

Abstract
The ability of bacteria expressing inhibitor resistant (IR) β-lactamases is stimulating the development of novel inhibitors. SA2-13 was previously designed to enhance the stabilization of the deacylation refractory, trans-enamine inhibitory intermediate. To test whether this mode of inhibition can overcome different IR mutations, we determined the binding mode of SA2-13 after soaking the inhibitor into crystals of the IR SHV β-lactamase variants S130G and M69V. The 1.45Å crystal structure of the S130G SHV: SA2-13 complex reveals that SA2-13 is still able to form the stable trans-enamine intermediate similar to the wild type complex structure yet with its carboxyl linker shifted deeper into the active site in the space vacated by the S130G mutation. In contrast, data from crystals of the M69V SHV variant soaked with SA2-13 at 1.3 Å did not reveal clear inhibitor density indicating that this IR variant disfavors the trans-enamine conformation, likely due to a subtle shift in A237.
Keywords: β-lactamase inhibitor, protein crystallography
Introduction
Inhibitor resistant (IR) β-lactamases threaten the current antibiotic armamentarium by overcoming the effectiveness of the β-lactamase inhibitors, consequently providing resistance to commercially available β-lactam/β-lactamase inhibitor combinations [1]. New inhibitors are currently being developed with the aim of overcoming resistance [1]. To date, clinically observed IR variants in the Class A SHV and TEM β-lactamases are found to have amino acid substitutions at Ambler positions 69, 130, 234, 244, 275, and 276 (www.lahey.org/studies/webt.asp)[1]. In this analysis we focus on S130G and M69V IR variants of SHV to study the intermediates of inactivation using protein crystallography. The SHV variants were probed with our novel SA2-13 inhibitor which we previously developed with the aim of exploiting stabilization of the trans-enamine intermediate in the inhibition pathway[2].
The IR S130G substitution occurs in the highly conserved SDN loop of Class A β-lactamases. Previous studies proposed that S130 plays several roles which include participating in substrate recognition, facilitating β-lactam ring opening during enzyme acylation, and initiating the irreversible step of inactivation by covalently cross-linking the inhibitor to S130[3–8]. To elucidate the details of inhibitor resistance caused by the S130G substitution, numerous studies were performed including molecular modeling, kinetics, protein crystallography, mass spectrometry, and Raman crystallography [9–12]. Molecular modeling and thermal denaturation studies indicated that the S130G TEM variant is stable compared to the corresponding apo enzyme[11]. Furthermore, kinetic studies suggested that S130 is actively involved in β-lactam hydrolysis, yet the S130G mutant remains active, albeit with a dramatically increased KI value [9,11]. A crystallographic study suggested that a water molecule can assume the role of the hydroxyl moiety of S130 in the S130G variant thus explaining the residual activity of S130G mutant β-lactamases [11]. The S130G mutation in SHV-1 was found to have a negative effect on the KI for as the KI for tazobactam increased from 0.07 to 4.2μM and increased from 0.14 to 46.5μM for clavulanic acid while the kinact and turnover numbers were not as drastically affected [9]. Microbiologically, the S130G mutation generated an inhibitor resistance phenotype as it increased the MIC for clavulanic acid 4-fold whereas the MIC for tazobactam was not altered upon the mutation.
Although Raman crystallographic studies indicated the presence of the trans-enamine intermediate of S130G SHV with tazobactam, clavulanate, and SA2-13[10], additional studies pointed toward a delayed appearance of the trans-enamine intermediate compared to wt and the formation of a cis-enamine and possible aldehyde species [12–14].
The second IR position studied herein, mutants at position M69 that can be found in either TEM or SHV-type β-lactamases, has a different mechanism of inhibitor resistance. Residue M69 is buried behind the active site[15], and therefore does not play a direct role in the enzyme-inhibitor interaction. Nevertheless, this residue accounts for the majority of IR TEM enzymes and several IR SHV enzymes (http://www.lahey.org/studies/). Evidence was presented that M69V/I/L mutants, that have a branched aliphatic sidechain, function through perturbation of S130 which is propagated through S70 as alternate conformations are seen for S130 in the structures of M69V and M69I [16,17]. One S130 conformation leads to S130 hydrogen-bonding to both K234 and K73, whereas the other conformation, as seen in wild-type structures, allows for bonding only to K234[17]. As mentioned above, S130 is potentially the residue responsible for irreversible inhibition and the alternate conformation of S130 may explain the IR phenotype of M69 mutants. In addition to observed changes in S130 conformation, the crystal structures of M69V/E166A SHV:inhibitor complexes also alluded to small yet significant shifts in the β3 strand. This strand, containing residue A237, flanks the active site and a shift in this region leads to a decrease in the size or configuration of the oxyanion hole [16]. This altered oxyanion hole could lead to diminished inhibitor binding and/or less efficient acylation or more efficient deacylation, and consequently, the IR phenotype. The net effect of the M69V mutation is that is leads to an inhibitor resistance phenotype as measured via the 2–4 fold increased minimum inhibitory concentration (MIC) values for the inhibitors clavulanic acid, sulbactam and tazobactam [18]. Although the M69V, as well as the M69I and M69L, mutation negatively affects the efficacy of the inhibitors, the kcat/Km for the ampicillin antibiotic only changes 0.7 fold [18]. Both the M69V and S130G IR mutations are intriguing due to their subtle effects in the active site conformation; however, their mode of IR action is not fully comprehended.
New β-lactamase inhibitors are needed that can overcome these different β-lactamase IR phenotypes. One relatively new approach to potentially overcome IR is to efficiently trap the β-lactamase inhibitor in a trans-enamine intermediate. This intermediate is more resistant to deacylation due to the conjugation of the trans-enamine double bond with the C=O carbonyl bond providing an increased energetic deacylation barrier. Such stabilization was achieved in the wt SHV-1 enzyme by adding a carboxyl linker to tazobactam, creating an inhibitor called SA2-13 (Figure 1A). The carboxyl moiety binds in the conserved carboxyl binding pocket after acylation, which led to a ~10-fold improvement in stabilization of this key inhibitory intermediate in wtSHV-1 β-lactamase[2]. In order to further test the potency of SA2-13 and possible resilience against IR variants, we determined the crystal structures of IR S130G and M69V SHV variants soaked with the inhibitor SA2-13. Probing these IR variants with an inhibitor that was designed to stabilize the trans-enamine intermediate could lead to additional insights into the mode of action of these IR enzymes and their ability to cope with this novel inhibition strategy.
Figure 1. SA2-13 and its active site interactions when bound to SHV-1 S130G.
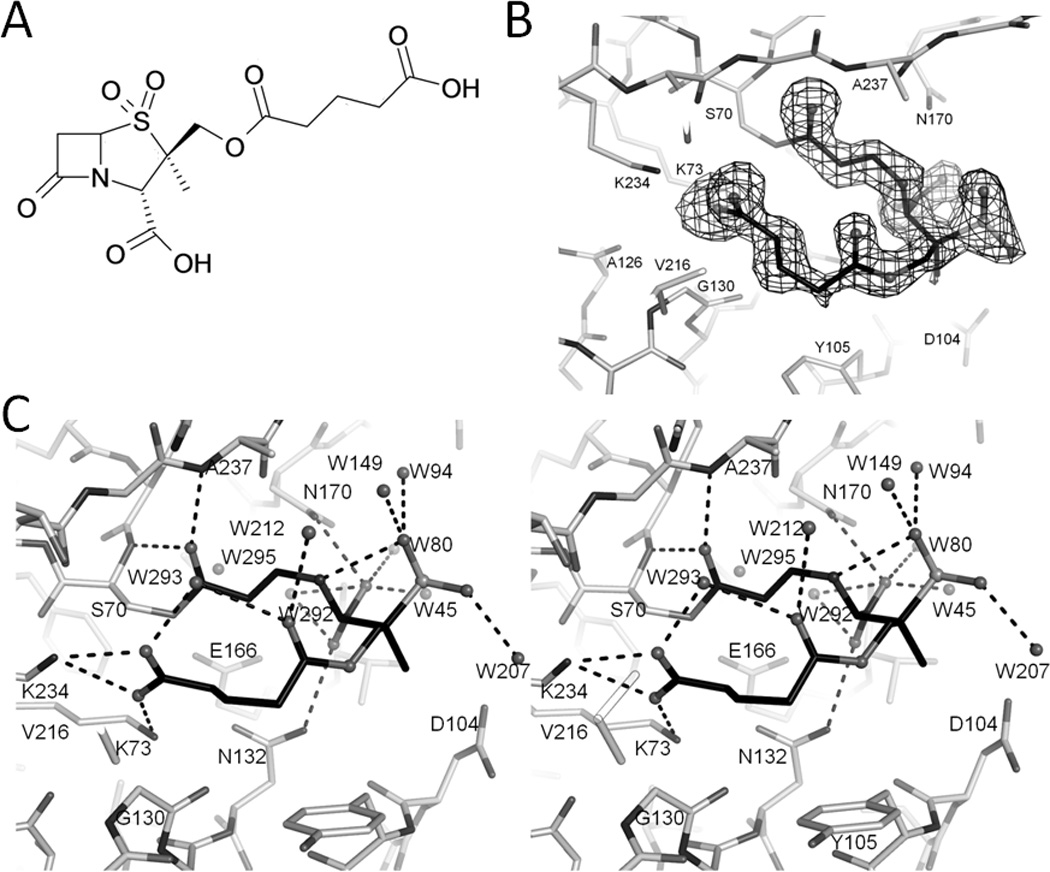
(A) Chemical structure of SA2-13. (B) Electron density of the SA2-13 compound in the active site of S130G SHV-1 β-lactamase. Unbiased omit Fo-Fc map is contoured at 2.5σ. SA2-13 carbon atoms are colored black and nitrogen, oxygen, and sulfur atoms in different shades of gray. (C) Stereo view of interactions of SA2-13 within S130G SHV-1 β-lactamase active site. Dashed black lines indicated hydrogen bonds. Water molecules are also depicted as spheres.
MATERIALS AND METHODS
Inhibitors
SA2-13 (Figure 1A) was synthesized as previously described[2].
Subcloning, expression and purification
The SHV-1 beta-lactamase gene was subcloned into pBC SK (-) vector (Stratagene). The S130G and M69V mutants of SHV β-lactamase were generated by site-directed mutagenesis using Stratagene’s Quick Change Mutagenesis Kit[9,16]. Both mutant constructs were transformed into Escherichia coli DH10B cells (Stratagene). The cells were allowed to grow overnight in lysogeny broth (LB) supplemented with 20µg/ml chloramphenicol to express the corresponding mutant protein. After cell lysis via stringent periplasmic fractionation, the S130G variant was purified to homogeneity by two steps using preparative isoelectric focusing and gel filtration HPLC[10,15,19]. The M69V SHV protein was purified similarly except that a Q-sepharose anion exchange column chromatography step was carried out prior to the preparative isoelectric focusing and gel filtration purification steps. β-Lactamase activity was detected using nitrocefin (Calbiochem), a chromogenic β-lactamase substrate, as previously described[20]. Protein purity was assessed using SDS-PAGE and the purified protein was concentrated to 5mg/ml using a 10K MWCO centrifugal concentrator (Amicon).
Crystallization and soaking experiments
The S130G and M69V SHV proteins were crystallized using a previously described protocol involving the sitting drop vapor diffusion method[10,15,16]. The well solution contained 21–30% PEG6000 and 0.1M HEPES pH 6.8–8.2. A 5mg/ml protein solution was combined with the detergent Cymal-6 (5.6mM, Hampton Research) to yield a final concentration of 0.56mM Cymal-6. This solution was used to set up 5 µl drops of a 1:1 well:protein ratio with crystals growing to full size after 1–3 weeks.
S130G crystals were soaked in 50mM SA2-13 in mother liquor soaking solution for 30 minutes followed by transferring the crystals to the cryo-protectant (soaking solution and 25% methyl-2,4-pentanediol) for 1 minute before being flash frozen in liquid nitrogen.
Crystals of M69V were soaked similarly in inhibitor soaking solution containing the mother liquor and 50mM SA-2-13 and then transferred briefly to a cryo-protectant solution with SA2-13 containing mother liquor and 20% 2-methyl-2,4-pentanediol prior to flash freezing. Due to lack of initial success of obtaining clear inhibitor density for this M69V mutant, a broad soaking time range was subsequently explored ranging from five minutes to overnight. All ~20 synchrotron datasets collected with resolutions up to 1.2Å revealed poor or absent inhibitor density and the results of a representative dataset is included below for a crystal that was soaked in 50mM SA2-13 containing mother liquor for 15 minutes.
Data collection and refinement
For S130G, X-ray diffraction data was collected at the Advanced Light Source (ALS) synchrotron (beamline 4.2.2) and processed using d*TREK[21]. The structure was solved using molecular replacement using the program PHASER[22] with the search model of uncomplexed SHV-1 structure (PDB ID: 1VM1; tazobactam and waters were removed and S at position 130 was altered to a G). Restrained refinement was carried out using REFMAC[23] with atoms being refined using an isotropic B-factor model; model building was done using COOT[24]. After initial refinement, strong density in the active site was found to extend from the hydroxyl group of S70, suggesting that an SA2-13 intermediate was covalently attached to S70. We included Cymal-6 and waters in the refinement prior to including the trans-enamine form of SA2-13 into the difference density and subsequent refinement. The PRODRG2 server was used to obtain the parameter and topology files for the modeled SA2-13 intermediate[25]. Crystallographic refinement was monitored using the program DDQ[26] and the final model quality was assessed using PROCHECK[27]. Data collection and refinement statistics are shown in Table 2.
For M69V, X-ray data collection was carried out at the Stanford Synchrotron Radiation Lights Source (SSRL), Stanford University, Menlo Park, CA. Data was integrated and scaled using HKL2000[28]. The structure was determined using an isomorphous structure of wild-type SHV-1 (PDB 1SHV). Restrained refinement was carried out in a similar manner as described above for the S130G mutant structure. One full and one partial Cymal-6 molecule were modeled in, followed by addition of waters. After several rounds of refinement (atoms refined using isotropic B-factors), the Fo-Fc density of the active site pocket was still poor such that SA2-13 could not be modeled in convincingly and the inhibitor was therefore not included in refinement. Data collection and refinement statistics are shown in Table 2. Coordinates and structure factors for the S130G SHV-1 complexed with SA2-13 and M69V SHV-1 have been deposited into Protein Data Bank (PDB ID: 3V50 and 3V5M, respectively).
Results and Discussion
S130G SHV:SA2-13 Complex
The crystal structure of SA2-13 bound to S130G SHV was determined to 1.45Å resolution. The unbiased omit |Fo|-|Fc| map revealed a well ordered intermediate covalently attached to Oγ of the catalytic S70 residue (Figure 1B). The overall S130G SHV:SA2-13 protein structure is similar to that of structures of apo wtSHV-1 (PDB ID: 1SHV) and apo S130G SHV (PDB ID: 1TDL) yielding a root-mean-square-deviation (r.m.s.d.) of 0.350 and 0.136 Å, respectively, for Cα atoms. Density in the active site of the S130G active site confirms the S to G mutation. Similar to what was observed in the apo S130G SHV structure, the S130G SHV:SA2-13 complex yielded a somewhat enlarged active site as evidenced by an increased Cα-Cα distance between G130 and S70; this distance increases from 6.8Å, in wt SHV-1, to 7.4Å in the S130G mutant structure. SA2-13 was observed in the trans-enamine intermediate conformation with the torsion angle of C7-C6=C5-N4 being refined to 154°. The inclusion of the SA2-13 trans-enamine intermediate brought down the R/Rfree considerably indicating its correct placement. The final R-factor is 18.05% and Rfree is 19.71%. Throughout the refinement, the occupancy of the modeled SA2-13 intermediate was kept at 1.0.
Interactions of SA2-13 in the active site
The functional groups of SA2-13 such as the sulfone, both carboxyl groups and the ester carbonyl are well-ordered as revealed by the density map (Figure 1B). The ester carbonyl moiety of SA2-13 resides in the oxyanion hole, hydrogen bonded to the backbone nitrogen atoms of S70 and A237. The C3 carboxylate is stabilized by N170 and N132; whereas, the sulfone group makes an intra-molecular hydrogen bond with N4 as well as hydrogen bonds with three water molecules (Figure 1C). The carboxylate tail makes salt bridge interactions with K73 and K234 and also makes a water mediated intra-molecular hydrogen bond with the carbonyl oxygen atom of the SA2-13 linker (Figure 1C). The carboxyl tail interaction is somewhat surprising since the hydroxyl group of S130 in the wt SHV-1 SA2-13 complex interacts with the carboxyl linker of SA2-13 thus stabilizing the trans-enamine species[2]. By mutating S130 to a glycine, it was anticipated that the engineered carboxylate tail of SA2-13 would lose its interaction with the hydroxyl group of S130, thereby reducing efficiency of forming the trans-enamine species. Nevertheless, despite the S130G mutation, SA2-13 retains the ability to adopt a similar trans-enamine intermediate conformation seen in the wt structure with only a slight adjustment of the carboxyl linker position. Surprisingly, SA2-13 managed to form a trans-enamine species with S130G SHV within 30min, which is not evident by soaking either tazobactam or clavulanic acid. In the S130G SHV:tazobactam complex, after a 3-hour soaking of 20mM tazobactam, the crystal structure revealed a cis-enamine intermediate and possibly also a S70-bound aldehyde[12]. Similar experiments were carried out using clavulanic acid soaked into S130G, but after 20 min and 30 min soaks with 50mM clavulanate the subsequent datasets did not reveal any convincing inhibitor density (crystallographic data not shown).
Comparison with SA2-13 complexed wt SHV-1 structure
When comparing the S130G SHV:SA2-13 complex to the wt SHV-1:SA2-13 complex (PDB ID: 2H5S), the protein is in a very similar conformation as evidenced by the low Cα superposition RMSD of 0.16Å. Moreover, SA2-13 is bound in both active sites in a similar fashion, with the exception of the repositioning of the carboxyl linker (Figure 2). In both structures, intermediates are refined to be a trans-enamine species and the positions of the ester carbonyl, the C3 carboxylate and the sulfone are similar. However, with S130 being substituted to a glycine, the carboxylate tail of SA2-13 has shifted 2.4 Å into the position normally occupied by the hydroxyl group of S130. As a result, the inhibitor carboxylate tail is stabilized by an additional salt bridge with K73 while retaining the original K234 salt-bridge (Figure 2).
Figure 2. Comparison of SA2-13 bound to wt SHV-1 and SHV-1 S130G.
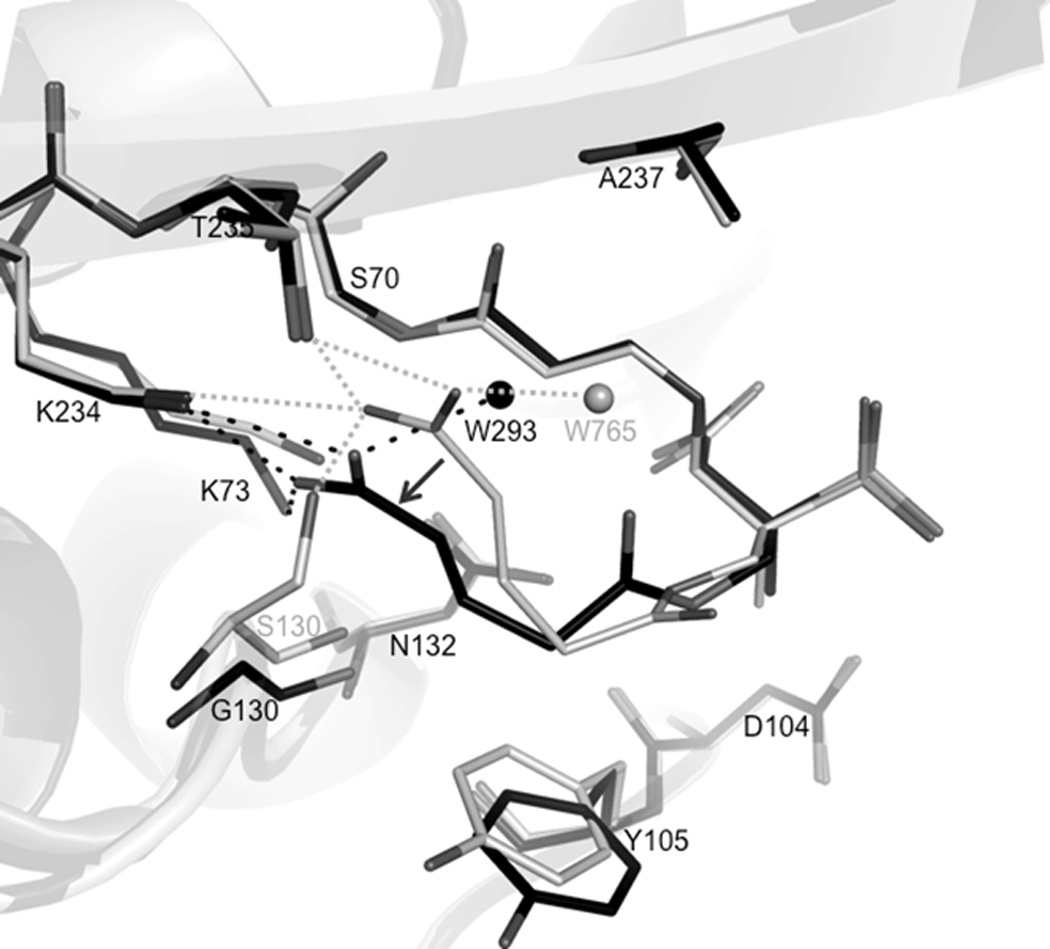
(A) All Cα superposition of S130G SHV:SA2-13 (black) with wtSHV-1:SA2-13 (PDBid 2H5S). Enzymes are shown as cartoon representation in the background. Only water molecules interacted with the carboxylate tail have been shown for simplicity. The shift of the SA2-13 carboxyl linker has been indicated via black arrows. Active site residues 70, 73, 104, 105, 130, 132, 170, 234, 235,237 are shown in lines. Both SA2-13 molecules are shown in ball-and-stick representation. Dashed lines indicated hydrogen bonds between wtSHV-1:SA2-13 (grey) or S130G SHV:SA2-13 (black).
The apparent plasticity of SA2-13 trans-enamine formation in S130G likely comes at an energetic cost as the trans-enamine dihedral torsion angle in the S130G SHV:SA2-13 structure is the one furthest away from ideality as compared to previous SA2-13 complexes. The measured trans-enamine torsion angle in the S130G SHV:SA2-13 complex was 154°; whereas, the trans-enamine torsion angles were 166° and 180° in the wt SHV-1[2] and R164 mutant SA2-13 complexes[29], respectively. Overall, these observations indicate the unique trans-enamine intermediate forming characteristics of SA2-13 as an inhibitor likely as a result of its designed carboxyl linker.
M69V SHV:SA2-13 Complex
M69 mutations introducing branched aliphatic side chains in TEM and SHV have been shown to cause an increased resistance against the three inhibitors[18]. The M69V mutant achieves this while having a minimal effect on the kcat/Km ratio for the substrate ampicillin as the lower kcat is compensated for by a decrease in Km[18]. The M69V structure was determined to 1.3Å resolution and the clear electron density confirmed the methionine to valine mutation at position 69. The RMSD between the M69V SHV structure and the wt SHV-1 structure is 0.32 Å indicating that the mutation has caused little change in the overall structure. Alternate conformations were assigned for ten residues, three of which are in the active site: S130 (40/60%), S70 (50/50%) and K234 (50/50%).
Regarding SA2-13, unlike for SA2-13 complexes with wt SHV-1, S130G SHV, and R164S/H[29], there is little density observed for the inhibitor in the active site of the M69V SHV after soaking this inhibitor (Figure 3). Although there is some density continuous with the density of S70, this additional density is likely that of a partially occupied water in the oxyanion hole with its density continuous with that of the other S70 conformation (Figure 3). This oxyanion hole is formed by the backbone nitrogens of S70 and A237. Some additional density is observed near S130 but it is neither large nor clear enough to model convincingly an inhibitor intermediate and is likely that of a partially ordered water network. Because of this lack of convincing interpretable density, the SA2-13 inhibitor was not included in refinement. We did however observe some active site shifts that likely form the molecular basis for why strong SA2-13 density is not observed in the M69V mutant and those will be discussed below.
Figure 3. Active site of SHV-1 M69V after soaking SA2-13.
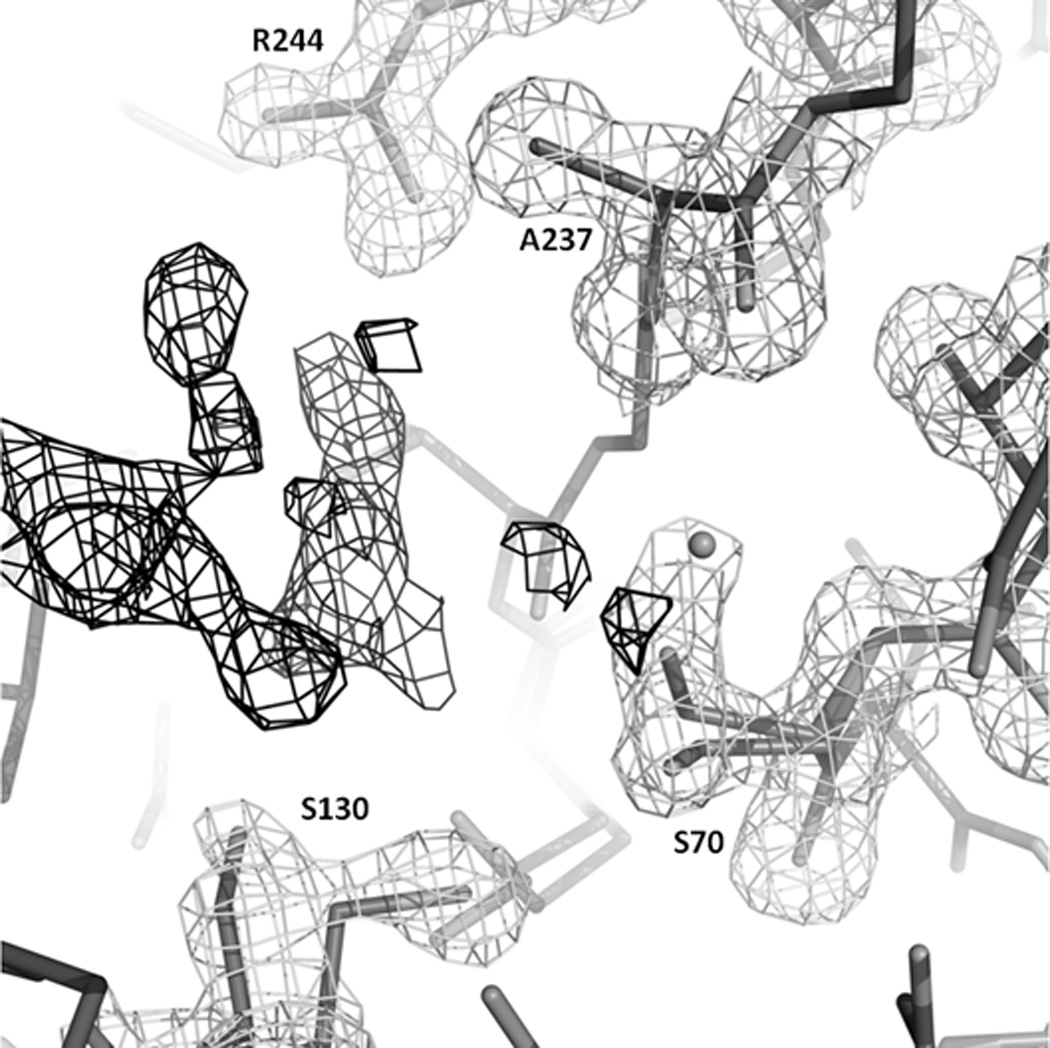
|Fo|-|Fc| positive difference electron density contoured at 3.5 σ depicted in black lines and 2 |Fo|-|Fc| electron density contoured at 1.25σ shown in gray lines. Some noisy density is present in the active site but could not be interpreted. It could perhaps be a mix of a HEPES buffer molecule, PEG, or a delocalized water network, made perhaps less interpretable due to some active site residues having multiple conformations. We did however rule out that this noisy floating density is a covalently bonded SA2-13 molecule nor an intact SA2-13 molecule.
Although residue 69 is not directly participating in ligand interactions in the active site, a mutation at this position that introduces a branched aliphatic side chain is likely able to propagate its effect through nearby residue 237. In the mutant M69V structure, the valine at the residue 69 position, being branched at Cβ, is closer to the A237 carbonyl than the methionine at the same position would be in the wt structure. Therefore, this M69V mutation forces the A237 carbonyl to be shifted towards the active site by 0.43Å as compared to the wild-type structure. This is a small shift yet it shortens the distance between the carbonyl and what would be the carbon atom position of the trans-enamine bond position (indicated by arrow) by 0.35Å (Figure 4). These shifts appear significant when compared to estimated coordinate error by ESU (estimated standard uncertainty), using R-free, of 0.05Å for the 1.3Å M69V structure. This carbonyl shift probably generated an unfavorable van der Waals distance between the trans-enamine carbon and the A237 carbonyl. This shortened distance therefore likely leads to destabilization of the trans-enamine conformation of SA2-13, which could explain the absence of strong inhibitor density in the M69V mutant. These A237 shifts were previously also observed in the M69V/E166A double mutant structure[16].
Figure 4. Comparison of M69V and wt SHV-1 structures.
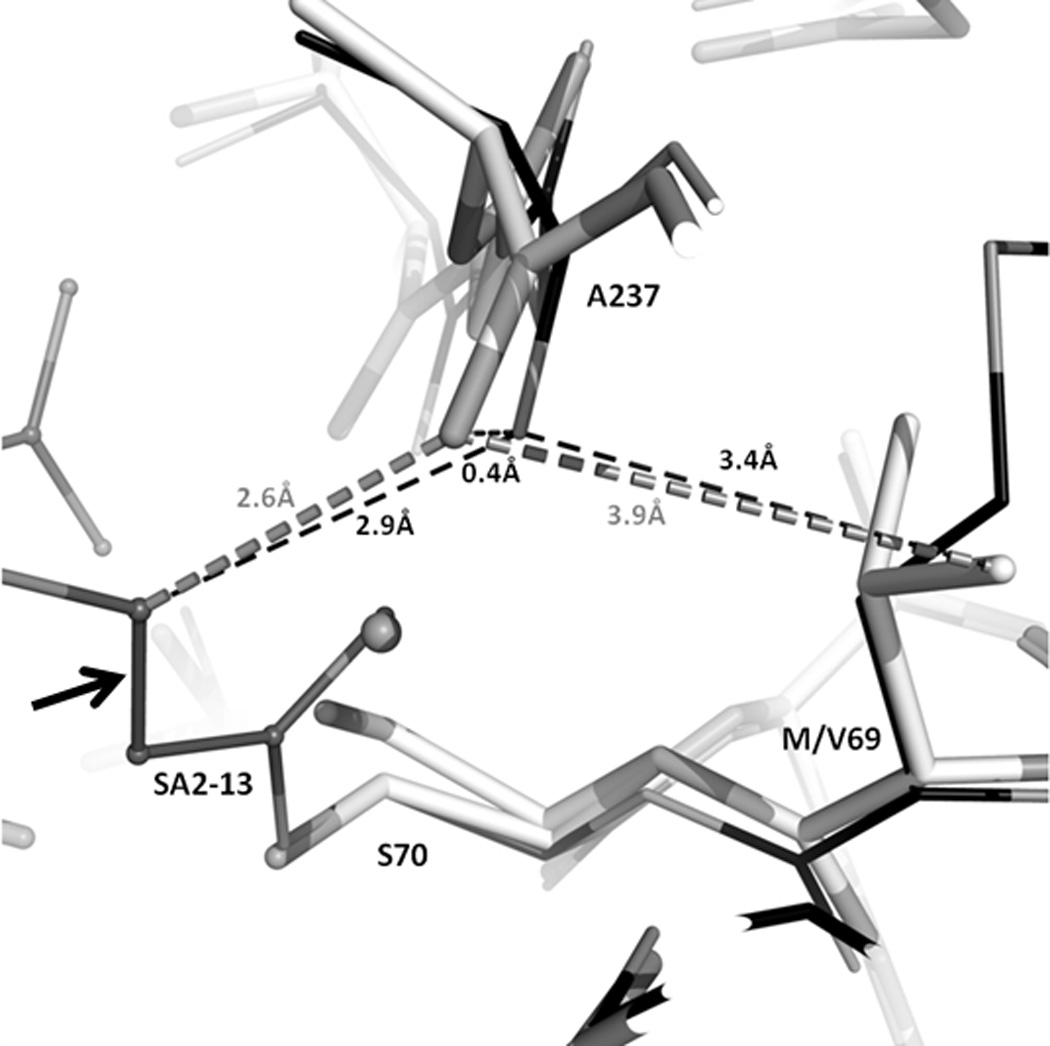
Superpositioning of M69V SHV:SA2-13 (grey) with wtSHV-1:SA2-13 (black, PDB: 2H5S). Enzymes are shown as cartoon representation in the background. Dashed lines indicate distances between atoms; distances colored grey are from M69V SHV:SA2-13 and those in black are from wtSHV-1:SA2-13. Arrow indicates the trans-enamine bond of SA2-13.
The net effect of these changes is that residue V69 pushes residue A237 outward towards the active site and the A237 carbonyl position would likely sterically hinder the trans-enamine moiety as extrapolated from the SHV-1:SA2-13 structure. Furthermore, the shift in A237 also causes the oxyanion hole, comprised of the backbone nitrogens of residues A237 and S70, to be affected since S70 itself has also moved as a result of the M69V substitution. In contrast to the SA2-13 soaked M69V crystals, soaking crystals of M69V/E166A with either tazobactam and sulbactam did result in a trans-enamine conformation of the inhibitor[16]. This observation is likely a result of these latter M69V complexes also containing the E166A mutation rendering the protein deacylation deficient or alternatively, the position of the trans-enamine bond in the tazobactam complex is 0.5Å further away from residue A237 compared to SA2-13. This latter is due to that tazobactam and also sulbactam have smaller C2 moieties compared to SA2-13, and are therefore able to shift the position of the trans-enamine moiety away from the carbonyl moiety of A237 [16]. Despite these differences, both the single and double M69V/E166A variant do show a common trend in that Raman studies did indicate a decrease in the trans-enamine population of the M69V/E166A double mutant for both tazobactam and clavulanic acid[16]. These above described small yet significant conformational changes could be the basis for M69V SHV’s resistance against being stably inhibited by trans-enamine inhibitory species of SA2-13.
An additional potential reason for SA2-13’s inability of forming a stable trans-enamine intermediate in the active site of M69V SHV could be due to the multiple conformations of active site residues. Three such residues were found to have alternate conformations in the M69V SHV structure: S130, S70 and K234. K234 has been implicated in both substrate and transition state binding[30] whereas S130 has been shown to be involved in the formation of the Michaelis-Menten complex during substrate binding (via the carboxylic acid group) and in acyl-enzyme formation[3]. S130 has also previously been shown to change conformation in M69 variants of TEM [17] and in M69V/E166A SHV complex structures[16]. These alternate conformations likely will cause a mixed population of inhibitor species as the inhibitor will experience a non-uniform active site environment. As such, these multiple alternate conformations for these residues could contribute to the absent inhibitor electron density for the trans-enamine intermediate of SA2-13.
In conclusion, the SA2-13 inhibitor and its mode of inhibition via the trans-enamine intermediate is resilient against the IR S130G SHV β-lactamase but not against the IR M69V SHV β-lactamase. SA2-13 is able to adopt a slightly different conformation in the S130G SHV mutant, thereby overcoming the loss of a S130 mediated hydrogen bond. This variant is able to do this via repositioning its carboxyl linker to generate additional interactions while still adopting a trans-enamine conformation. However, SA2-13 trans-enamine forming ability cannot overcome the IR M69V mutation, likely due to close proximity of the A237 carbonyl with respect to the trans-enamine bond of SA2-13 and/or possible alternate conformations of key active site residues. Our results have yielded key molecular insights into the functioning of a trans-enamine based inhibitor when in context of different IR mutations. These results extend our previous work, including on other different inhibitor-resistant mutants of SHV β-lactamase, including Arg244 and Asn276, that point to that different inhibitor resistant-resistant mutations can have different modes of action, as they for example can involve both first and second shell residues, that do not have a unified mechanism for their acquired inhibitor-resistance. Overall, the insights provided in this study can be used to design new inhibitors that have additional trans-enamine bond stabilizing attributes or have the trans-enamine bond shifted away from residue A237.
Table 1.
Data collection and refinement statistics
| SA2-13 : S130G SHV | SA2-13:M69V | ||
|---|---|---|---|
| Data collection | |||
| space group | P212121 | P212121 | |
| cell dimensions a, b, c (Å) α, β, γ (deg) |
49.67, 55.48, 83.62 90.00, 90.00, 90.00 |
49.5, 55.2, 83.9 | |
| wavelength (Å) | 0.9790 | 0.9793 | |
| resolution (Å) a | 46.23-1.45 (1.50-1.45) | 50.00-1.30 (1.35-1.30) | |
| Rsym | 5.5 (37.9) | 8.2 (75.3) | |
| I/σI | 9.9 (2.0) | 21.5/11.1 | |
| Completeness (%) | 99.3 (97.5) | 99.7 (99.7) | |
| Redundancy | 3.17 (2.35) | 4.6 (4.2) | |
| Refinement | |||
| Resolution range (Å) | 33.84-1.45 (1.488-1.450) | 50.00-1.30 | |
| no. of reflections | 39329 | 57438 | |
| Rwork/Rfree | 18.05/19.71 | 16.4/18.6 | |
| no. of atoms: protein, ligands, water | 2112, 68, 248 | 2119, 47, 287 | |
| r.m.s.d. | |||
| bond length (Å) | 0.009 | 0.012 | |
| bond angles (deg) | 1.311 | 1.53 | |
| average B-factors (Å2) | |||
| protein | 12.914 | 12.5 | |
| SA2-13 / cymal-6 | 22.589 / 21.4 | - / 23.0 | |
| water | 28.321 | 30.8 | |
| Ramanchandran plot statistics (%) | |||
| core regions | 92.2 | 98.9 | |
| additional allowed regions | 7.4 | 0.8 | |
| generously allowed regions | 0.4 | ||
| disallowed regions | 0.0 | 0.4 | |
Numbers in parentheses refer to the highest resolution shell.
Acknowledgements
JDB is supported by the Robert A. Welch Foundation, Grant N-0871. FVDA is supported by National Institutes of Health (R01 AI062968). The Veterans Affairs Merit Review Program, Geriatric Research Education and Clinical Care (GRECC) and the National Institutes of Health (RO1 AI063517-01) supported RAB. The Veterans Affairs Merit Review Program supported MJS. We thank the beamline support personnel at SSRL (Stanford, CA) and ALS (Berkeley, CA) for help with data collection.
References
- 1.Drawz SM, Bonomo RA. Clin. Microbiol. Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Padayatti PS, Sheri A, Totir MA, Helfand MS, Carey MP, Anderson VE, Carey PR, Bethel CR, Bonomo RA, Buynak JD, van den Akker F. J. Am. Chem. Soc. 2006;128:13235–13242. doi: 10.1021/ja063715w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamotte-Brasseur J, Dive G, Dideberg O, Charlier P, Frere JM, Ghuysen JM. Biochem. J. 1991;279(Pt 1):213–221. doi: 10.1042/bj2790213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vakulenko SB, Geryk B, Kotra LP, Mobashery S, Lerner SA. Antimicrob. Agents Chemother. 1998;42:1542–1548. doi: 10.1128/aac.42.7.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imtiaz U, Billings EM, Knox JR, Mobashery S. Biochemistry. 1994;33:5728–5738. doi: 10.1021/bi00185a009. [DOI] [PubMed] [Google Scholar]
- 6.Kuzin AP, Nukaga M, Nukaga Y, Hujer A, Bonomo RA, Knox JR. Biochemistry. 2001;40:1861–1866. doi: 10.1021/bi0022745. [DOI] [PubMed] [Google Scholar]
- 7.Atanasov BP, Mustafi D, Makinen MW. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3160–3165. doi: 10.1073/pnas.060027897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imtiaz U, Billings E, Knox JR, Manavathu EK, Lerner SA, Mobashery S. J. Am. Chem. Soc. 1993;115:4435–4442. [Google Scholar]
- 9.Helfand MS, Bethel CR, Hujer AM, Hujer KM, Anderson VE, Bonomo RA. J. Biol. Chem. 2003;278:52724–52729. doi: 10.1074/jbc.M306059200. [DOI] [PubMed] [Google Scholar]
- 10.Helfand MS, Taracila MA, Totir MA, Bonomo RA, Buynak JD, van den Akker F, Carey PR. Biochemistry. 2007;46:8689–8699. doi: 10.1021/bi700581q. [DOI] [PubMed] [Google Scholar]
- 11.Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, Shoichet BK. Biochemistry. 2005;44:9330–9338. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun T, Bethel CR, Bonomo RA, Knox JR. Biochemistry. 2004;43:14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 13.Pagan-Rodriguez D, Zhou X, Simmons R, Bethel CR, Hujer AM, Helfand MS, Jin Z, Guo B, Anderson VE, Ng LM, Bonomo RA. J. Biol. Chem. 2004;279:19494–19501. doi: 10.1074/jbc.M311669200. [DOI] [PubMed] [Google Scholar]
- 14.Sulton D, Pagan-Roderiguez D, Zhou X, Liu Y, Hujer AM, Bethel CR, Helfand MS, Thomson JM, Anderson VE, Buynak JD, Ng LM, Bonomo RA. J. Biol. Chem. 2005;280:35528–35536. doi: 10.1074/jbc.M501251200. [DOI] [PubMed] [Google Scholar]
- 15.Kuzin AP, Nukaga M, Nukaga Y, Hujer AM, Bonomo RA, Knox JR. Biochemistry. 1999;38:5720–5727. doi: 10.1021/bi990136d. [DOI] [PubMed] [Google Scholar]
- 16.Totir MA, Padayatti PS, Helfand MS, Carey MP, Bonomo RA, Carey PR, van den Akker F. Biochemistry. 2006;45:11895–11904. doi: 10.1021/bi060990m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Minasov G, Shoichet BK. J. Biol. Chem. 2002;277:32149–32156. doi: 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- 18.Helfand MS, Hujer AM, Sonnichsen FD, Bonomo RA. J. Biol. Chem. 2002;277:47719–47723. doi: 10.1074/jbc.M207271200. [DOI] [PubMed] [Google Scholar]
- 19.Hujer AM, Hujer KM, Bonomo RA. Biochim. Biophys. Acta. 2001;1547:37–50. doi: 10.1016/s0167-4838(01)00164-9. [DOI] [PubMed] [Google Scholar]
- 20.Lin S, Thomas M, Mark S, Anderson V, Bonomo RA. Biochim. Biophys. Acta. 1999;1432:125–136. doi: 10.1016/s0167-4838(99)00025-4. [DOI] [PubMed] [Google Scholar]
- 21.Pflugrath JW. Acta Crystallogr. D. Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 22.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr. D. Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Schuttelkopf AW, van Aalten DM. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 26.van den Akker F, Hol WG. Acta Crystallogr. D Biol. Crystallogr. 1999;55(Pt 1):206–218. doi: 10.1107/S0907444998007161. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J. Appl. Cryst. 2001;26:283–291. [Google Scholar]
- 28.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Sampson JM, Ke W, Bethel CR, Pagadala SR, Nottingham MD, Bonomo RA, Buynak JD, van den Akker F. Antimicrob. Agents Chemother. 2011 doi: 10.1128/AAC.01360-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellerby LM, Escobar WA, Fink AL, Mitchinson C, Wells JA. Biochemistry. 1990;29:5797–5806. doi: 10.1021/bi00476a022. [DOI] [PubMed] [Google Scholar]