

Abstract
When complex biological materials are analyzed without an adequate sample preparation technique, MS signal and response undergo significant alteration and result in poor quantification and assay. This problem generally takes place due to the presence of several endogenous materials component in samples. One of the major causes of ion suppression in bioanalysis is the presence of phospholipids during LC-MS analysis. The phospholipid-based matrix effect was investigated with a commercially available electro spray ionization (ESI) source coupled with a triple quadrupole mass spectrometer. HybridSPE dramatically reduced the levels of residual phospholipids in biological samples, leading to significant reduction in matrix effects. This new procedure that combines the simplicity of precipitation with the selectivity of SPE allows obtaining much cleaner extracts than with conventional procedures. HybridSPE-precipitation procedure provides significant improvement in bioanalysis and a practical and fast way to ensure the avoidance of phospholipids-based matrix effects. The present review outlines the HybridSPE technique to minimize phospholipids-based matrix effects on LC–ESI-MS bioanalysis.
KEYWORDS: Bioanalytical, electro spray ionization, HybridSPE, matrix effects, phospholipids
Introduction
High-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) due to its high degree of sensitivity and selectivity is widely used in bioanalysis of small molecules. When complex materials are analyzed without an adequate sample preparation technique, MS signal and response undergoes significant alteration and result in poor quantification and assay. This problem generally takes place due to the presence of several endogenous materials component in samples.[1–7] Matrix effect is defined as the effect of co-eluting residual matrix components on the ionization of the target analyte. When a rather long isocratic or gradient chromatographic program is used in the quantitative assay, matrix effect may not be present at the retention time for an analyte. However, in the case of high-throughput LC-MS/MS analysis, matrix effect is one of the major issues to be addressed in method development and validation, especially when analyte is not well separated from the LC-front. One problem brought by matrix suppression effect is reduced sensitivity when analyte signal is suppressed. Detailed studies on matrix effects revealed that the ion suppression or enhancement are frequently accompanied by significant deterioration of the precision of the analytical method as demonstrated by Matuszewski et al.[8] The authors studied the precision (%R.S.D.) upon repetitive injection of post-extraction spiked plasma samples as a function of the analyte concentration for a single lot and for five different lots of plasma. While for the single plasma lot the precision is acceptable, it may not be when different plasma lots are taken into account. Generally, matrix effect impacts more on the low end of calibration curve than the mid range or highly end. When discussing matrix effects, it is useful to discriminate between ion suppression (or enhancement) by the matrix on one hand, and different matrix effects exerted by different sample lots on the other hand. The difference in response between a neat solution sample and the post-extraction spiked sample is called the absolute matrix effect, while the difference in response between various lots of post-extraction spiked samples is called the relative matrix effect. If no counteraction is taken, an absolute matrix effect will primarily affect the accuracy of the method, while a relative matrix effect will primarily affect the precision of the method.[2] One of the best tools for observing matrix effects is the post column infusion technique.
Matrix Effect
Ion-suppression, sometimes referred to as matrix effect, is a common problem in atmospheric pressure ionization (API) mass spectrometer. Phospholipids, and in particular glycerophosphocholines and lysophosphatidylcholine [Figure 1] represent the major class of endogenous compounds causing significant matrix effects.[2–9] Phospholipids are important class of biological compounds containing one or more phosphate groups.
Figure 1.
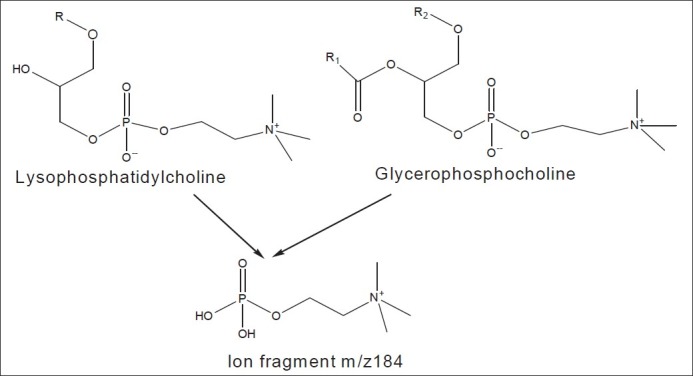
Structural formula of phospholipids and their fragment contributing major matrix effect problem
For their inherent characteristics these polar lipids are of great importance for the structure and function of cell membranes and are the most abundant of membrane lipids. Although not stored in large amounts in the system,[10] they are particularly abundant in plasma samples. Their molecular structure exhibit two major functional group regions: a polar head group substituent, which includes an ionizable organic fatty acid ester group which impart considerable hydrophobicity to the molecule. In particular, the highly ionic nature of phospholipids makes them responsible for influencing the ionization in electrospray MS sources,[11,12] and desolvation of the LC effluent droplets in electrospray MS analysis.[13]
Therefore, the removal of phospholipids represents phosphate moiety as well as other polar groups of various types, and one or two long chain being an extremely important step in the sample preparation process.[14] Several techniques have been applied toward the removal of phospholipids in order to obtain cleaner sample extracts. Most of them are based on solid-phase extraction procedures using strong cation exchange sorbents,[12] automated on-line SPE.[15] Matrix effect is a common problem in atmospheric pressure ionization (API) mass spectrometry.[16] There is difference in opinion as to amount of ion-suppression that is acceptable for an analytical method. In some laboratories, ion-suppression in an analytical method is not acceptable, but in other cases, it is tolerated if there is no significant effect on the validity of the analytical data with appropriate QCs. The phenomenon of ion-suppression results in sporadic reduction of signal intensity. Consequently, the lower limit of quantitation (LLOQ) for highly sensitive bioanalytical methods could be difficult to achieve. The problem can further be complicated by differential ion-suppression due to inter subject variability, and the use of blank bio-matrix with varying lot numbers (i.e., control blood obtained from different patients/subjects) in preparing the calibrators. Differences in ion-suppression between the analyte and structurally different internal standards may also be problematic. This issue can be mostly mitigated by the use of stable isotopically labeled (SIL) internal standards. The extent of ion-suppression is dependent on the sample preparation method and chromatographic separation. The use of protein precipitation (PPT) is most likely to cause an ion-suppression in ESI due to the lack of selectivity (co-elution of endogenous compounds such as lipids, phospholipids, fatty acids, etc., that affect the ESI droplet de-salvation process; however, in comparison, the extracts obtained from solid phase extraction (SPE) and liquid–liquid extraction (LLE) are relatively cleaner.[9] Further studies on the molecular identities of co-eluting endogenous compounds are required to clearly delineate their contributions to ion-suppression. Ion-suppression can lead to random variations in the internal standard signal, standard curve slope and intercept, LLOQ, and overall reproducibility of an assay. Lastly, with an exception, the use of APCI instead of ESI can eliminate or minimize ion-suppression. However, the analyst should perform preliminary experiments on assay sensitivity as well as thermal stability of the test article during APCI optimization to ascertain its structural integrity during analysis.[17]
How to Diagnose that it is Matrix Effect or Poor Recovery
Recovery is determined by comparing the peak area of extracted sample with those neat solutions spiked into the blank matrix. Because both samples have the matrix ingredients present, the matrix can be considered same for the extracted sample. Any difference in response is due to the extraction recovery. The extraction efficiency (or recovery) was determined by measuring an extracted sample against a (post-extraction) spiked sample:
Where, A is the response for the extracted samples and B the response for post- extracted spiked samples.
Now the matrix effect is determined by comparing the peak area of the neat solution spiked into blank (post-extracted) with those of the neat solution. Because analyte is not extracted, it should have same response in the post-extraction spiked sample and in neat solution. The matrix effect will be responsible for difference in response. A useful method to assess matrix suppression is post-column infusion of analyte into mass detector. The matrix effect was measured by referring the post-extracted spiked sample to the unextracted sample, using the following equation:
Where, C is the peak area of a neat standard and B is the corresponding peak area for standards spiked into plasma after extraction.
A Post-Column Infusion Experiment That Can be Used to Detect Matrix Effects in an HPLC–MS–MS Assay
Here the extracted blank matrix is injected by an auto sampler into the analytical column. The purpose of post-column infusion with analyte is to raise the background level so that the suppression matrix will show negative peak. This method [Figure 2] has been successfully used to identify matrix suppression peak.[18]
Figure 2.

Schematic of post column infusion experiment commonly used for the qualitative assessment of ion suppression
Problems/Difficulties in Removing Phospholipids from Biological Samples
Due to the nature of phospholipids, none of the mainstream sample preparation techniques; protein precipitation technique (PPT), liquid–liquid extraction (LLE), and solid phase extraction (SPE) offer the proper selectivity to distinguish phospholipids from analytes of interest. Their molecular structure exhibit two major functional group regions: a polar head group substituent, which includes an ionizable organic phosphate moiety as well as other polar groups of various types, and one or two long chain fatty acid ester groups [Figure 3], which impart considerable hydrophobicity to the molecule. Phospholipids co-extract with analytes of interest when using traditional sample preparation technique.[19]
Figure 3.
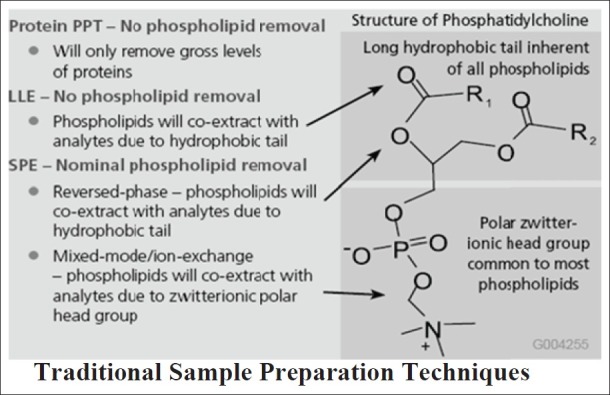
Phospholipids co-extracts with analytes of interest when using traditional techniques (Courtesy of Sigma-Aldrich; HybridSPE-ppt, USA)
Protein precipitation technique
Protein precipitation is the simplest means of sample pretreatment. Due to the selectivity of MS detectors, it is thought that sample pretreatment for LC–MS/MS assays is redundant. Infact, the largely polar and hydrophilic character of some natural products makes it difficult to extract analytes from plasma with solvents, and PPT technique was therefore often used.[20,21] The results showed the deproteinization by acetonitrile gave a good resolution and high recovery. Based on the above mentioned reasons, many natural products in biological fluids were extracted by this method.[22–30] It is also worth noting that precipitation of proteins with acids may catalyze the hydrolysis of some conjugates such as glucuronides and sulphates.[31] It removes gross levels of proteins but no phospholipids.
Liquid–liquid extraction
Liquid–liquid extraction is especially suited for lipophilic compounds. The selection of sample pretreatment techniques for each bio-fluid depends on expected analyte concentrations and required detection limits. Acetonitrile protein precipitation provides sufficient pre-concentration and protein removal for quantitative analysis of analytes in bio-fluids. Matrix suppression data indicate that SPE or LLE is required prior to LC–MS bioanalysis.[3] In LLE phospholipids will co-extract with analytes due to hydrophobic tail.[19]
Solid-phase extraction
In the bioanalysis solid-phase extraction is a frequently used technique for sample pre-treatment. Compared with the PPT procedure, the SPE method reduces the serum background greatly [Figure 4]. SPE is chosen for the extraction and purification of analytes due to its high selectivity, speed of extraction, the potential for automation, and the fact that much lower volumes of organic solvents are required than those for liquid–liquid extraction.[32] In some cases, it is necessary to acidify or alkalify the biological fluids containing analytes before transferring samples to the SPE column.[33,34] SPE can be performed off-line manually; it is known that the efficiency of SPE depends on the type of sorbents, the sample volume and pH, the content of organic modifier. In SPE, nominal phospholipids are removed (in reversed-phase SPE-phospholipids will co-extract with analytes due to hydrophobic tail while in mixed-mode/ion-exchange SPE-phospholipids will co-extract with analytes due to the zwitterionic polar head group).[19]
Figure 4.
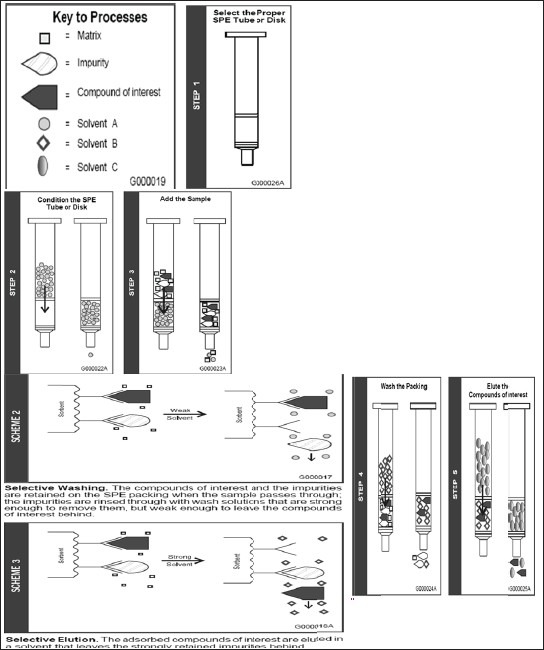
Different step of SPE (Courtesy of Sigma-Aldrich; HybridSPE-ppt, USA)
Novel-Hybrid SPE-Precipitation Technology
A pitfall of all the above-discussed methods is the matrix effect since all of these methods, except one, used pure neat analyte solutions to model the SPE characteristics for use with real matrix samples. Since plasma, serum, and urine are complex matrices, containing many compounds that could interfere with analyte retention, care must be taken in applying the models to real samples. To diminish the effect of the matrix, it can be diluted as far as possible before applying it to the sorbent. Also, it is wise to take some margin into account in selecting organic modifier concentrations used in wash and elution steps. The matrix effect is compound dependent and rather unpredictable. Therefore, the recovery of a proposed SPE method should always be verified by using a spiked matrix sample.
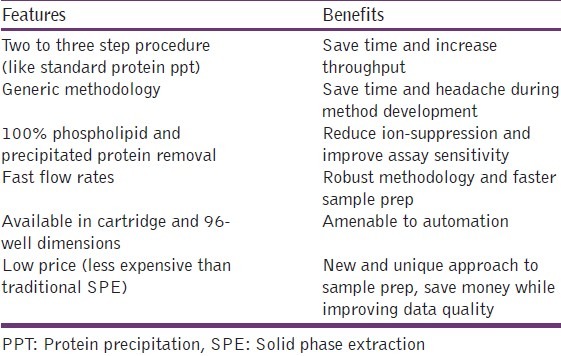
HybridSPE-PPT technology is a simple and generic sample prep platform designed for the gross level removal of endogenous protein and phospholipids interferences from biological plasma and serum prior to LC-MS or LC-MS-MS analysis. The important feature and benefits of this technique are mentioned in Table 1.
Table 1.
Important feature and benefits of the HybridSPEPPT technique
Procedure
Biological plasma or serum is first subjected to protein precipitation in which biological plasma/serum is first added to the 96-well plate followed by acidified acetonitrile (precipitation agent). An upper PTFE frit impedes premature flow of the sample before vacuum application. After a brief mixing/vortexing step to facilitate protein ppt, vacuum is applied to the 96-well plate.
The 96-well version contains a series of low porosity hydrophobic filters/frits. The packed-bed filter/frit assembly acts as a depth filter facilitating the concurrent removal of both phospholipids and precipitated proteins during the extraction process. After the protein-precipitated sample passes through the HybridSPE-PPT device, the resulting eluent is ready for immediate LC-MS or LC-MS-MS analysis [Figure 5].
Figure 5.
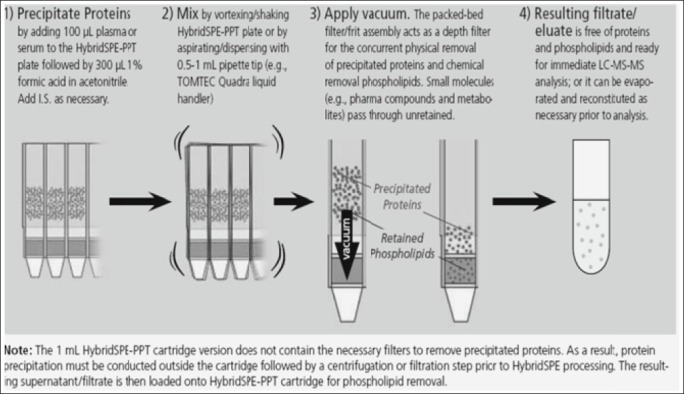
Different step of HybridSPE-PPT (Courtesy of Sigma-Aldrich; Hybrid SPE-ppt, USA)
Working mechanism
Once the plasma/serum sample is subjected to protein precipitation via the addition of acetonitrile (containing 1% formic acid), it is passed through the HybridSPE-PPT packed bed. The packed bed consists of proprietary zirconia-coated silica particles [Figure 6]. The zirconia sites exhibit Lewis acid (electron acceptor) properties that will interact strongly with Lewis bases (electron donor). Phospholipids structurally consist of a polar head group (zwitterionic phosphonate moiety) and a large hydrophobic tail (two fatty acyl groups that are hydrophobic). The phosphate group inherent with all phospholipids acts as a very strong Lewis base that will interact strongly with zirconia atoms functionalized on the particle surface. Formic acid is a critical modifier used in the precipitation agent to improve the recovery of many analytes of interest (e.g., acidic compounds). It plays a critical role in preventing analyte retention without affecting phospholipids retention/removal.
Figure 6.
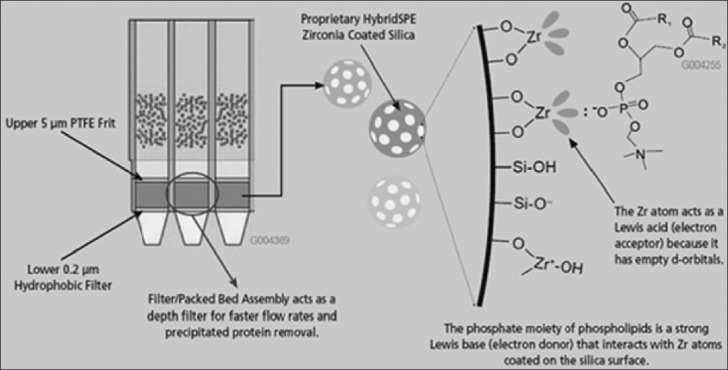
HybridSPE-ppt 96-well schematic and phospholipids retention mechanism (Courtesy of Sigma-Aldrich; HybridSPE-ppt, USA)
Method Development and Recovery Optimization
Zr atoms bonded to the HybridSPE silica surface act as Lewis acid because it contains vacant d-orbital and interacts with sample component by Lewis acid-base interaction. Phospholipids are retained/removed from the sample through a highly selectivity Lewis acid-base interaction between the phosphate group (Lewis base), inherent of all phospholipids, and Zr atoms bonded to the HybridSPE silica surface. When developing new HybridSPE-PPT methods, there are three precipitation agents recommended for researchers are citric acid, formic acid and ammonium form a tea modifier that inhibits analyte(s) of interest from co-retaining with phospholipids on the Zr-Si HybridSPE stationary phase, potentially resulting in low recovery.
Formic acid is used as a modifier for most of the acidic, basic, and neutral compound. Because the formate ion is stronger lewis base that can prevent the interaction of most of acid compound to Zerconiasites and H+ produced by formic acid ionization neutralized silanol effect, it can improve recovery [Figure 7].
Figure 7.
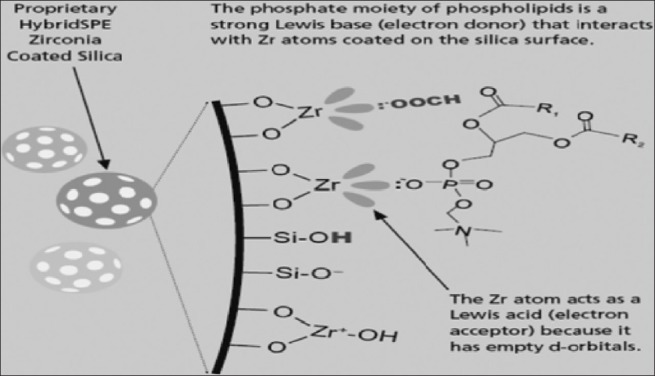
Interaction of formic acid modifier with zerconia-coated stationary phases (Courtesy of Sigma-Aldrich; HybridSPE-ppt, USA)
Ammonium formate in methanol recommended for low recovery basic and neutral compounds. Methanol combined with ammonium formate is a powerful protein precipitation agent, providing precipitants similar in consistency to acetonitrile protein precipitation. The formate ion (HCOO-) is a strong enough Lewis base to inhibit most acidic compounds from interacting with Zr sites, improving recovery of acidic compounds. NH4+ is a stronger counter-ion than H+, able to inhibit a wider range of basic compounds from interacting with exposed silanol groups.
Citric acid in acetonitrile recommended for low recovery chelator and acidic chelator compounds (citric acid) is a stronger Lewis base than format, inhibiting most chelator compounds from interacting with Zr ions on the HybridSPE stationary phase [Figure 8]. Note that citrate is not strong enough to disrupt phosphate (phospholipids) binding.[19] The flow chart [Figure 9] is given here under for the convenience of reader.
Figure 8.
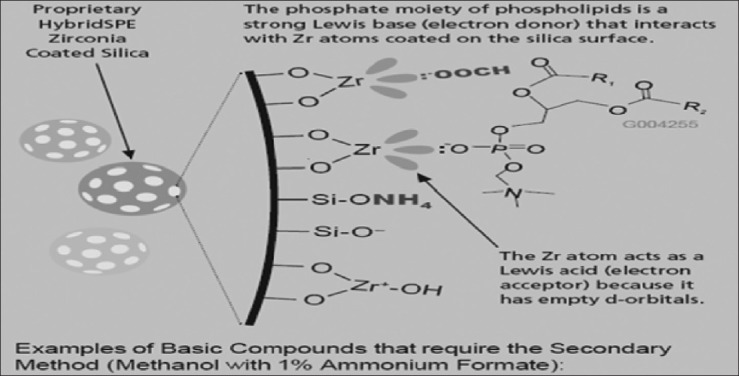
Interaction of ammonia formate modifier with zerconia-coated stationary phases (Courtesy of Sigma-Aldrich; HybridSPE-ppt, USA)
Figure 9.
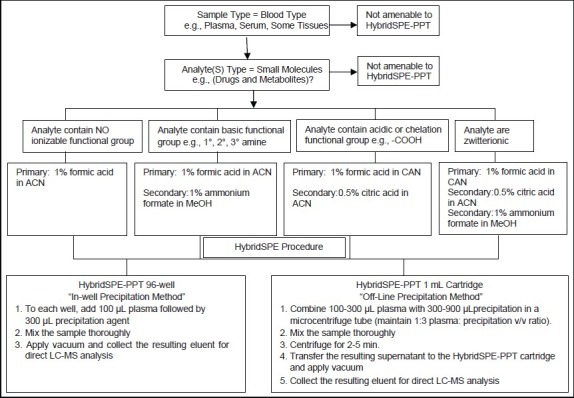
HybridSPE-PPT method development (recovery optimization) flow chart
Comparison of sample preparation techniques
HybridSPE-PPT combines the simplicity of protein precipitation with the selectivity of SPE targeting the specific removal of both phospholipids and precipitated proteins from biological plasma. Both phospholipids and protein represent the two largest contributors of ion-suppression in pharmaceutical bioanalysis. If adequately removed prior to LC-MS, analysts can more easily and quickly achieve targeted LLOQ for most applications. Comparison of sample preparation techniques are given in Table 2.
Table 2.
Comparison of sample prep techniques
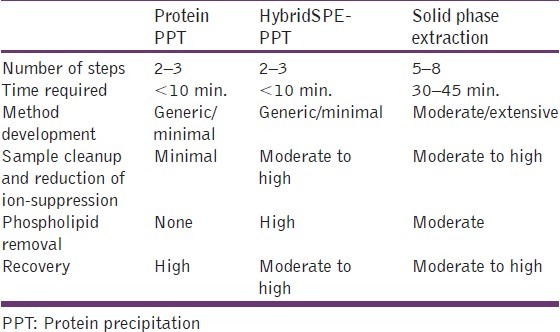
Comparison of HybridSPE-PPT, protein precipitaiton, and SPE
In this application example, rat plasma samples were spiked with clenbuterol (R(-) and S(+) enantiomers) at the level of 10 ng/mL and extracted using three different procedures: HybridSPE-PPT, Protein PPT, and a 9-step SPE procedure optimized for trace level clenbuterol analysis. The analysis was performed using a chiral stationary phase containing a macrocyclic glycopeptide covalently bound to silica and detection via MS-MS. Comparisons of sample preparation methods were made in terms of the amount of phospholipids in the sample extract and the overall effect on signal response of clenbuterol enantiomers. Absolute recovery was determined against an external standard. Representative chromatograms of each the sample preparation techniques are depicted in [Figure 10]. From the results, it was found that phospholipid contamination levels were highest for protein precipitation resulting in signal suppression levels 70% and 25% for the R(-) and S(+) enantiomers of clenbuterol, respectively. For the SPE procedure, phospholipid contamination was still evident after multiple wash steps, and overall absolute recovery was less than 50%. In contrast, HybridSPE-PPT offered 100% removal of phospholipids resulting in absolute recovery levels of 95%.
Figure 10.
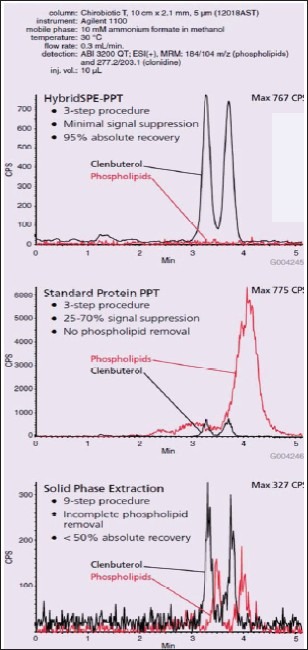
Comparative extraction and LC-MS-MS of 10 ng/mL Clenbuterol (R(-) and S(+) enantiomers) in rat plasma (Courtesy of Sigma-Aldrich; HybridSPE-ppt, USA)
Summary and Conclusion
The aforementioned discussion indicated that although plasma protein precipitation can sometimes fulfill the requirements of extraction yield but not adequate to sufficiently remove phospholipids. It removes gross levels of proteins. Moreover, the monitoring of the MRM transitions of phospholipids and the matrix effect evaluation clearly indicated that endogenous phospholipids in both rat and human plasma PPT extracts are a significant tool in order to evaluate phospholipids-based matrix effects in LC–MS/MS analyses leading to ion suppression in bioanalytical determinations. While in Liquid–liquid extraction phospholipids will co-extract with analytes due to hydrophobic tail. A systematic comparison of the different traditional sample preparation techniques to the new sample preparation platform HybridSPE-Precipitation showed to represent a useful and effective tool in providing cleaner extract and in reducing or eliminating phospholipids-based matrix effects.
Acknowledgments
It is great pleasure for us to acknowledge all those who have contributed towards the conception, origin and nurturing of this review article. With a deep sense of gratitude and the respect, we thank to Dr. R.S. Gaud, Dean School of pharmacy and Technology Management, NMIMS University, Mumbai (India), for his constant encouragement and co-operation. We extend thanks to Sigma-Aldrich for giving us permission to use their copyrighted material.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared
References
- 1.Shou WZ, Naidong W. Post-column infusion study of the ‘dosing vehicle effect’ in the liquid chromatography/tandem mass spectrometric analysis of discovery pharmacokinetic samples. Rapid Commun Mass Spectrom. 2003;17:589–97. doi: 10.1002/rcm.951. [DOI] [PubMed] [Google Scholar]
- 2.Ismaiel OA, Halquist MS, Elmamly MY, Shalaby A, Karnes HT. Monitoring phospholipids for assessment of matrix effects in a liquid chromatography-tandem mass spectrometry method for hydrocodone and pseudoephedrine in human plasma. Analyt Technol Biomed Life Sci. 2007;859:84–93. doi: 10.1016/j.jchromb.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Dams R, Huestis MA, Lambert WE, Murphy CM. Matrix effect in bio-analysis of illicit drugs with LC-MS/MS: Influence of ionization type, sample preparation, and biofluid. J Am Soc Mass Spectrom. 2003;14:1290–4. doi: 10.1016/S1044-0305(03)00574-9. [DOI] [PubMed] [Google Scholar]
- 4.Taylor PJ. Matrix effects: The achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin Biochem. 2005;38:328–34. doi: 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 5.van De Steene JC, Mortier KA, Lambert WE. Tackling matrix effects during development of a liquid chromatographic-electrospray ionization tandem mass spectrometric analysis of nine basic pharmaceuticals in aqueous environmental samples. J Chromatogr A. 2006;1123:71–81. doi: 10.1016/j.chroma.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Jemal M, Xia YQ. LC-MS Development strategies for quantitative bioanalysis. Curr Drug Metab. 2006;7:491–502. doi: 10.2174/138920006777697927. [DOI] [PubMed] [Google Scholar]
- 7.Souverain S, Rudaz S, Veuthey J. Matrix effect in LC-ESI-MS and LC-APCI-MS with off-line and on-line extraction procedures. J Chromatogr A. 2004;1058:61–6. [PubMed] [Google Scholar]
- 8.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC–MS/MS. Anal Chem. 2003;75:3019–30. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 9.Little JL, Wempe MF, Buchanan CM. Liquid chromatography-mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. J Chromatogr B. 2006;833:219–30. doi: 10.1016/j.jchromb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 10.Myher JJ, Kuksis A, Pind S. Molecular species of glycerophospholipids and sphingomyelins of human plasma: Comparison to red blood cells. Lipids. 1989;24:408–18. doi: 10.1007/BF02535148. [DOI] [PubMed] [Google Scholar]
- 11.Mei H, Korfmacher M. Using Mass Spectrometry for Drug Metabolism Studies. Boca Raton, FL: CRC Press; 2005. pp. 103–50. [Google Scholar]
- 12.Shen JX, Motyka RJ, Roach JP, Hayes RN. Minimization of ion suppression in LC-MS/MS analysis through the application of strong cation exchange solid-phase extraction (SCX-SPE) J Pharm Biomed Anal. 2005;37:359–67. doi: 10.1016/j.jpba.2004.10.035. [DOI] [PubMed] [Google Scholar]
- 13.King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom. 2000;11:942–50. doi: 10.1016/S1044-0305(00)00163-X. [DOI] [PubMed] [Google Scholar]
- 14.Chambers E, Wagrowski-Diehl DM, Lu Z, Mazzeo JR. Systematic and comprehensive strategy for reducing matrix effects in LC/MS/ MS analyses. J Chromatogr B. 2007;852:22–34. doi: 10.1016/j.jchromb.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Marchese A, McHugh C, Kehler J, Bi H. Determination of pranlukast and its metabolites in human plasma by LC/MS/MS with PROSPEKT™ on-line solid-phase extraction. J Mass Spectrom. 1998;33:1071–9. doi: 10.1002/(SICI)1096-9888(1998110)33:11<1071::AID-JMS719>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 16.Bonfiglio R, King RC, Olah T, Merkle K. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun Mass Spectrom. 1999;13:1175–85. doi: 10.1002/(SICI)1097-0231(19990630)13:12<1175::AID-RCM639>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 17.Liu DQ, Pereira T. Interference of a carbamoylglucuronides metabolite in quantitative liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:142–6. doi: 10.1002/rcm.549. [DOI] [PubMed] [Google Scholar]
- 18.Pucci V, Palma SD, Alferi A, Bonelli F, Monteagudo E. A novel strategy for reducing phospholipids-based matrix effect in LC-ESI-MS bioanalysis by means of HybridSPE. J Pharm Biomed Anal. 2009;50:867–71. doi: 10.1016/j.jpba.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 19.Sigmaaldrich.com. USA: Sigma-Aldrich. [Last accessed on 2010 Jan 12]. Available from: http://www.sigmaaldrich.com/Graphics/Supelco/objects/4600/4538.pdf .
- 20.Guo JF, Zhao YM, Zhao L, Zhang WQ, Zhang AJ, Xu B. Simultaneous quantification of CTN986 and its deglycosylation products in rat serum using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:1701–8. doi: 10.1002/rcm.2491. [DOI] [PubMed] [Google Scholar]
- 21.Xing J, Chen X, Zhong D. Stability of baicalin in biological fluids in vitro. J Pharm Biomed Anal. 2005;39:593–600. doi: 10.1016/j.jpba.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 22.Ji HY, Lee HW, Kim HK, Kim HH, Chang SG, Sohn DH, et al. Simultaneous determination of ginsenoside Rb(1) and Rg(1) in human plasma by liquid chromatography-mass spectrometry. J Pharm Biomed Anal. 2004;35:207–12. doi: 10.1016/j.jpba.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 23.Soepenberg O, de Bruijn P, Verweij J, Atsumi R, Sparreboom A. Liquid chromatographic assays for DE-310, a novel camptothecinanalog, and two major enzymatic products in human matrices. J Chromatogr B. 2004;799:15–22. doi: 10.1016/j.jchromb.2003.09.047. [DOI] [PubMed] [Google Scholar]
- 24.Zhao HQ, Qu Y, Wang XY, Lu XY, Zhang XH, Hattori M. Determination of trigonelline by HPLC and study on its pharmacokinetics. Yao XueXueBao. 2003;38:279–82. [PubMed] [Google Scholar]
- 25.Kaneko R, Hattori S, Furuta S, Hamajima M, Hirata Y, Watanabe K, et al. Sensitive analysis of aconitine, hypaconitine, mesaconitine and jesaconitine in human body fluids and Aconitum-tubers by LC/ ESI- TOF-MS. J Mass Spectrom. 2006;41:810–4. doi: 10.1002/jms.1038. [DOI] [PubMed] [Google Scholar]
- 26.Guo W, Ahmad A, Khan S, Dahhani F, Wang YF, Ahmad I. Determination by liquid chromatography with fluorescence detection of total 7-ethyl-10-hydroxy-camptothecin (SN-38) in beagle dog plasma after intravenous administration of liposome-based SN-38 (LE–SN38) J Chromatogr B. 2003;79:85–92. doi: 10.1016/s1570-0232(03)00210-1. [DOI] [PubMed] [Google Scholar]
- 27.Zhai S, Sausville E, Figg WD. A high-performance liquid chromatography method using ultraviolet detection for the quantitation of flavopiridol from human plasma. Biomed Chromatogr. 2002;16:379–82. doi: 10.1002/bmc.166. [DOI] [PubMed] [Google Scholar]
- 28.Wang XL, Xing DM, Wang W, Su H, Tao JL, Du LJ. Pharmacokinetics of Berberine in Rat Thalamus after Intravenous Administration of Coptidis Rhizoma Extract. Am J Chin Med. 2005;33:935–43. doi: 10.1142/S0192415X05003557. [DOI] [PubMed] [Google Scholar]
- 29.Cheng X, Shin YG, Levine BS, Smith AC, Tomaszewski JE, van Breemen RB. Quantitative analysis of betulinic acid in mouse, rat and dog plasma using electrospray liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2089–92. doi: 10.1002/rcm.1155. [DOI] [PubMed] [Google Scholar]
- 30.Xu RN, Fan L, Rieser MJ, El-Shourbagy TA. Recent advances in high-throughput quantitative bioanalysis by LC-MS/MS. J Pharm Biomed Anal. 2007;44:342–55. doi: 10.1016/j.jpba.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Tan ZR, Ouyang DS, Zhou G, Wang LS, Li Z, Wang D, et al. Sensitive bioassay for the simultaneous determination of pseudoephedrine and cetirizine in human plasma by liquid-chromatography-ion trap spectrometry. J Pharm Biomed Anal. 2006;42:207–12. doi: 10.1016/j.jpba.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 32.Murphy CM, Huestis MA. LC-ESI-MS/MS analysis for the quantification of morphine, codeine, morphine-3-β-D-glucuronide, morphine-6-β-D-glucuronide, and codeine-6-β-D-glucuronide in human urine. J Mass Spectrom. 2005;40:1412–6. doi: 10.1002/jms.921. [DOI] [PubMed] [Google Scholar]
- 33.Samanidou VF, Evaggelopoulou EN, Papadoyannis IN. Simultaneous determination of quinine and chloroquine anti-malarial agents in pharmaceuticals and biological fluids by HPLC and fluorescence detection. J Pharm Biomed Anal. 2005;38:21–8. doi: 10.1016/j.jpba.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Xing J, Xie C, Lou H. Recent application of liquid chromatography-mass spectrometry in natural product. J Pharm Biomed Anal. 2007;44:368–78. doi: 10.1016/j.jpba.2007.01.010. [DOI] [PubMed] [Google Scholar]