

Abstract
Background:
Fruit extracts of Xylopia aethiopica are used traditionally in the management of pain disorders including rheumatism, headache, colic pain, and neuralgia. Little pharmacological data exists in scientific literature of the effect of the fruit extract and its major diterpene, xylopic acid, on pain. The present study evaluated the analgesic properties of the ethanol extract of X. aethiopica (XAE) and xylopic acid (XA), in murine models.
Materials and Methods:
XAE and XA were assessed in chemical (acetic acid-induced abdominal writhing and formalin tests), thermal (Tail-flick and Hargreaves thermal hyperalgesia tests), and mechanical (Randall-Selitto paw pressure test) pain models.
Results:
XAE and XA exhibited significant analgesic activity in all the pain models used. XAE (30-300 mg kg-1, p.o.) and XA (10-100 mg kg-1, p.o.) inhibited acetic acid-induced visceral nociception, formalin- induced paw pain (both neurogenic and inflammatory), thermal pain as well as carrageenan-induced mechanical and thermal hyperalgesia in animals. Morphine (1-10 mg kg-1, i.p.) and diclofenac (1-10 mg kg-1, i.p.), used as controls, exhibited similar anti-nociceptive activities. XAE and XA did not induce tolerance to their respective anti-nociceptive effects in the formalin test after chronic administration. Morphine tolerance did not also cross-generalize to the analgesic effects of XAE or XA.
Conclusions:
These findings establish the analgesic properties of the ethanol fruit extract of X. aethiopica and its major diterpene, xylopic acid.
KEYWORDS: Formalin test, kaurene diterpenes, opioid tolerance, pain, Xylopic acid
Pain is associated with most disease states, and it is usually the major factor of the disease that alerts the patient to seek medical treatment.[1] Currently, pain treatment is far from perfect and although some of the reasons for this are attributable to inappropriate or insufficient use of existing therapies,[2–4] there is clearly a need for more effective analgesics. Again, most available analgesics have considerable side effects, especially on long term use, hence the need for the development of new analgesics with minimum side effects. Since natural products offer a huge potential for new pharmacological agents, the assessment of medicinal plants as alternative/complementary therapies is justified.
The fruit of Xylopia aethiopica (Dunnal) A. Rich (Family: Annonaceae), commonly known as African pepper in West Africa, is used for the treatment of rheumatism, headache, neuralgia, and colic pain.[5] It was employed in government hospitals in Ghana to induce placental discharge postpartum due to its abortifacient effect.[5,6] The ethanolic extract of the fruits has been shown to exert antimicrobial action against Gram-positive and Gram-negative bacteria,[7] and this was attributed to xylopic acid and the kaurene derivatives in the extract. The fruit of Xylopia aethiopica has been found to contain kauranes, a class of diterpenes, namely kaurenoic and xylopic acid.[8] Kauranes are compounds of rigid tetracyclic skeleton, and they form intermediates in the biosynthesis of plant growth hormones such as gibberellins.[9] Many biological activities have been reported for different kauranes including antimicrobial, cytotoxic, anti-parasitic, insect antifeedant, anti-HIV, and anti- inflammatory activities.[10,11] The anti-inflammatory activity of the kauranes has been shown to involve the impairment of inflammation signaling through inhibition of NF-κB activity.[12] Kaurenoic acid (ent-kaur-16-en-19-oic acid), a well-known ent-kaurene diterpene, is reported to have different biological activities such as analgesia,[13] diuretic, vasorelaxant, anti-inflammatory and anti-pyretic effects in rodents.[14,15] Xylopic acid [15β-acetoxy-(-)-kaur-16-en-19-oic acid; Figure 1] and its epimer, acetylgrandifloric acid [15α-acetoxy-(-)-kaur-16-en-19-oic acid), are reported to exhibit anti-bacterial activity.[16] Some studies have also shown that xylopic acid and the ethanol fruit extract of X. aethiopica have a low toxicity profile.[14,17] Despite the traditional use of the fruits of Xylopia aethiopica in some painful conditions as well as the reported analgesic property of many of the kaurene diterpenes, there is little pharmacological data on the analgesic effect of the fruits and xylopic acid. In this study, therefore, we evaluated the analgesic properties of the ethanol extract of the fruits of Xylopia aethiopica as well as the kaurene diterpene constituent, xylopic acid.
Figure 1.
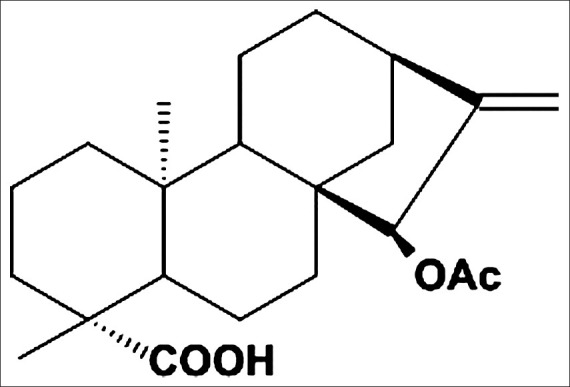
Chemical structure of 15β-Acetoxy-(-)-kaur-16-en-19-oic acid (Xylopic acid)
Materials and Methods
Collection of plant material
The dried fruits of Xylopia aethiopica were collected from the Botanical Gardens (06°41’6.39”N; 01°33’45.35”W) of Kwame Nkrumah University of Science and Technology (KNUST), Kumasi, Ghana between the months of August and December, 2008. The fruits were authenticated by Dr. Kofi Annan at the Department of Pharmacognosy, Faculty of Pharmacy and Pharmaceutical Sciences, College of Health Sciences, KNUST. A voucher specimen (No. FP/09/77) has been kept at the herbarium of the Faculty.
Preparation of the ethanol extract of Xylopia aethiopica
A quantity of the fruit was pulverized into fine powder. About 0.36 kg of the powdered material was placed in cylindrical jars and was macerated with 70% (v/v) ethanol for 3 days. The filtrate was concentrated using rotary evaporator at a temperature of 60°C. This resulted in a greenish-solid mass of ethanol extract of Xylopia aethiopica with a percentage yield of 34.8% (w/w).
Isolation and purification of xylopic acid (15β-Acetoxy-(-) - kaur-16-en-19-oic Acid)
The extraction process was carried out similar to that described by Ekong and Ogan.[8] A quantity of the fruit (0.36 kg) was pulverized and placed in cylindrical jars. This was soaked with 5 L of petroleum ether (40-60°C) and was allowed to stand for 3 days. The petroleum ether extract was collected and concentrated using rotary evaporator at a temperature of 50°C. Ethyl acetate was added to the concentrate to facilitate crystallization of xylopic acid. Crystals (xylopic acid), formed after the concentrate had been allowed to stand for a couple of days, were washed with petroleum ether (40-60°C).
The xylopic acid obtained was purified by recrystallization. The process involved the dissolution of the non-purified xylopic acid in 96% ethanol. The resulting concentrated solution was filtered while hot, and crystals of xylopic acid were deposited after the solution cooled and stood for a couple of days. The yield of the isolated/purified xylopic acid [Figure 1] was 1.41% (w/w).
Purity of the isolated xylopic acid was determined using high performance liquid chromatography (HPLC) . The chromatograph consisted of LC-10AT Shimadzu pump with programmable absorbance detector (783A Applied Biosystems) and Shimadzu CR501 Chromatopac. Phenomenex Hypersil 20 micron C18 200 × 3.20 mm column was used. The mobile phase consisted of methanol and water (9:1) eluted isocratically at 0.5 ml min-1. Portions of 20 μl of a suitable concentration of pure XA were loaded and injected unto the column after dissolving in the mobile phase at 60°C. The eluent was monitored at 206 nm. Portions of the XAE and XA were loaded and injected. The peak(s) were noted as component(s) of the XAE and XA. The purity of the isolated xylopic acid was 95%.
Drugs and chemicals
The following drugs and chemicals were used: Diclofenac sodium (Troge Medical GmbH, Hamburg, Germany); morphine hydrochloride (Phyto-Riker, Accra, Ghana); carrageenan sodium salt, formalin and acetic acid (Sigma-Aldrich Inc., St. Louis, MO, USA). XAE and XA were dissolved in an emulsion of tween 80.
Animals
Sprague-Dawley rats (150-200 g) and ICR mice (20-25 g) were purchased from Noguchi Memorial Institute for Medical Research, University of Ghana, Legon, Ghana and housed in the animal facility of the Department of Pharmacology, Kwame Nkrumah University of Science and Technology (KNUST). The animals were housed in groups of 6 in stainless steel cages (34 × 47 × 18 cm) with soft wood shavings as bedding, fed with normal commercial pellet diet (GAFCO, Tema, Ghana), given water ad libitum and were maintained under laboratory conditions (temperature 24-25°C, relative humidity 60-70%, and 12 h light-dark cycle). All procedures and techniques used in these studies were in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85 - 23, 1985, revised 1996). All protocols used were approved by the Departmental Ethics Committee.
Acetic acid-induced writhing test
The test was carried out as described earlier[18,19] with modifications. Separate groups of female mice received vehicle (10 ml kg-1 of 0.9% NaCl, i.p.), XAE (30-300 mg kg-1, p.o.), XA (10-100 mg kg-1, p.o.), morphine (1-10 mg kg-1, i.p.) or diclofenac (1-10 mg kg-1, i.p.) 60 min (p.o.) or 30 min (i.p.) before intraperitoneal administration of acetic acid (0.6%, 10 ml kg-1). Mice were then placed individually in a testing chamber (a Perspex chamber, 15 × 15 × 15 cm). A mirror inclined at 45° below the floor of the chamber allowed a complete view of the mice.
Injection of acetic acid induced a nociceptive behavior, writhing, an exaggerated extension of the abdomen combined with the outstretching of the hind limbs. Responses were captured (30 min) for analysis by a camcorder (Everio™, model GZ-MG1300, JVC, Tokyo, Japan) placed directly opposite the mirror and attached to a computer. Tracking of the behavior was done using public domain software JWatcher, version 1.0 (University of California, LA, USA, and Macquarie University, Sidney, Australia, available at http://www.jwatcher.ucla.edu/) to obtain the frequency and duration of writhes per 5 min, starting immediately after acetic acid administration. A nociceptive score was determined for each 5-min time block by multiplying the frequency and duration of writhes. These data were expressed in a time course, which enabled the observance of changes in the writhing induced.
Formalin-induced nociception
The formalin test was carried out as described previously.[20,21] Each animal was assigned and acclimatized to 1 of 20 formalin test chambers (a Perspex chamber 15 × 15 × 15 cm) for 30 minutes before formalin injection. Male mice were then pre- treated with vehicle (10 ml kg-1 of 0.9% NaCl, i.p), XAE (30-300 mg kg-1, p.o.), XA (10-100 mg kg-1, p.o.) or morphine (1-10 mg kg-1, i.p.) 60 min (p.o.) or 30 min (i.p.) before intraplantar injection of 10 μl of 5% formalin. The animals were immediately returned individually into the testing chamber, and their nociceptive behaviors captured (1 h) for analysis in the same way as that described previously in the writhing test above. The pain response was scored for 1 h, starting immediately after formalin injection. A nociceptive score was determined for each 5-min time block by measuring the amount of time spent biting/licking of the injected paw. Tracking of the behavior was done using public domain software JWatcher™, version 1.0. The average nociceptive score for each time block was calculated by multiplying the frequency and time spent in biting/licking. Data were expressed as the mean ± SEM of scores between 0-10 min (first phase) and 10–60 min (second phase) after formalin injection.
Tail-flick test
Tail-flick latencies were determined with the IITC Analgesia Meter (Model 336, IITC Life Science Inc., Woodland Hills, CA, USA), similar to that described earlier.[22] A focused beam of radiant light (Active intensity: 50% of maximum) was delivered to the mice's tail. Basal reaction times of mice (Males; 8 per group) were taken before the administration of XAE (30-300 mg kg-1, p.o), XA (10-100 mg kg-1, p.o), morphine (1-10 mg kg-1, i.p), or vehicle (10 ml kg-1 of 0.9% NaCl, i.p). Tail-flick reflex latencies were then measured at 1 h, 2 h, 3 h, and 4 h. A cut-off time of 25 s was used to prevent tissue damage. The analgesic effects exerted by drugs were calculated, from the tail-flick latencies, as a percentage of the maximum possible effect (% MPE) using the following formula: [(T2-T1)/(T0-T1) × 100], where T1 and T2 are the pre- and post-drug latencies, respectively, and T0 is the cut-off time.
Carrageenan-induced mechanical hyperalgesia
Mechanical nociceptive thresholds were measured in the rat paw pressure test[23] using an analgesimeter (Model No.15776, Ugo Basile, Comerio, Varese, Italy), which is based on the Randall-Selitto test.[24] This was used to apply a linearly-increasing pressure, by means of a blunt Perspex cone, to the dorsal region of the right hind paw until the rat withdrew the paw. Rats (Males) received 2 training seasons before the day of testing. Pressure was gradually applied to the right hind paw, and paw withdrawal thresholds (PWTs) were assessed as the pressure (grams) required eliciting paw withdrawal. A cut-off point of 250 g was used to prevent any tissue damage to the paw. A change in hyperalgesic state was calculated as a percentage of the maximum possible effect (% MPE). On the test day, a baseline measurement was taken before animals were administered carrageenan (100 μl of a 20 mg ml-1 solution) into the right hind paw. PWTs were determined again 2.5 h after carrageenan to establish that mechanical hyperalgesia had developed. XA (10-100 mg kg-1, p.o), XAE (30-300 mg kg-1, p.o.), morphine (1-10 mg kg-1, i.p.), diclofenac (1-10 mg kg-1, i.p.), and vehicle (10 ml kg-1 of 0.9% NaCl, i.p) were then administered 3-h post-carrageenan, and PWTs were taken again at 3.5, 4, 4.5, 5, 5.5, and 6 h post-carrageenan.
Hargreaves thermal hyperalgesia
Possible anti-hyperalgesic effect of XAE and XA was also assessed by the Hargreaves model of thermal hyperalgesia.[25,26] Hind paw sensitivity to a noxious thermal stimulus was measured with the radiant heat source method using the IITC Analgesia Meter (Model 336, IITC Life Science Inc., Woodland Hills, CA, USA). Mice (males) were individually placed in Plexiglas cages on a clear glass platform and were allowed to acclimatize for 15 min to the testing environment. The test head of the paw simulator was used to present a focused beam of radiant light on to the mid plantar region of the left and right hind paws. The idle intensity of the light was set at 10% of the maximum intensity, and it was used to accurately direct the beam of light to the appropriate region of the hind paw: The active intensity of the light was set at 50% maximum. The thermal nociceptive stimulus was manually positioned under the foot pad before and after the intraplantar injection of carrageenan (100 μl of a 2% solution) in to the right hind paw. A timer was automatically actuated with the light source, and the paw withdrawal latencies (PWLs) measured was defined as the time required for the paw to show an abrupt withdrawal. A cut-off time of 25 s was used to prevent tissue damage. Baseline measurements were taken preceding administration of carrageenan into the right hind paw. Animals received vehicle (10 ml kg-1 of 0.9% NaCl, i.p), XAE (30-300 mg kg-1, p.o), XA (10-100 mg kg-1, p.o), or morphine (1-10 mg kg-1, i.p) 1 hour post-carrageenan injection. PWLs were taken again hourly for 4 hours.
Tolerance studies
The formalin test was used to ascertain the possibility of tolerance developing to the anti-nociceptive activity of XAE and XA after chronic treatment. Male mice were divided randomly into 9 groups (n = 8) and were treated once-daily for 8 days as follows: 4 groups with saline i.p., 3 groups with morphine 6 mg kg-1, i.p. and 2 groups with either XAE 600 mg kg-1, p.o. or XA 200 mg kg-1, p.o. On day 9, these groups were treated in the following manner: 1 saline-pretreated group was treated with saline i.p.; 3 saline-pretreated groups were treated with morphine (3 mg kg-1, i.p., 30 min before formalin), XAE (300 mg kg-1, p.o.), or XA (100 mg kg-1, p.o., 60 min before formalin), the 3 groups pretreated with morphine 6 mg kg-1 chronically also received morphine (3 mg kg-1, i.p., 30 min before formalin), XAE (300 mg kg-1, p.o.), or XA (100 mg kg-1, p.o., 60 min before formalin) and the 2 groups pretreated with XAE (600 mg kg-1, p.o.) and XA 200 mg kg-1, p.o. received XAE (300 mg kg-1, p.o.) and XA (100 mg kg-1, p.o.), respectively on the test day.
Rotarod
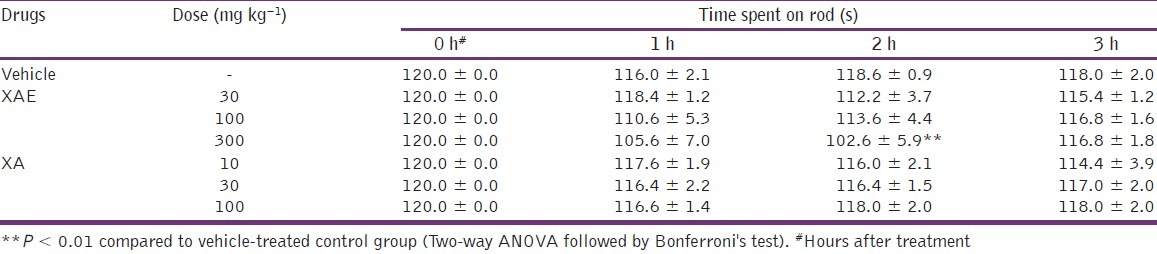
The test was carried out as described previously[27] but with modifications. Naive male mice were trained on 3 successive days on the rotarod (Ugo Basile, model 7600, Comerio, Varese, Italy) at the speed of 25 rpm. On the test day (day 4), the animals (8 per group) received XAE (30-300 mg kg-1, p.o.), XA (10-100 mg kg-1, p.o.) or vehicle. The animals were then repeatedly tested for their motor coordination performance on the rotarod (cut of time 120 s) at 1, 2, and 3 h after drug injection.
Statistical analysis
All data are presented as mean ± S.E.M (n=7-8). Raw data for the tail-flick, carrageenan-induced mechanical and thermal hyperalgesia tests were calculated as the percentage change in maximum possible effect (% MPE).
The time-course curves were subjected to 2-way (treatment × time) repeated measures analysis of variance (ANOVA) with Bonferroni's post hoc test. Total nociceptive score for each treatment was calculated in arbitrary unit as the area under the curve (AUC). To determine the percentage inhibition for each treatment, the following equation was used:

Differences in AUCs were analyzed using one-way ANOVA with drug treatment as a between-subjects factor. Further comparisons between vehicle- and drug-treated groups were performed using the Newman–Keuls test.
Doses for 50% of the maximal effect (ED50) for each drug were determined by using an iterative computer least squares method, with the following non-linear regression (3-parameter logistic) equation:

Where, X is the logarithm of dose and Y is the response. Y starts at a (the bottom) and goes to b (the top) with a sigmoid shape.
The fitted midpoints (ED50s) of the curves were compared statistically using F test.[28,29] GraphPad Prism for Windows version 5 (GraphPad Software, San Diego, CA, USA) was used for all statistical analyzes and ED50 determinations. P < 0.05 was considered statistically significant.
Results
High performance liquid chromatography
HPLC was used for the determination of the purity of the isolated xylopic acid. Several peaks were observed after loading XAE indicating the presence of several compounds in the fruits [Figure 2a]. A single peak was observed for XA indicating the presence of a single compound [Figure 2b]. The percentage of XA in XAE was 1.15%.
Figure 2.
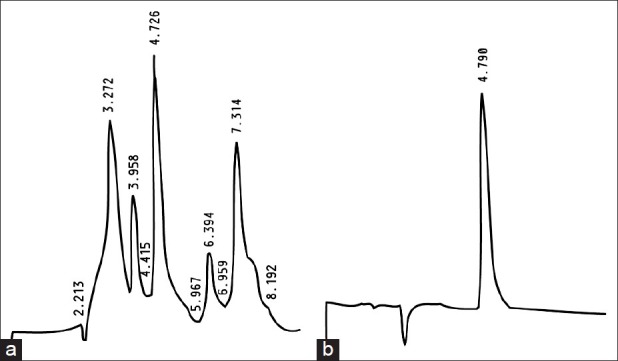
HPLC chromatograms of Xylopia aethiopica extract (a) and xylopic acid (b)
Acetic acid-induced writhing assay
Figure 3 shows the effect of various treatments on acetic acid-induced writhing during the 30-min observation period. XAE (30-300 mg kg-1, p.o.) significantly (F3,27 = 14.37, P < 0.0001; [Figure 3a] reduced abdominal writhes induced by acetic acid in mice with the highest dose causing a reduction of 98.8 ± 0.8% [Figure 3b]. XA (10 -100 mg kg-1, p.o.) also significantly (F3,28 = 20.56, P < 0.0001; Figure 3c) and dose-dependently reduced the writhes, with the highest dose causing a reduction of 93.8 ± 1.4% Figure 3d. Morphine dose-dependently reduced (F3,27 = 9.77, P = 0.00016; Figure 3e) acetic acid-induced nociception with maximal percentage inhibition of 97.4 ± 2.5 [Figure 3f] at the dose of 10 mg kg-1. Additionally, diclofenac reduced the abdominal writhes dose-dependently and significantly (F3,28 = 4.04, P = 0.0165; Figure 3g]; with the highest dose (10 mg kg-1) producing an inhibition of 90.4 ± 2.6% [Figure 3h].
Figure 3.
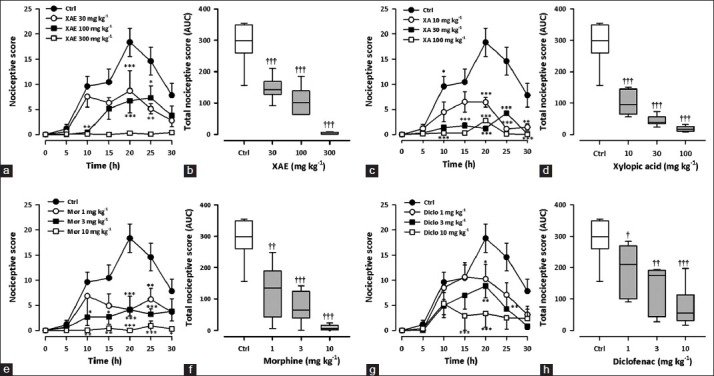
Effect of XAE (30-300 mg kg-1), XA (10-100 mg kg-1), morphine (1-10 mg kg-1) and diclofenac (1-10 mg kg-1) on the time course curves (a, c, e, g) and the total nociceptive score (calc. as AUCs) (b, d, f, h) of acetic acid-induced writhing in mice. Nociceptive scores are shown in 5 min time blocks up to 30 min for the time course curves. Data are presented as mean ± SEM (n = 7-8). The lower and upper margins of the boxes (b, d, f, h) represent the 25th and 75th percentiles, with the extended arms representing the 10th and 90th percentiles, respectively. The median is shown as the horizontal line within the box.*P < 0.05, **P < 0.01, ***P < 0.001 compared to control group (ctrl) (Two-way repeated measures ANOVA followed by Bonferroni's post hoc). †P < 0.05, ††P < 0.01, †††P < 0.001 compared to control group (ctrl) (one-way ANOVA followed by Newman-Keuls post hoc)
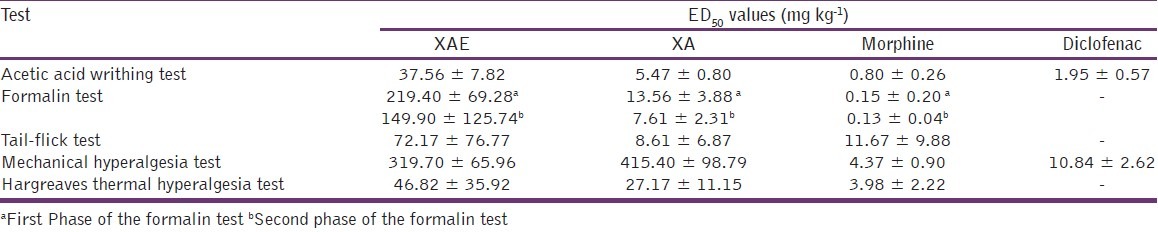
From the ED50 values obtained by non-linear regression [Table 1], XA (5.472 ± 0.80 mg kg-1) was more potent than XAE (37.56 ± 7.82 mg kg-1). However, morphine was the most potent (0.80 ± 0.26 mg kg-1).
Table 1.
ED50 values of drugs in the nociceptive tests
Formalin-induced nociception
Administration of formalin produced a typical pattern of biting and licking behavior in mice. The first phase lasted for 10 min, and the second phase started at about 15 min and lasted until 1 h. Oral treatment of mice with X. aethiopica extract (30-300 mg kg-1, p.o.) produced significant attenuation of formalin-induced nociception in both phases (Phase 1: F3,28= 4.29, P = 0.0131; Phase 2: F3,28 = 5.18, P = 0.0056; Figure 4a); with maximal inhibition of 57.3 ± 25.0% and 52.9 ± 34.1%, respectively at the dose of 300 mg kg-1 [Figure 4b]. Xylopic acid (10-100 mg kg-1) also dose-dependently inhibited both phases of formalin-induced nociception phases (Phase 1: F3,28 = 7.85, P = 0.0006; Phase 2 F3,28 = 7.68, P = 0.0007; Figure 4c], with the highest dose giving a maximal inhibition of 79.6 ± 17.5% and 90.2 ± 4.9% [Figure 4d] in the early and late phases, respectively. Morphine (1-10 mg kg-1, i.p.) produced a significant inhibition of both neurogenic phase (F3,28 = 22.51, P = P < 0.0001) and inflammatory (F3,28 = 4.76, P = 0.0083; Figure 4e) phases of the formalin test. Morphine reduced formalin-evoked nociceptive behavior by maxima of 85.6 ± 12.7% and 98.3 ± 3% in the early and late phases, respectively [Figure 4f].
Figure 4.
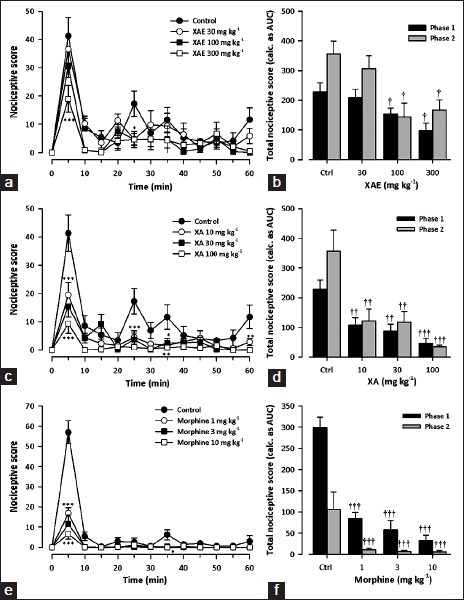
Effect of XAE (30-300 mg kg- 1), XA (10-100 mg kg-1) and morphine (1-10 mg kg-1) on the time course curves (a, c, e) and the total nociceptive score (calc. as AUCs) (b, d, f) of formalin-induced nociception in mice. Nociceptive scores are shown in 5 min time blocks up to 60 min for the time course curves. Data are presented as mean ± SEM (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001 compared to control group (ctrl) (Two-way repeated measures ANOVA followed by Bonferroni's post hoc). †P < 0.05, ††P < 0.01, †††P < 0.001 compared to control group (ctrl) (one-way ANOVA followed by Newman-Keuls post hoc)
Comparison of ED50s obtained [Table 1] by F-test revealed that both XAE and XA were more potent in the second phase than the first. The potency of morphine in the second phase (ED50 = 0.13 ± 0.04 mg kg-1) was comparable to the first phase (ED50 = 0.15 ± 0.20 mg kg-1). The rank order of potency was: Morphine > XA > XAE for both phases of the formalin test.
Tail-flick test
All test drugs caused an increase in tail-flick latency, calculated as % MPE [(XAE: F3,28 = 3.33, P = 0.0336; XA: F3,28 = 5.89, P = 0.003; morphine: F3,28 = 14.70; P < 0.0001; 2-way ANOVA (treatment × time); Figure 5a, c and e]. XAE (30-300 mg kg-1, p.o.) caused a significant, dose-dependent, attenuation of thermal nociception (F3,28 = 3.19, P = 0.0387) with maximum effect at the dose of 300 mg kg-1 Figure 5b]. Similarly, XA (10-100 mg kg-1, p.o.) also dose-dependently inhibited (F3,28 = 5.16, P = 0.0058) thermal pain in the mice [Figure 5d]. The reference analgesic, morphine (1-10 mg kg-1, i.p.), showed significant, dose-dependent anti-nociception (F3,28 = 13.23, P < 0.0001) with a maximum effect at 10 mg kg-1 [Figure 5f].
Figure 5.
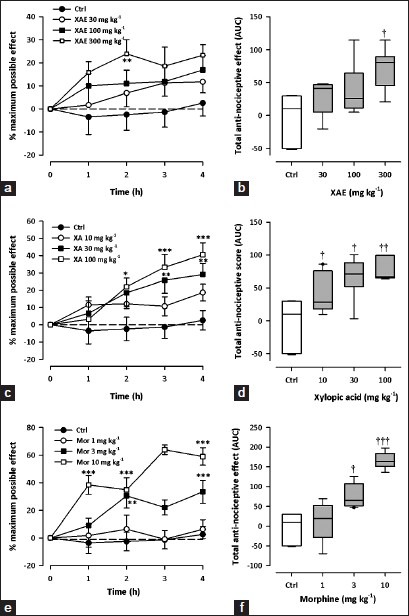
Effect of XAE (30-300 mg kg-1), XA (10-100 mg kg-1) and morphine (1-10 mg kg-1) on the time course curves (a, c, e) and the total anti-nociceptive effect (calc. as AUCs) (b, c, f) in the tail-flick test in mice. Data was presented as mean ± S.E.M. (n = 8). The lower and upper margins of the boxes (B, D, F) represent the 25th and 75th percentiles, with the extended arms representing the 10th and 90th percentiles, respectively. The median is shown as the horizontal line within the box. .*P < 0.05, **P < 0.01, ***P < 0.001 compared to control group (ctrl) (Two-way repeated measures ANOVA followed by Bonferroni's post hoc). †P < 0.05, ††P < 0.01, †††P < 0.001 compared to control group (ctrl) (one-way ANOVA followed by Newman-Keuls post hoc)
By comparing the ED50 values [Table 1] of effects in the tail-flick test, XAE (72.17 ± 76.77 mg kg-1) was less potent than XA (8.61 ± 6.87 mg kg-1) and morphine (11.67 ± 9.88 mg kg-1). Morphine was, however, the most effective [Figure 5].
Carrageenan-Induced mechanical hyperalgesia
Two hours after carrageenan injection, the ipsilateral paw exhibited marked mechanical hyperalgesia and inflammation. A change in hyperalgesic state was calculated as a percentage of the maximum possible effect. XAE (30-300 mg kg-1, p.o.) and XA (10-100 mg kg-1, p.o.) administered 3 h after carrageenan produced a significant and dose-dependent attenuation of mechanical hyperalgesia (XAE: F3,28 = 6.93, P = 0.0012; XA: F3,28 = 4.86, P = 0.0076; Figure 6a–d) with maximal effects at 300 and 100 mg kg-1, respectively. Morphine (1-10 mg kg-1, i.p.) and diclofenac (1-10 mg kg-1, i.p.) also dose-dependently attenuated mechanical hyperalgesia with maximal effects at 10 mg kg-1 (Morphine: F3,28 =1 0.67, P < 0.0001; diclofenac: F3,28 = 5.86, P < 0.0031; Figure 6e–h).
Figure 6.
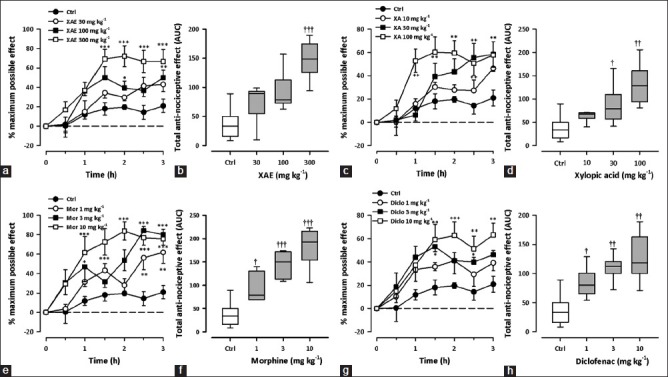
Effect of XAE (30-300 mg kg-1), XA (10-100 mg kg-1), morphine (1-10 mg kg-1) and diclofenac (1-10 mg kg-1) on the time course curves (a, c, e, g) and the total nociceptive score (AUCs) (b, d, f, h) of carrageenan-induced mechanical hyperalgesia in rats. Data was presented as mean ± S.E.M. (n = 8). The lower and upper margins of the boxes (b, d, f, h) represent the 25th and 75th percentiles, with the extended arms representing the 10th and 90th percentiles, respectively. The median is shown as the horizontal line within the box.*P < 0.05, **P < 0.01, ***P < 0.001 compared to control group (ctrl) (Two-way repeated measures ANOVA followed by Bonferroni's post hoc). †P < 0.05, ††P < 0.01, †††P < 0.001 compared to control group (ctrl) (one-way ANOVA followed by Newman-Keuls post hoc)
From the ED50 values [Table 1], XAE was more potent than XA but less than diclofenac and morphine.
Hargreaves thermal hyperalgesia
All test drugs caused an increase in paw withdrawal latency, calculated as % MPE [(XAE: F3,28 = 5.44, P = 0.0045; XA: F3,28 = 13.35, P < 0.0001; morphine: F3,28 = 10.24; P = 0.0001; 2-way ANOVA (treatment × time); Figure 7a, c and e]. XAE (30-300 mg kg-1, p.o.) significantly and dose-dependently attenuated thermal hyperalgesia (F3,28 = 4,73, P = 0.0086) with maximal effect at the dose of 300 mg kg-1 [Figure 7b]. Similarly, XA (10-100 mg kg-1, p.o.) also dose-dependently inhibited (F3,28 = 12.56, P < 0.0001; Figure 7b) carrageenan-induced thermal hyperalgesia in the mice. The reference analgesic, morphine (1–10 mg kg-1, i.p.), also showed significant, dose-dependent anti-hyperalgesic activity (F3,28 = 9.30, P = 0.0002; Figure 7f) against the carrageenan-induced thermal hyperalgesia.
Figure 7.
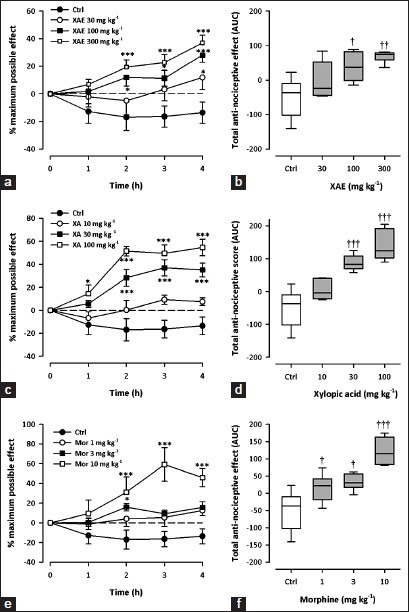
Effect of XAE (30-300 mg kg-1), XA (10-100 mg kg-1) and morphine (1-10 mg kg-1) on the time course curves (a, c, e) and the total anti-nociceptive effect (AUCs) (b, d, f) in carrageenan-induced thermal hyperalgesia test in mice. Data was presented as mean ± S.E.M. (n = 8). The lower and upper margins of the boxes (b, d, f) represent the 25th and 75th percentiles, with the extended arms representing the 10th and 90th percentiles, respectively. The median is shown as the horizontal line within the box.*P < 0.05, ***P < 0.001 compared to control group (ctrl) (Two-way repeated measures ANOVA followed by Bonferroni's post hoc). †P < 0.05, ††P < 0.01, †††P < 0.001 compared to control group (ctrl) (one-way ANOVA followed by Newman-Keuls post hoc)
From the ED50 values [Table 1], XAE (46.82±35.92 mg kg-1) was less potent than XA (27.17 ± 11.15 mg kg-1) and morphine (3.98 ± 2.22 mg kg-1).
Tolerance studies
Morphine (3 mg kg-1, i.p.) significantly attenuated the basal nociceptive response in both phases [Figure 8] of formalin test in animals chronically-treated with vehicle for 8 days. However, the same dose of morphine administered on day 9 in animals chronically-treated with 6 mg kg -1, i.p. morphine failed to show such effect, indicating the development of tolerance in first phase [Figure 8a] and second phase [Figure 8a] of the formalin test. XAE (300 mg kg-1, p.o.) and XA (100 mg kg-1, p.o.) exhibited anti-nociceptive activity in mice chronically-treated with XAE (600 mg kg-1, p.o.) and XA (200 mg kg-1, p.o.) respectively or vehicle for 8 days, indicating failure of tolerance development to the anti-nociceptive effects of XAE and XA in both phases [Figure 8] of the formalin test. XAE (300 mg kg-1, p.o.) and XA (100 mg kg-1, p.o.) still demonstrated anti-nociceptive activity in mice chronically-treated with morphine (6 mg kg-1 i.p.) for 8 days, indicating the absence of cross-tolerance development with morphine in the formalin test [Figure 8].
Figure 8.
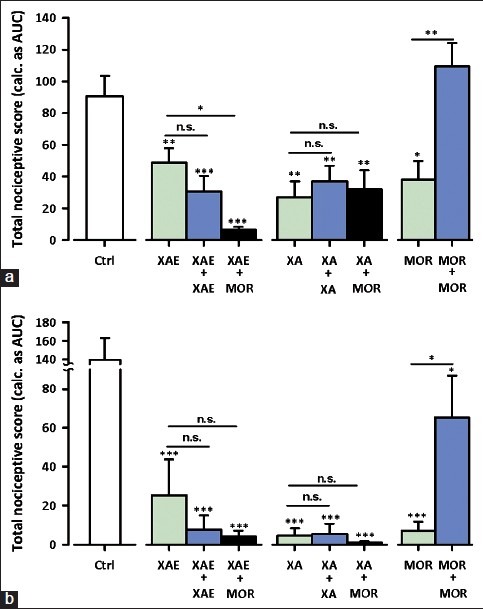
Effect of XAE (300 mg kg-1), XA (100 mg kg-1) and morphine (MOR; 3 mg kg-1) challenge on mice chronically treated with vehicle, XAE (600 mg kg-1), XA (200 mg kg-1) or morphine (6 mg kg- 1) for 8 days on the total nociceptive score in Phase 1 (a) and Phase 2 (b) of formalin-induced pain in mice. Each column represent the mean ± S.E.M. (n = 8); *P 0.05, ***P < 0.01, ***P < 0.001 compared to vehicle-treated control group (ctrl) (one-way ANOVA followed by Newman-Keuls post hoc test)
Effect of XAE and XA on motor coordination on the rotarod
Effect on motor coordination was measured as duration of stay on the rotating drum of the rotarod. The results obtained indicated that there were no significant differences between XA-treated (F3,28 = 0.39, P = 0.7635; Table 2) groups compared to the control group. ANOVA did not reveal any significant effect of XAE on the time spent on the rotarod (F3,28 = 2.44, P = 0.0855; Table 2). However, Bonferroni's post hoc analysis revealed significant effects (P < 0.01, Table 2) for XAE at the dose of 300 mg kg-1 when compared to vehicle-treated controls.
Table 2.
Effect of XAE and XA on motor coordination measured as time spent on the rod
Discussion
The present study clearly demonstrates that the oral administration of XAE and xylopic acid has significant analgesic activity against acetic acid-induced visceral nociception, formalin-induced paw pain (both neurogenic and inflammatory), thermal pain as well as carrageenan-induced mechanical and thermal hyperalgesia in rodents. High performance liquid chromatographic analysis of XAE revealed several peaks. These may correspond to any of the various compounds such as essential oil, volatile oil, resin, arocene, a rutheroside fat, bitter principles, alkaloids, glycosides, saponnis, tanins, stereols, carbohydrate, protein-free fatty acid, mucilages, kaurenoic and xylopic acid,[5,6,14] which have been reported to be present in the fruits. XA, however, showed a single peak, indicating the purity of the isolated xylopic acid. In this study, doses of 10-100 mg kg-1 (xylopic acid) and 30-300 mg kg-1 (extract) were used and were selected on the basis of pilot experiments in our laboratory. Preliminary acute toxicity studies in our laboratory showed that xylopic acid and the extract produced significant adverse effects beyond doses of 100 mg kg-1 and 300 mg kg-1, respectively.
In the present study, the analgesic effect of XAE and XA was first evaluated in the acetic acid-induced writhing test. The results obtained indicate that oral administration of XAE and XA produced marked, dose-related anti-nociception in this test model. The writhing test is useful in investigating visceral anti-nociceptive activity of drugs because it shows good sensitivity and allows for the detection of the effects of weak analgesics.[30] Aside the role of acetic acid-derived protons that can directly activate non-selective cation channels located at primary afferent pathways to cause pain,[31] the actions of acetic acid are known to be the indirect cause of the release of nociceptive endogenous mediators such as bradykinin, substance P, serotonin, histamine, sympathomimetic amines, prostaglandins (PGE2 and PGF2α) and pro-inflammatory cytokines such as TNF-α, IL-1β and IL-8.[32–35] Peritoneal inflammation resulting from the local release of these pro-inflammatory substances result in the activation and sensitization of peripheral nociceptive afferent neurons.[36] The anti-nociceptive activity exhibited by XAE and XA in this model may be due to the inhibition of local synthesis, release and/or action of pro-inflammatory mediators.[37–39] In fact, a significant number of kaurenes diterpenes, including kaurenoic acid, are known to inhibit iNOS, COX-2 and TNF- α.[12,15,40,41]
To further confirm the analgesic properties of XAE and XA, the formalin test was employed. The test is considered the most predictive of acute pain and is believed to be a more valid model for clinical pain.[20,42] Additionally, formalin test can be used to elucidate the possible mechanism(s) of analgesia produced by XAE and XA.[18,43] The formalin test produces a distinct biphasic nociceptive response. The first transient (neurogenic) phase is caused by the direct effect of formalin on nociceptors, and the second prolonged (inflammatory) phase is associated with an inflammatory reaction in the peripheral tissue, causing a release of nociceptive mediators (e.g. serotonin, histamine, bradykinin and prostaglandins), which subsequently cause sensitization of the central neurons leading to changes in central pain processing.[44,45] It is known that centrally-acting drugs, such as opioids, inhibit both phases equally,[46] however, many NSAIDs and corticosteroids inhibit only the late phase. Both XAE and XA inhibited the neurogenic and inflammatory phases of the mouse formalin test. Morphine, an opioid analgesic used as a control, also blocked both phases. Therefore, XAE and XA, like morphine, acted both peripherally and centrally to produce anti-nociception in the formalin test. The inhibitory effect in the second phase also suggests anti-inflammatory action of XAE and XA. Indeed, XAE has been shown to have anti-inflammatory properties in our laboratory (unpublished results).
The results obtained from the tail-flick test confirmed the involvement of central mechanisms in the anti-nociceptive effects of XAE and XA. The tail-flick response is believed to be a spinally-mediated reflex,[47] but the mechanism of response could also involve higher neural structures.[48] The activity of XAE and XA in this model shows they act, at least in part, by spinally-mediated central mechanisms.
Our results also show that oral administration of XAE and XA inhibited carrageenan-induced inflammatory pain triggered by both mechanical and thermal stimuli in rats. The effect of XAE and XA on mechanical hyperalgesia was assessed with the Randall-Selitto paw pressure test, an inflammatory pain model, widely used for quantification of thresholds of the rat hind paw withdrawal reflex to nociceptive pressure stimulation. The Hargreaves model[25,26] was also used to evaluate the effect of XAE and XA on thermal hyperalgesia. Carrageenan-induced inflammatory pain is well-known to involve inflammatory mediators like cyclooxygenase products (PGE2), leukotrienes, mast cells products (histamine, 5-HT), neuropeptides, cytokines (IL-1β and TNFα), nitric oxide, nerve growth factor (NGF), and transcription factors (nuclear factor-kappa B; NF-κB),[49–52] which are released as a result of tissue injury. Most of these mediators (especially prostaglandins) sensitize peripheral nociceptors to noxious stimuli and subsequently release other mediators in spinal cord, resulting in thermal and mechanical hyperalgesia.[53–55] Therefore, the effectiveness of XAE and XA against inflammatory pain in this model suggests that XAE and XA may be acting against some of these mediators. The results obtained also corroborate the observed activity of the XAE and XA in the second phase (inflammatory pain) of the formalin test.
Activation of the transcription factor NF-κB has been shown to be a key component in the expression of genes involved in the production of inflammatory mediators, which may exacerbate pain, hyperalgesia, and nociception.[56–58] Therefore, the inhibition of activation or action of NF-κB can effectively control inflammation and associated inflammatory pain. Castrillo et al.[12 have shown that kaurene diterpenes inhibit NF-κB activation through a mechanism that involves an impairment of IκB kinase (IKK) activity as a result of the inhibition of NF-κB-inducing kinase (NIK) and the lack of a coordinate activation of p38 and/or extracellular signal-regulated kinase 1/2 (ERK1/2). This mechanisms possibly explains, at least in part, the observed analgesic actions by xylopic acid, a kaurene diterpene, and XAE (which contains at least two kaurenes: kaurenoic acid and xylopic acid) against inflammatory pain.
The development of tolerance that necessitates dose escalation regardless of disease progression, greatly limit the effectiveness and usage of some important analgesics such as the opioids.[59,60] The present study, therefore, determined if repeated administration of XAE and XE could lead to the development of analgesic tolerance and also if morphine tolerance could cross-generalize to the analgesic effects of XAE and XA. The results suggest that, unlike morphine, XAE and XA do not induce tolerance to their analgesic effects after chronic administration in the formalin test. Also, morphine tolerance does not cross-generalize to the effects of XAE and XA. These are significant findings and imply that XAE and XA can be used to treat pain in opioid-tolerant individuals.
In order to eliminate the possibility that decreased motor function is confounding the results obtained in the nociceptive test models, motor performance of the mice was evaluated using the rotarod. XAE at the highest dose tested (i.e. 300 mg kg-1) exerted some effect on motor coordination, which could be due to muscle relaxation and/or sedation. It cannot, however, be concluded that the analgesic activity of XAE in this study is a false positive since XA, a constituent of XAE, did not cause any such motor impairments. The XAE-induced motor impairment effect observed could be attributed to other constituents of the extract such as kaurenoic acid. In fact, kaurenoic acid has been shown to have sedative properties.[61]
Many phytochemical constituents including essential oils, volatile oil, resin, alkaloids, glycosides, saponnis, tanins, and diterpenes have been identified in the dried fruit extract of X. aethiopica. From the results obtained in this and other studies,[13] it can be said that the analgesic effects of XAE are partly due to the kaurenoic acid and xylopia acid it contains. Other constituents may also contribute to its analgesic effects. The ED50 values obtained [Table 1] also revealed that XAE and XA were effective against neurogenic, nociceptive, and inflammatory pain although it appears they were more potent in reducing inflammatory-related pain. Studies are currently ongoing in our laboratory to assess the effect of XAE and XA on neuropathic pain.
The current study also revealed that XA was generally more potent than XAE in relieving pain [Table 1]. This is, however, not surprising since XAE, as a crude extract, comprises several chemical constituents which could be acting via contradicting mechanisms. Studies are currently underway to determine the exact mechanism(s) of analgesic action of XAE and XA.
Conclusion
In conclusion, the results have shown that the ethanol extract of Xylopia aethiopica and its constituent, xylopic acid, have analgesic properties.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared
References
- 1.Schim JD, Stang P. Overview of pain management. Pain Pract. 2004;4(Suppl 1):S4–18. doi: 10.1111/j.1533-2500.2004.04010.x. [DOI] [PubMed] [Google Scholar]
- 2.McMahon SB, Koltzenburg M. Wall and Melzack's textbook of pain. 5th ed. Edinburgh: Elsevier/Churchill Livingstone; 2006. pp. 300–350. [Google Scholar]
- 3.Chen CH, Tang ST, Chen CH. Meta-analysis of cultural differences in Western and Asian patient-perceived barriers to managing cancer pain. Palliat Med. 2012;26:206–21. doi: 10.1177/0269216311402711. [DOI] [PubMed] [Google Scholar]
- 4.Bandolier Bandolier: Acute pain. Bandolier Extra February. 2003:1–22. [Google Scholar]
- 5.Igwe SA, Afonne JC, Ghasi SI. Ocular dynamics of systemic aqueous extracts of Xylopia aethiopica (African guinea pepper) seeds on visually active volunteers. J Ethnopharmacol. 2003;86:139–42. doi: 10.1016/s0378-8741(02)00371-9. [DOI] [PubMed] [Google Scholar]
- 6.Burkill HM. The useful plants of west tropical africa. A revision of dalziel. 2nd ed. II. Kent: Families A–D Royal Botanical Garden; pp. 130–132. [Google Scholar]
- 7.Boakye-Yiadom K, Fiagbe NI, Ayim JS. Antimicrobial properties of some West African medicinal plants iv.Antimicrobial activity of xylopic acid and other constituents of the fruits of Xylopia aethiopica (Annonaceae) Lloydia. 1977;40:543–5. [PubMed] [Google Scholar]
- 8.Ekong DE, Ogan AU. Chemistry of the constituents of Xylopia aethiopica. The structure of xylopic acid, a new diterpene acid. J Chem Soc C: Organic. 1968:311–2. [Google Scholar]
- 9.Bresciani LF, Yunes RA, Burger C, De Oliveira LE, Bof KL, Cechinel-Filho V. Seasonal variation of kaurenoic acid, a hypoglycemic diterpene present in Wedelia paludosa (Acmela brasiliensis) (Asteraceae) Z Naturforsch C. 2004;59:229–32. doi: 10.1515/znc-2004-3-419. [DOI] [PubMed] [Google Scholar]
- 10.Ghisalberti EL. The biological activity of naturally occurring kaurane diterpenes. Fitoterapia. 1997;68:303. [Google Scholar]
- 11.Garcia PA, de Oliveira AB, Batista R. Occurrence, biological activities and synthesis of kaurane diterpenes and their glycosides. Molecules. 2007;12:455–83. doi: 10.3390/12030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castrillo A, de Las Heras B, Hortelano S, Rodriguez B, Villar A, Bosca L. Inhibition of the nuclear factor kappa B (NF-kappa B) pathway by tetracyclic kaurene diterpenes in macrophages. Specific effects on NF-kappa B-inducing kinase activity and on the coordinate activation of ERK and p38 MAPK. J Biol Chem. 2001;276:15854–60. doi: 10.1074/jbc.M100010200. [DOI] [PubMed] [Google Scholar]
- 13.Block LC, Santos AR, de Souza MM, Scheidt C, Yunes RA, Santos MA, et al. Chemical and pharmacological examination of antinociceptive constituents of Wedelia paludosa. J Ethnopharmacol. 1998;61:85–9. doi: 10.1016/s0378-8741(98)00019-1. [DOI] [PubMed] [Google Scholar]
- 14.Somova LI, Shode FO, Moodley K, Govender Y. Cardiovascular and diuretic activity of kaurene derivatives of Xylopia aethiopica and Alepidea amatymbica. J Ethnopharmacol. 2001;77:165–74. doi: 10.1016/s0378-8741(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 15.Sosa-Sequera MC, Suarez O, Dalo NL. Kaurenic acid: An in vivo experimental study of its anti-inflammatory and antipyretic effects. Indian J Pharmacol. 2010;42:293–6. doi: 10.4103/0253-7613.70205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davino SC, Giesbrecht AM, Roque NF. Antimicrobial activity of kaurenoic acid derivatives substituted on carbon-15. Braz J Med Biol Res. 1989;22:1127–9. [PubMed] [Google Scholar]
- 17.Abaidoo CS, Woode E, Alhassan A. An evaluation of the effect of ethanolic fruit extracts of Xylopia aethiopica on haematological and biochemical parameters in male rats. Der Pharmacia Sinica. 2011;2:39–45. [Google Scholar]
- 18.Tang L, Chen Y, Chen Z, Blumberg PM, Kozikowski AP, Wang ZJ. Antinociceptive pharmacology of N-(4-chlorobenzyl)-N’-(4-hydroxy-3-iodo-5-methoxybenzyl) thiourea, a high-affinity competitive antagonist of the transient receptor potential vanilloid 1 receptor. J Pharmacol Exp Ther. 2007;321:791–8. doi: 10.1124/jpet.106.117572. [DOI] [PubMed] [Google Scholar]
- 19.Koster R, Anderson M, Beer EJ. Acetic acid for analgesic screening. Fed Proc. 1959;18:412–8. [Google Scholar]
- 20.Dubuisson D, Dennis SG. The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 1977;4:161–74. doi: 10.1016/0304-3959(77)90130-0. [DOI] [PubMed] [Google Scholar]
- 21.Woode E, Poku RA, Ainooson GK, Boakye-Gyasi E, Abotsi WK, Mensah TL, et al. An evaluation of the anti-inflammatory, antipyretic and antinociceptive effects of ficus exasperata (Vahl) leaf extract. J Pharmaco Toxicol. 2009;4:138–51. [Google Scholar]
- 22.Fecho K, Nackley AG, Wu Y, Maixner W. Basal and carrageenan-induced pain behavior in Sprague-Dawley, Lewis and Fischer rats. Physiol Behav. 2005;85:177–86. doi: 10.1016/j.physbeh.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Stohr T, Krause E, Selve N. Lacosamide displays potent antinociceptive effects in animal models for inflammatory pain. Eur J Pain. 2006;10:241–9. doi: 10.1016/j.ejpain.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–19. [PubMed] [Google Scholar]
- 25.Galbraith JA, Mrosko BJ, Myers RR. A system to measure thermal nociception. J Neurosci Methods. 1993;49:63–8. doi: 10.1016/0165-0270(93)90109-5. [DOI] [PubMed] [Google Scholar]
- 26.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 27.Gareri P, Condorelli D, Belluardo N, Citraro R, Barresi V, Trovato-Salinaro A, et al. Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice) Neuropharmacology. 2005;49:551–63. doi: 10.1016/j.neuropharm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Miller JR. Step-by-Step Examples. San Diego, CA: GraphPad Software Inc; 2003. GraphPad Version 4.0; pp. 55–69. [Google Scholar]
- 29.Motulsky HJ, Christopoulos A. A practical guide to curve fitting. San Diego, CA: GraphPad Software Inc; 2003. Fitting model to biological data using linear and nonlinear regression; pp. 13–46. [Google Scholar]
- 30.Pietrovski EF, Rosa KA, Facundo VA, Rios K, Marques MC, Santos AR. Antinociceptive properties of the ethanolic extract and of the triterpene 3beta,6beta,16beta-trihidroxilup-20(29)-ene obtained from the flowers of Combretum leprosum in mice. Pharmacol Biochem Behav. 2006;83:90–9. doi: 10.1016/j.pbb.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–10. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 32.dos Santos DA, Fukui Mde J, Dhammika Nanayakkara NP, Khan SI, Sousa JP, Bastos JK, et al. Anti-inflammatory and antinociceptive effects of Baccharis dracunculifolia DC (Asteraceae) in different experimental models. J Ethnopharmacol. 127:543–50. doi: 10.1016/j.jep.2009.09.061. [DOI] [PubMed] [Google Scholar]
- 33.Ribeiro RA, Vale ML, Thomazzi SM, Paschoalato AB, Poole S, Ferreira SH, et al. Involvement of resident macrophages and mast cells in the writhing nociceptive response induced by zymosan and acetic acid in mice. Eur J Pharmacol. 2000;387:111–8. doi: 10.1016/s0014-2999(99)00790-6. [DOI] [PubMed] [Google Scholar]
- 34.Deraedt R, Jouquey S, Delevallee F, Flahaut M. Release of prostaglandins E and F in an algogenic reaction and its inhibition. Eur J Pharmacol. 1980;61:17–24. doi: 10.1016/0014-2999(80)90377-5. [DOI] [PubMed] [Google Scholar]
- 35.Duarte ID, Nakamura M, Ferreira SH. Participation of the sympathetic system in acetic acid-induced writhing in mice. Braz J Med Biol Res. 1988;21:341–3. [PubMed] [Google Scholar]
- 36.Raja SN, Meyer RA, Campbell JN. Peripheral mechanisms of somatic pain. Anesthesiology. 1988;68:571–90. doi: 10.1097/00000542-198804000-00016. [DOI] [PubMed] [Google Scholar]
- 37.Panthong A, Norkaew P, Kanjanapothi D, Taesotikul T, Anantachoke N, Reutrakul V. Anti-inflammatory, analgesic and antipyretic activities of the extract of gamboge from Garcinia hanburyi Hook f. J Ethnopharmacol. 2007;111:335–40. doi: 10.1016/j.jep.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 38.Ferreira SH, Nakamura M. II - Prostaglandin hyperalgesia: The peripheral analgesic activity of morphine, enkephalins and opioid antagonists. Prostaglandins. 1979;18:191–200. doi: 10.1016/0090-6980(79)90104-7. [DOI] [PubMed] [Google Scholar]
- 39.Levine JD, Taiwo YO. Involvement of the mu-opiate receptor in peripheral analgesia. Neuroscience. 1989;32:571–5. doi: 10.1016/0306-4522(89)90279-0. [DOI] [PubMed] [Google Scholar]
- 40.Park H-J, Kim I-T, Won J-H, Jeong S-H, Park E-Y, Nam J-H, et al. Anti-inflammatory activities of ent-16[alpha]H,17-hydroxy-kauran-19-oic acid isolated from the roots of Siegesbeckia pubescens are due to the inhibition of iNOS and COX-2 expression in RAW 264.7 macrophages via NF-[kappa]B inactivation. Eur J Pharmacol. 2007;558:185–93. doi: 10.1016/j.ejphar.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 41.Leung CH, Grill SP, Lam W, Han QB, Sun HD, Cheng YC. Novel mechanism of inhibition of nuclear factor-kappa B DNA-binding activity by diterpenoids isolated from Isodon rubescens. Mol Pharmacol. 2005;68:286–97. doi: 10.1124/mol.105.012765. [DOI] [PubMed] [Google Scholar]
- 42.Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: An evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- 43.Hunskaar S, Hole K. The formalin test in mice: Dissociation between inflammatory and non-inflammatory pain. Pain. 1987;30:103–14. doi: 10.1016/0304-3959(87)90088-1. [DOI] [PubMed] [Google Scholar]
- 44.Santa-Cecilia FV, Vilela FC, da Rocha CQ, Dias DF, Cavalcante GP, Freitas LA, et al. Anti-inflammatory and antinociceptive effects of Garcinia brasiliensis. J Ethnopharmacol. 2011;133:467–73. doi: 10.1016/j.jep.2010.09.036. [DOI] [PubMed] [Google Scholar]
- 45.da Rocha CQ, Vilela FC, Cavalcante GP, Santa-Cecilia FV, Santos-e-Silva L, dos Santos MH, et al. Anti-inflammatory and antinociceptive effects of Arrabidaea brachypoda (DC.) Bureau roots. J Ethnopharmacol. 2011;133:396–401. doi: 10.1016/j.jep.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 46.Shibata M, Ohkubo T, Takahashi H, Inoki R. Modified formalin test: Characteristic biphasic pain response. Pain. 1989;38:347–52. doi: 10.1016/0304-3959(89)90222-4. [DOI] [PubMed] [Google Scholar]
- 47.Chapman CR, Casey KL, Dubner R, Foley KM, Gracely RH, Reading AE. Pain measurement: An overview. Pain. 1985;22:1–31. doi: 10.1016/0304-3959(85)90145-9. [DOI] [PubMed] [Google Scholar]
- 48.Jensen TS, Yaksh TL. Comparison of antinociceptive action of morphine in the periaqueductal gray, medial and paramedial medulla in rat. Brain Res. 1986;363:99–113. doi: 10.1016/0006-8993(86)90662-1. [DOI] [PubMed] [Google Scholar]
- 49.Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: Peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–7. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 50.Dray A. Inflammatory mediators of pain. Br J Anaesth. 1995;75:125–31. doi: 10.1093/bja/75.2.125. [DOI] [PubMed] [Google Scholar]
- 51.Morris CJ. Carrageenan-induced paw edema in the rat and mouse. Methods Mol Biol. 2003;225:115–21. doi: 10.1385/1-59259-374-7:115. [DOI] [PubMed] [Google Scholar]
- 52.Peskar BM, Trautmann M, Nowak P, Peskar BA. Release of 15-hydroxy-5,8,11,13-eicosatetraenoic acid and cysteinyl-leukotrienes in carrageenin-induced inflammation: Effect of non-steroidal anti-inflammatory drugs. Agents Actions. 1991;33:240–6. doi: 10.1007/BF01986569. [DOI] [PubMed] [Google Scholar]
- 53.Rueff A, Dray A. Sensitization of peripheral afferent fibres in the in vitro neonatal rat spinal cord-tail by bradykinin and prostaglandins. Neuroscience. 1993;54:527–35. doi: 10.1016/0306-4522(93)90272-h. [DOI] [PubMed] [Google Scholar]
- 54.Singh VP, Patil CS, Kulkarni SK. Differential effect of zileuton, a 5-lipoxygenase inhibitor, against nociceptive paradigms in mice and rats. Pharmacol Biochem Behav. 2005;81:433–9. doi: 10.1016/j.pbb.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 55.Collier HO, Schneider C. Nociceptive response to prostaglandins and analgesic actions of aspirin and morphine. Nat New Biol. 1972;236:141–3. doi: 10.1038/newbio236141a0. [DOI] [PubMed] [Google Scholar]
- 56.Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, et al. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur J Neurosci. 2004;19:3375–81. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- 57.Haddad JJ. Cellular and molecular regulation of inflammatory pain, nociception and hyperalgesia - The role of the transcription factor NF-B as the lynchpin nocisensor: Hyperalgesic or analgesic effect? Curr Immunol Rev. 2007;3:117–31. [Google Scholar]
- 58.Baeuerle PA. IkappaB-NF-kappaB structures: At the interface of inflammation control. Cell. 1998;95:729–31. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 59.Tang L, Shukla PK, Wang LX, Wang ZJ. Reversal of morphine antinociceptive tolerance and dependence by the acute supraspinal inhibition of Ca(2+)/calmodulin-dependent protein kinase II. J Pharmacol Exp Ther. 2006;317:901–9. doi: 10.1124/jpet.105.097733. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Geis C, Sommer C. Activation of TRPV1 contributes to morphine tolerance: Involvement of the mitogen-activated protein kinase signaling pathway. J Neurosci. 2008;28:5836–45. doi: 10.1523/JNEUROSCI.4170-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dalo NL, Sosa-Sequera MC, Usubillaga A. On the anticonvulsant activity of kaurenic acid. Invest Clin. 2007;48:349–58. [PubMed] [Google Scholar]