-5-(methoxymethyl)benzene (see Supporting Information Scheme SI2). The desired isopropyl containing analog (6d) was synthesized through the known 5-bromo-1-isopropyl-2,3-dimethoxybenzene18 (see Supporting Information Scheme SI3). The phenyl-substituted compound 6e was obtained through a route involving Suzuki coupling of phenylboronic acid with the above-mentioned 1-bromo-2,3-dimethoxy-5-(methoxymethyl)benzene (see Supporting Information Scheme SI4). Finally, the trimethoxyphenyl-containing analog 6f was obtained from commercially-available (3,4,5-trimethoxyphenyl)methanol by methylation using iodomethane and sodium hydride (see Supporting Information Scheme SI5).
Scheme 1.

Reagents and conditions: i) nBuLi, ClCO2Me; ii) AcCl, ZnCl2, Et2O; iii) 3-chloro-4-fluoro-benzylamine, Et3N, CH3CN; iv) BBr3, CH2Cl2.
Treatment of the benzyl methyl ethers 6(a – f) with n-butyl lithium followed by methyl chloroformate provided the corresponding methyl esters 7(a – f) (Scheme 1). Transformation to the benzyl chlorides 8(a – f) was then accomplished using acetyl chloride in the presence of a catalytic amount of zinc chloride. Subsequent ring closure to the isoindolin-1-ones 9(a – f) was achieved by coupling with 3-chloro-4-fluorobenzylamine. The final 5-substituted 6,7-dihydroxyisoindolin-1-ones 2, 4(b – e, g, h) were obtained by demethylation using boron tribromide in dichloromethane (Scheme 1).
Introduction of sulfonyl functionality at the indolin-1-one 4-position was achieved by reacting the methyl ether-protected intermediates 9(a – f) with chlorosulfonic acid gave the sulfonyl chlorides 10(a – f, h) (Scheme 2). Further treatment with a variety of primary and secondary amines yielded the corresponding sulfonamides 11(a-n) – 11(f-n) and 11(h-n), where “n” designates variation in the sulfonamido group. Finally, deprotection of the methyl ethers was accomplished by cleavage with boron tribromide in dichloromethane to produce the final products 5(a-n) – 5(e-n), 5(g-n) and 5(h-n) (Scheme 2).
Scheme 2.

Reagents and conditions: i) ClSO3H; ii) R2R3NH, Et3N, CH2Cl2; iii) BBr3, CH2Cl2.
Our previous structural studies on 6,7-dihydroxyoxoisoindolin-1-one-based IN inhibitors (2) were limited to analogs that were unsubstituted at the 4- and 5-positions.12–14 An important objective of our current work was to explore the effects of incorporating functionalities at the 5-position of the isoindolinone ring system (compounds 4). MK-0536 (3) is one example of a diverse range of IN inhibitors that have substituents in this region.5 We were also interested in determining the effects of placing sulfonamido groups at the 4-position. Highly potent IN inhibitors have been reported that have sulfonamide groups located in this region. However, in almost all cases the sulfonamides are either in “reversed” orientations, having the amine rather than the sulfur attached to the metal-chelating aryl ring (for example19,20), or the sulfonamide is attached indirectly through intervening structures (for example21). In some instances, carboxamide rather than sulfonamide functionality is employed, as in MK-0536 (3).
The IN inhibitory IC50 values of the synthetic analogs against both 3′-P and ST reactions were determined using an in vitro assay employing Mg2+ cofactor as previously described.12 Introduction of substituents at the 5-position uniformly decreased ST inhibitory potency relative to the unsubstituted parent (2; ST IC50 = 0.16 μM), in most cases by more than one order of magnitude (Table 1). The least deleterious substituents were phenyl (4e; ST IC50 = 0.87 μM) and 4-dimethylsulfonamidophenyl (4h; ST IC50 = 0.4 μM). With the exception of the 5-hydroxy analog (4g; 3′-P IC50 = 36.0 μM), all the substituents we tested reduced the 3′-P inhibitory potency by at least an order of magnitude (3′-P IC50 >111 μM) relative to the parent 2 (3′-P IC50 = 13.2 μM).
Table 1.
Inhibitory potencies using an in vitro IN assay.a
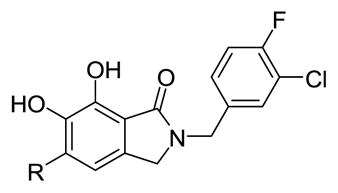
| |||
|---|---|---|---|
| No. | R | IC50 Values (μM)
|
|
| 3′-Processing | Strand transfer | ||
| 2b | H | 13.2 ± 3.1 | 0.16 ± 0.08 |
| 4b | Me | >111 | 11.5 ± 1.6 |
| 4c | nBu | >111 | 6.8 ± 0.9 |
| 4d | iPr | >111 | 5.4 ± 0.6 |
| 4e | Ph | >111 | 0.87 ± 0.08 |
| 4g | OH | 36.0 ± 6.0 | 1.5 ± 0.3 |
| 4h |
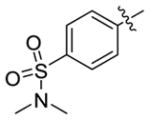
|
>111 | 0.4 ± 0.04 |
In contrast, introducing sulfonamide groups at the 4-position of parent compound 2 had little effect on inhibitory potencies in 3′-P reactions relative to 2 (Table 2). However, ST inhibitory potencies were generally increased (5a-1 to 5a-3 and 5a-9 to 5a-12) with ST IC50 values ranging from 0.047 μM to 0.19 μM). These results are similar to what has been reported for the effects of sulfonamido functionality in this general structural region of other IN inhibitors.19–21 A recent report showed that introduction of 5-sulfonamido groups onto N-(4-fluorobenzyl)-2,3-dihydroxybenzylamide increased inhibitory potency.22 This is of note, since we had originally designed the 6,7-dihydroxyisoindolin-1-ones as conformationally constrained variants of N-(4-fluorobenzyl)-2,3-dihydroxybenzylamide.12 We found that this constraint increased the inhibitory potency of the compounds in the ST reaction by approximately ten-fold. The enhancing effects of sulfonamido substituents in our current conformationally constrained analogs is consistent with enhancing effects reported for the ring-open compounds.22
Table 2.
Inhibitory potencies using an in vitro IN assay.a
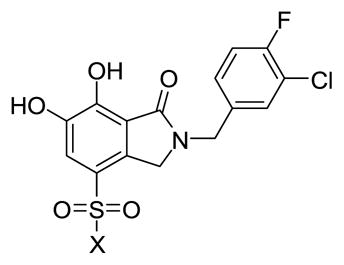
| |||
|---|---|---|---|
| No. | X | IC50 Values (μM)
|
|
| 3′-Processing | Strand transfer | ||
| 5a-1 | NHMe | 17.5 ± 3.0 | 0.12 ± 0.04 |
| 5a-2 | NEt2 | 9.5 ± 1.2 | 0.054 ± 0.009 |
| 5a-3 |
|
14.7 ± 1.7 | 0.11 ± 0.01 |
| 5a-4 |
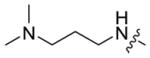
|
15.9 ± 1.3 | 0.24 ± 0.02 |
| 5a-5 |
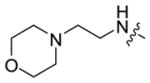
|
30.2 ± 2.3 | 0.32 ± 0.05 |
| 5a-6 |
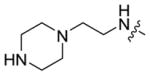
|
19.3 ± 0.6 | 0.31 ± 0.03 |
| 5a-7 |
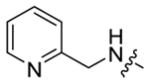
|
23 ± 3 | 0.37 ± 0.04 |
| 5a-8 |
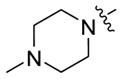
|
23 ± 4 | 0.4 ± 0.08 |
| 5a-9 |
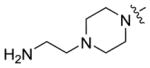
|
21.2 ± 1.9 | 0.19 ± 0.08 |
| 5a-10 |
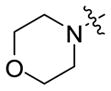
|
6.8 ± 1.1 | 0.047 ± 0.007 |
| 5a-11 |
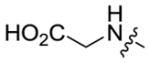
|
11.7 ± 0.7 | 0.08 ± 0.01 |
| 5a-12 |
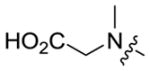
|
6.9 ± 0.6 | 0.058 ± 0.01 |
Assays were performed using a gel-based protocol with Mg2+ cofactor as described in reference 12.
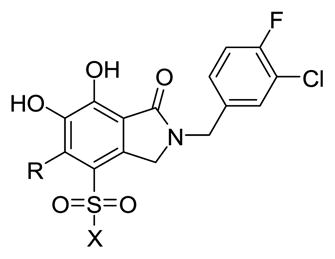
Although substituents at the 5-position (4b – 4h) decreased inhibitory potencies in the ST reactions compared to the parent compound 2, these potentially represented biased examples, since the compounds lacked functionalities at the adjoining 4-position (Table 1). IN inhibitors often have substituents at both the 4 and 5-position, as exemplified by raltegravir (1) and MK-0536 (3). Therefore, a series of analogs was prepared that combined sulfonamido functionality at the 4-position with various substituents at the 5-position (Table 3). Adding a 4-sulfonamido group increased the inhibitory potencies in the ST reaction relative to the compounds described in Table 1. The increase was most pronounced in the case of 5d-2 (ST IC50 = 0.19 μM), which has an i-Pr group at the 5-position. This led to a 20-fold enhancement in the ST reaction relative to the parent i-Pr-containing 4d (Table 1 ST IC50 = 5.4 μM). However, for a given 4-sulfonamido group, introducing substituents at the 5-substituent decreased ST inhibitory potency (Table 3).
Table 3.
Inhibitory potencies using an in vitro IN assay.a
| ||||
|---|---|---|---|---|
| No. | R | X | IC50 Values (μM)
|
|
| 3′-Processing | Strand transfer | |||
| 5b-2 | Me | NMe2 | 49.7 ± 4.0 | 2.8 ± 0.6 |
| 5c-2 | nBu | NMe2 | >111 | 2.3 ± 0.4 |
| 5d-2 | iPr | NMe2 | >111 | 0.19 ± 0.03 |
| 5d-10 | iPr |
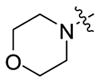
|
>111 | 0.41 ± 0.06 |
| 5g-2 | OH | NMe2 | 7.6 ± 0.8 | 0.26 ± 0.08 |
| 5h-2 |
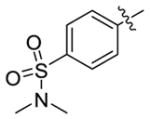
|
NMe2 | 35.2 ± 2.6 | 0.25 ± 0.06 |
Assays were performed using a gel-based protocol with Mg2+ cofactor as describe in reference 12.
Clinical resistance to raltegravir (1) is associated primarily with IN mutations at amino acids Y143, Q148 and N155.23 In order to examine the effects of these resistance mutations on the potency of the new compounds, antiviral efficacy (EC50 values) were determined for select inhibitors using HIV vectors that replicated using either WT IN or vectors carrying Y143R, N155H, or the G140S-Q148H double mutant (designated as “SH”) (Table 4).24,25 Sub-micromolar EC50 values were obtained against the wild-type vector for several of the sulfonamide derivatives that lacked a substituent at the 5-position (“5a” compounds, Table 4) and for certain compounds, nanomolar potencies (17 ~ 40 nM) were observed. Comparing in vitro IC50 values obtained for inhibition of ST reactions with the cellular EC50 values revealed differing degrees of concordance. Compounds 5a-1, 5a-2, 5a-3 and 5a-10 gave EC50 values (0.04 μM, 0.017 μM, 0.93 μM and 0.022 μM, respectively, Table 4) that were slightly better than the corresponding in vitro ST IC50 data (0.12 μM, 0.054 μM, 0.11 μM and 0.047 μM, respectively, Table 2). Compound 5a-8 showed a 10-fold enhancement in its cellular antiviral EC50 value relative to its in vitro ST IC50 data (0.043 μM and 0.4 μM, respectively). This is somewhat unexpected, because 5a-8 contains a piperazine nitrogen that should be protonated at physiological pH, and this charged species would be expected to reduce cell membrane transit.
Table 4.
Antiviral potencies in cell infected with HIV-1 containing wild-type or mutant integrase enzymes.a
| No. | CC50 (μM)b | EC50 (μM)c | SId | Integrase Mutantse
|
||
|---|---|---|---|---|---|---|
| Y143R | N155H | SHf | ||||
| 2 | 5.2 | 0.447 | 12 | 1x | 2x | 8x |
| 5a-1 | 4.3 | 0.040 | 108 | 1x | 19x | 95x |
| 5a-2 | 8.4 | 0.017 | 494 | 3x | 57x | 300x |
| 5a-3 | 4.6 | 0.093 | 49 | 1x | 68x | 132x |
| 5a-8 | 6.2 | 0.043 | 144 | 1x | 44x | 430x |
| 5a-10 | 5.3 | 0.022 | 241 | 2x | 23x | 218x |
| 5a-11 | 106.6 | 0.929 | 115 | 3x | ND | ND |
| 5a-12 | 95.2 | 0.140 | 680 | 2x | 272x | ND |
| 5b-2 | 6.3 | 0.322 | 20 | 2x | 12x | 30x |
| 5c-2 | 7.0 | 3.8 | 2 | 1x | 4x | 1x |
| 5d-2 | 12.1 | 3.6 | 3 | 3x | 2x | 4x |
| 5d-10 | 5.9 | 3.4 | 2 | 4x | 2x | 3x |
| 5g-2 | 4.6 | 0.159 | 29 | 2x | 10x | ND |
| 5h-2 | 2.8 | 0.132 | 21 | 4x | 16x | 25x |
Assays were performed as described in reference 25;
Cytotoxic concentration resulting in 50% reduction in the level of ATP in human osteosarcoma (HOS) cells;
Values obtained from cells infected with lentiviral vector harboring WT IN;
Selectivity index calculated as the ration of CC50 to EC50;
Cells were infected with viral constructs carrying IN mutantion as described in reference 25 and indicated values correspond to the fold-change in EC50 relative to WT;
G140S-Q148H double mutant.
It is not uncommon for cell-based antiviral efficacy to be greater than the IC50 data obtained in vitro in ST inhibition assays.12–14,26 This may be due in part to the fact that IC50 values obtained in vitro in ST inhibition assays depend on factors, such as the concentration of substrate DNA, that may be different in the cellular environment.27 Two compounds (5a-11 and 5a-12) provided antiviral EC50 values that were higher than their corresponding in vitro ST IC50 values. These compounds contained anionic carboxylic acid functionality that would be expected to reduce cellular bioavailability.
In cell-based assays performed with HIV vectors carrying the Y143R mutant, raltegravir (1) has been reported to exhibit an approximate 40-fold loss of potency relative to the WT enzyme.24,25 This loss of potency can be understood on the basis of X-ray crystallographic studies with the prototype foamy virus (PFV) intasome, which show that the oxadiazole ring of raltegravir makes extensive interactions through π-stacking with the side chain aryl ring of Y212 of PFV IN (which is equivalent to Y143 of HIV-1 IN). Replacement of Tyr with Arg eliminates these interactions and is generally understood to account for the reduced potency of raltegravir against the Y143R mutant.28 The retention of relative good potency against the Y143R mutant shown by all sulfonamides in Table 4 is consistent with the absence of aromatic functionality equivalent to raltegravir’s oxadiazole ring;29 none of these compounds rely on a π-π interaction with Y143.
Raltegravir has also been reported to exhibit an approximate 40-fold loss of antiviral potency in cellular studies employing HIV vectors bearing the N155H mutant.25 Crystallographic studies with the PFV intasome show that the side chain of N224 (equivalent to N155 of HIV-1 IN) forms a hydrogen bond with the carboxylate group of E221, which is part of the catalytic “DDE” triad chelating the divalent Mg2+ ions. In the case of N224H mutant, the His imidazole ring makes electrostatic interactions with the phosphate group of the 3′ adenosine of the viral DNA, causing a shift in the position of the DNA backbone and the catalytic Asp and Glu carboxlylates (PFV D128 and E221).28 Binding of INSTIs requires the energetically unfavorable breaking of the His-phosphate interaction. The sulfonamides shown in Table 4 exhibit a range of sensitivities to the N155H mutation that are both greater and less than what has been reported for raltegravir. Interestingly, compounds having bulky n-butyl or i-propyl groups at the 5-position were significantly less affected by the mutation (5c-2, 5d-2 and 5d-10, Table 4). Although the absolute potencies of these latter compounds against the WT enzyme is not as good as some of the other inhibitors shown in Table 4, their ability to retain inhibitory efficacy against the N155H mutant may reflect an enhanced capacity to break the H155 imidazole – phosphate interaction and re-order the DNA needed to accommodate ligand binding.
Raltegravir has also been reported to exhibit an approximate 500-fold loss of antiviral potency in cellular studies employing HIV vectors having the 140S/Q148H [designated as “SH”] double mutant.25 G140 and Q148 correspond to PFV residues S209 and S217, respectively, which occupy the termini of the “flexible loop” bearing the Y212 residue (Y143 of HIV-1 IN), which is critical for raltegravir binding. Crystallographic studies with the PFV intasome show that the S217H mutation requires a shift in the His side chain to accommodate binding of the fluorbenzyl ring of raltegravir.28 Due to their positions sat the termini of the flexible loop, the side chains of the S140 and H148 residues are in close proximity. In the “SH” double mutant, movement of the bulky H148 side chain is sterically impeded by the side chain of S140. In antiviral studies using the SH mutant, sulfonamides lacking substituents at the 5-position exhibited two-orders-of-magnitude reductions in potency (5a-n, Table 4). The 5-substituted sulfonamides showed less relative loss of potency. However, similar to what was observed against the N155H mutant, in most cases the absolute efficacy of these inhibitors against wildtype IN was also reduced.
Except for those compounds bearing free carboxylic acid groups (5a-11 and 5a-12), which may adversely affect cellular uptake, cytotoxicity CC50 values for all sulfonamides in Table 4 were uniformly in the low micromolar range and very similar to the value of 5.2 μM reported for the parent 6,7-dihydroxyisoindolinone (2). Although cytotoxicity remained unchanged, several of the sulfonamide-containing analogs exhibit cellular EC50 values that are approximately two orders-of-magnitude better than the parent 2. The overall effect was to improve the “selectivity index” (SI = CC50 / EC50) for several sulfonamides by two orders-of-magnitude relative to 2.
Our current report describes the effects of introducing substituents into the 4- and 5- positions of the parent 6,7-dihydroxyoxoisoindolin-1-one platform. We found that several sulfonamide-containing analogs exhibit potencies in cell-based HIV assays that are enhanced by more than two orders-of-magnitude. The overall effect of these efforts has been to generate several compounds exhibiting better absolute efficacy than raltegravir against the clinically relevant Y143R IN mutant and with SI values exceeding 100. Although cytotoxicity remains an issue, our current work has advanced the development of the 6,7-dihydroxyisoindolinone platform.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, Frederick National Laboratory for Cancer Research and the National Cancer Institute, National Institutes of Health and the Joint Science and Technology Office of the Department of Defense. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supplementary data (synthetic experimental procedures and analytical data for synthetic products) associated with this article can be found in Electronic supplementary information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Lewinski MK, Bushman FD. In: Advances in Genetics. Jeffrey C, Hall J, CDTF, van Veronica H, editors. Vol. 55. Academic Press; 2005. pp. 147–181. [DOI] [PubMed] [Google Scholar]
- 2.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Nature. 2010;464:232. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelman A, Cherepanov P. Nat Rev Microbiol. 2012;10:279. doi: 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchand C, Maddali K, Metifiot M, Pommier Y. Curr Top Med Chem. 2009;9:1016. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao C, Marchand C, Burke TR, Jr, Pommier Y, Nicklaus MC. Future Med Chem. 2010;2:1107. doi: 10.4155/fmc.10.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. J Med Chem. 2008;51:5843. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 7.Pommier Y, Marchand C. Nat Rev Drug Discov. 2012;11:25. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charpentier C, Karmochkine M, Laureillard D, Tisserand P, Belec L, Weiss L, Si-Mohamed A, Piketty C. HIV Med. 2008;9:765. doi: 10.1111/j.1468-1293.2008.00628.x. [DOI] [PubMed] [Google Scholar]
- 9.Quashie PK, Sloan RD, Wainberg MA. BMC Med. 2012;10:34. doi: 10.1186/1741-7015-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metifiot M, Marchand C, Maddali K, Pommier Y. Viruses. 2010;2:1347. doi: 10.3390/v2071347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katlama C, Murphy R. Expert Opin Inv Drug. 2012;21:523. doi: 10.1517/13543784.2012.661713. [DOI] [PubMed] [Google Scholar]
- 12.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr J Med Chem. 2008;51:251. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 13.Zhao XZ, Maddali K, Vu BC, Marchand C, Hughes Stephen H, Pommier Y, Burke Terrence R., Jr Bioorg Med Chem Lett. 2009;19:2714. doi: 10.1016/j.bmcl.2009.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao XZ, Maddali K, Marchand C, Pommier Y, Burke TR., Jr Bioorg Med Chem. 2009;17:5318. doi: 10.1016/j.bmc.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egbertson MS, Wai JS, Cameron M, Hoerrner RS. In: Antiviral Drugs. Kazmierski WM, editor. John Wiley & Sons, Inc; 2011. pp. 163–180. [Google Scholar]
- 16.Métifiot M, Johnson B, Smith S, Zhao XZ, Marchand C, Burke TR, Jr, Hughes S, Pommier Y. Antimicrob Agents Chemother. 2011;55:5127. doi: 10.1128/AAC.05288-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinhababu AK, Bochardt RT. Synth Commun. 1983;13:677. [Google Scholar]
- 18.Chin CL, Tran DDP, Shia KS, Liu HJ. Synlett. 2005;2005:417. [Google Scholar]
- 19.Jin H, Wright M, Pastor R, Mish M, Metobo S, Jabri S, Lansdown R, Cai R, Pyun P, Tsiang M, Chen X, Kim CU. Bioorg Med Chem Lett. 2008;18:1388. doi: 10.1016/j.bmcl.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Johns BA, Weatherhead JG, Allen SH, Thompson JB, Garvey EP, Foster SA, Jeffrey JL, Miller WH. Bioorg Med Chem Lett. 2009;19:1807. doi: 10.1016/j.bmcl.2009.01.089. [DOI] [PubMed] [Google Scholar]
- 21.Muraglia E, Kinzel O, Gardelli C, Crescenzi B, Donghi M, Ferrara M, Nizi E, Orvieto F, Pescatore G, Laufer R, Gonzalez-Paz O, Di Marco A, Fiore F, Monteagudo E, Fonsi M, Felock PJ, Rowley M, Summa V. J Med Chem. 2008;51:861. doi: 10.1021/jm701164t. [DOI] [PubMed] [Google Scholar]
- 22.Fan X, Zhang FH, Al-Safi RI, Zeng LF, Shabaik Y, Debnath B, Sanchez TW, Odde S, Neamati N, Long YQ. Bioorg Med Chem. 2011;19:4935. doi: 10.1016/j.bmc.2011.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, Lazzarin A, Clotet B, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Zhao J, Chen J, Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Teppler H, Nguyen BY. New Engl J of Med. 2008;359:355. [Google Scholar]
- 24.Metifiot M, Maddali K, Naumova A, Zhang X, Marchand C, Pommier Y. Biochemistry. 2010;49:3715. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao XZ, Maddali K, Metifiot M, Smith SJ, Vu BC, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr Chem Biol Drug Des. 2012;79:157. doi: 10.1111/j.1747-0285.2011.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. Proc Natl Acad Sci USA. 2000;97:11244. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grobler Jay A, Stillmock K, Hu B, Witmer M, Felock P, Espeseth Amy S, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai John S, Young S, Vacca J, Hazuda Daria J. Proc Natl Acad Sci USA. 2002;99:6661. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Proc Nat Acad Sci USA. 2010;107:20057. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, Marchand C, Pommier Y. AIDS. 2011;25:1175. doi: 10.1097/QAD.0b013e3283473599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.