

Abstract
Mitochondrial DNA (mtDNA) haplogroups may contribute to the development of aging-related diseases. A reliable in vitro cellular system for investigating the physiologic significance of mtDNA haplogroups is essential. This study aims to construct and characterize a series of cybrid cell lines harboring variant mtDNA haplogroups collected from healthy Taiwanese volunteers. Cybrid cells harboring different mtDNA haplogroups like B4a, B4b, B4c, B4d, B5, R, F1a, F2, D4e, D4a, D5b, D5a, E, M8, C, and N9a were prepared. Luminex 1000 and full-length mtDNA sequencing were used to confirm that mtDNA haplogroups of transmitochondrial cybrids were identical to their original donors. Cybrid B4b had a significantly lower oxygen consumption rate and higher mitochondrial membrane potential compared to F1a, B5, D5a, D4a, and N9a but had more susceptibility to H2O2-induced oxidative stress than cybrid F1a, D4a, and N9a. Cybrid N9a had better oxygen consumption and H2O2-challenged viability compared to B4b, F1a, B5, D5a, and D4a. A series of cybrid cells harboring the main haplogroups of the Taiwanese population with ethnic Chinese background has been developed in vitro. With this mtDNA haplogroup population, the underlying mechanisms of aging-related diseases may be better understood, and therapeutic interventions can be accelerated.
1. Introduction
A double-membrane structure and a circular mitochondrial genome with its own transcription, translation, and protein assembly systems support the endosymbiotic origin of the mitochondria [1]. While most mitochondrial proteins are encoded by nuclear DNA and imported into the organelle, 13 are encoded by mitochondrial DNA. Human mitochondrial DNA (mtDNA) is a 16,568 bp circular double-stranded DNA molecule coding for 13 polypeptide components of the mitochondrial oxidative phosphorylation (OXPHOS) machinery, 2 ribosomal RNA molecules, and a set of 22 transfer RNA molecules [1–4]. More than 99% of mitochondrial proteins are encoded by the nuclear genome, translated on cytoplasmic ribosomes, and selectively imported into the appropriate mitochondrial compartment [5].
Mitochondrial DNA is inherited maternally, and each mitochondrion contains 2–10 mtDNA molecules [6]. Mitochondrial DNA has a very high mutation rate and when a mutation occurs, normal and mutant mitochondrial DNA can coexist within the same cell, known as heteroplasmy [5, 7]. Mitochondrial DNA defects are linked with a wide range of diseases known as mitochondrial encephalomyopathies. Over the last two decades, more than 100 pathologic defects of mtDNA have been characterized, many due to gross structural rearrangements (i.e., single or multiple deletions or duplications) or from point mutations in the mtDNA [7–9]. Large-scale deletions of mtDNA in cancer cells imply that deficits in the mitochondrial genome can change cellular behavior similar to cross-talking with nuclear genome [10].
Phylogeographic studies on human mtDNA have revealed a remarkable correlation between mtDNA lineages and the geographic origins of indigenous populations. Regional mtDNA lineages are groups of related individual mtDNA sequences (haplotypes) known as haplogroups [11]. Unique sets of common SNPs define groups of mtDNA (mtDNA haplogroups) that have evolved from the same ancestors [12]. Cann et al. found the mtDNA evolution originated from a common African ancestor, named “mitochondrial Eve” [11]. Haplogroups L1, L2, and L3 are specific for the African population, whereas all non-African mtDNA haplogroups are descended from L3 and belong to either the M or the N superclades. European populations belong to nine haplogroups (i.e., H, I, J, K, T, U, V, W, and X) from the N superclades [13], while the haplogroups A, B, C, D, and E are specific to Asian populations.
Recently, mtDNA epidemiologic studies have shown that mtDNA haplogroups are associated with various phenotypes. There are significant epidemiologic associations between mtDNA haplogroup and longevity among the Japanese and Chinese [14–16], neurodegenerative [17–19] psychiatric [20, 21], cardiovascular [22, 23], and metabolic diseases [24, 25]. An association between human mtDNA variants and diseases has also been reported [26–28].
To further confirm the molecular pathogenesis and screen effective treatment for mtDNA-associated diseases, optimal cell models for reproducing the progress of diseases are valuable. A cybrid model that carries the same nuclear background and harbors variant mtDNA can exclude variations from the nuclear genome. Within analogous nuclear background, the physiologic function of mitochondrial genome can be reproduced in vitro, enabling the validation of epidemiologic observations. Thus, a complete set of cybrid cells carrying different mtDNA haplogroups are warranted to exclusively investigate the impact of mtDNA common variations on mitochondrial function. The present study successfully developed the most commonly occurring cybrids harboring mtDNA haplogroups from the Taiwanese population with ethnic Chinese background.
2. Materials and Methods
2.1. Cell Culture and Generation of ρ 0 Cells
Human 143B osteosarcoma (BCRC 60439) (Bioresource Collection and Research Center, Taiwan, Republic of China) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (growth medium) at 37°C in 5% CO2. The FBS was thawed at 37°C and then heated to 56°C for 30 min for heat inactivation. Production and culturing of ρ 0 cells required growth medium containing 1 mM pyruvate and 50 μg/mL uridine (ρ 0 medium) to support growth [29]. The ρ 0 cells were produced by culturing 143B osteosarcoma cells in the presence of ethidium bromide (EtBr, 50 ng/mL) for 8 weeks. The cells were subcultured every 2-3 days and kept in 50–70% of confluence.
2.2. Isolation and Transfusion of Platelets to ρ 0 Cells
Healthy Taiwanese volunteers donated their peripheral blood. All of them provided written informed consent in accordance with the protocols approved by the institutional review board of the contributing center. The studies were conducted according to the guidelines of the Declaration of Helsinki and adopted the methods previously reported by Chomyn [30]. Briefly, platelets were isolated and fused with ρ 0 cells in the presence of polyethylene glycol 1500 (50% w/v) (Roche, Nutley, NJ, USA). After 24 h of incubation, fresh ρ 0 medium was introduced. The cells were allowed to recover for 1 week in ρ 0 medium, with medium changed every 2 days. After 1 week of recovery in ρ 0 medium, the cells were placed on a 96-well plate in a 1 : 2 limited dilution in DMEM-based selection medium composed of 10% dialyzed FBS (Gemini), which lacks uridine and pyruvate. After 10–14 days, the cybrid populations forming single colonies were screened and transferred to a 35 mm dish. With continuing growth, the cybrids were transferred to 100 mm dish for further 20 generations of culture in order to ensure that nuclear-encoded mitochondrial proteins were incorporated into the newly introduced mitochondria.
2.3. MtDNA Copy Number Analysis for ρ 0 and Cybrid Cells
Total DNA was isolated from peripheral blood leucocytes using the PUREGENE DNA Purification kit (Qiagen, Chatsworth, CA, USA). The extracted DNA samples were frozen at −20°C until the assay without repeated freeze-thawing cycles. The relative mtDNA copy numbers were measured using real-time PCR and corrected by simultaneous measurement of nuclear DNA. The forward and reverse primers for nuclear gene that were complementary to chromosome 1 genomic contig (accession no. NT_004487.19) were 5′-GGCTCTGTGAGGGATATAAAGACA-3′ and 5′-CAAACCACCCGAGCAACTAATCT-3′. The forward and reverse primers for mtDNA that were complementary to the sequence of the ND2 gene (accession no. NC_012920.1) were 5′-CACAGAAGCTGCCATCAAGTA-3′ and 5′-CCGGAGAGTATATTGTTGAAGAG-3′. The PCR was performed by a LightCycler 480 Real-Time PCR System (Roche Diagnostics; F. Hoffmann-La Roche Ltd.) using the SYBR GREEN I PCR MASTER MIX (Roche Diagnostics; F. Hoffmann-La Roche Ltd.). The DNA (10 ng) was mixed with 10 μL SYBR GREEN I PCR MASTER MIX containing 400 nmol of forward and reverse primers in a final volume of 20 μL. The PCR conditions were 10 min at 95°C, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 20 s and primer extension at 72°C for 15 s. The amplified products were denatured and reannealed at different temperatures to detect their specific melting points. Samples showing primer dimmers or unspecific fragments were excluded.
The threshold cycle number (Ct) values of the chromosome 1 sequence and the mitochondrial ND2 gene were determined for each individual in the same quantitative PCR run. Each measurement was carried out at least three times and normalized in each experiment against a serial dilution of control DNA sample. There was good reproducibility within and between runs. The intraassay coefficients of variation of Ct values were 2.2% and 3.8% for ND2 and chromosome 1, respectively. The Ct values were used as a measure of the input copy number, and Ct value differences were used to quantify the mtDNA copy number relative to the chromosome 1 using the following equation:
| (1) |
where results were expressed as the copy number of mtDNA/cell.
2.4. Aerobic Metabolism
Cultured cells used glucose as substrates for glycolysis to generate ATP. By replacing galactose with glucose in the culture medium, energy production shunted glycolysis to oxidation phosphorylation depending on the mitochondria [31, 32]. To examine the aerobic capacity of mitochondria, cells were grown in DMEM containing 10 mM galactose and 10% FBS in the absence of glucose.
2.5. Oxygen Consumption Analysis for ρ 0 and Cybrid Cells
Detection of O2 consumption in permeabilized cells was as described previously with some modifications [33–35]. O2 consumption was monitored by a Clark electrode (Mitocell S200 micro respirometry system; Strathkelvin Instruments, Motherwell, UK). Cells (100 μL at 5 × 106 cells/mL) in KCl medium (100 mM KCl, 3 mM MgCl2, 20 mM HEPES, 1 mM EDTA, and 5 mM KH2PO4; pH 7.4) were permeabilized by digitonin (optimal concentration 32.5 μg/mL as determined by trypan blue staining) and loaded into a 200 μL MT200 Respirometer Chamber, suspended by a fixed-speed solid-state magnetic stirrer inside the chamber and maintained at 37°C by a circulating water bath. Glutamate/malate (10 mM) and succinate (10 mM), combined with ADP (0.1 mM), were used as substrates to test the function of complex I- and complex II-linked respiration. Changes in O2 levels were calculated by respirometry software. For control recordings, 100 μL of buffer was injected into the chamber.
2.6. Measurement of Mitochondrial Membrane Potential
Cells (5 × 105) were placed in six-well plates and allowed to attach for 16–18 h and then incubated in 100 nM TMRE (Sigma) for 30 min at 37°C. They were then washed twice with PBS, harvested by trypsinization, and resuspended in 500 μL PBS containing 2% FBS. Cytofluorimetric analysis was conducted using a fluorescence-activated cell scanner machine (BD Biosciences FACScan system).
2.7. Cellular Viability Assay
Cells (8 × 103) were seeded into 96-well plates 24 h prior to treatment. Cell survival was determined using cell counting kit 8 (Wako) with WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2-H-tetrazolium, monosodium salt] as substrate. The WST-8 was reduced by dehydrogenase to form formazan that was proportional to living cells. The amount of formazan was assayed by spectrophotometry at OD450.
2.8. Western Blot
Cells were lysed in RIPA lysis buffer (50 mM Tris, pH 7.4; 150 mM NaCl; 1 mM PMSF (phenylmethanesulfonyl fluoride); 1 mM EDTA (ethylenediaminetetraacetic acid); 1% Triton X-100; 1% sodium deoxycholate; 0.1% SDS) with the addition of Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail I (Sigma). The antibodies used included anti-COX II antibody (Abcam), peroxidase-labeled anti-rabbit IgG (H + L) secondary antibody (Abcam), and anti-β-actin (Cell Signaling, Danvers, MA). The signals were developed by ECL plus (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) using X-ray films.
2.9. Genotyping of Mitochondrial Haplogroups
The selected polymorphic sites were based on Nishigaki's and Kong's work [36] (Table 1). Venous blood (7 mL) was collected from each subject into tubes containing 50 mmol/L EDTA (disodium salt). Genomic DNA was isolated using a commercial kit (Qiagen, Chatsworth, CA, USA). For amplifying mtDNA fragments, 24 pair primers for the mitochondrial DNA were used for gene amplification by PCR using primer sequences described previously [28]. Amplicon size ranged from 190 to 300 bp. The PCR amplification was conducted in a final reaction volume of 20 μL, containing 10 ng human genomic DNA, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.2 mM dNTP (i.e., dATP, dTTP, dCTP, and dGTP), 0.5 μM of each primer, and 1 unit of ExTaq DNA polymerase (Takara Bio Inc.). The PCR conditions were as follows: an initial denaturation step at 94°C for 10 min, followed by 40 cycles of denaturation at 94°C for 35 s, annealing at 60°C for 45 s, and extension at 72°C for 45 s, with a final extension of 10 min at 72°C and kept at 25°C using an Eppendorf Mastercycler gradient.
Table 1.
Polymorphism sites characteristic [B4a, B4b, B4c, B5, R9, F1a, F2, D4a, D4e, D5a, D5b, E, M8, C, and N9a].
| Haplogroup | Polymorphism |
|---|---|
| B4a | 8272 (9-bp deletion), G9123A |
| B4b | 8272 (9-bp deletion), C15535T, G4820A |
| B4c | 8272 (9-bp deletion), G15346A |
| B4d | 8272 (9-bp deletion), C15535T, A13942G |
| B5 | 8272 (9-bp deletion), T9950C, A10398G |
| R | G13928C, C3970T |
| F1a | C3970T, G10310A, G13928C, G12406A, C4086T |
| F2 | G13928C, C3970T, G10310A, T12338C |
| D4a | G3010A, C5178A, C4883T, T14979C |
| D4e | G3010A, C5178A, C4883T, A15924G |
| D5a | C5178A, C4883T, A10397G |
| D5b | T681C, C5178A, C4883T, A10397G |
| E | G4491A, T3027C |
| M8 | A4715G, C8684T, G6179A, A15487T |
| C | A4715G, A15487T, A13263G |
| N9a | G5231A, G12372A, A12358G |
After PCR amplification, 60 probes were used for mitochondrial haplogroup definition. Probes for haplotyping were as described previously [28]. Briefly, synthesized oligonucleotide probes were modified at the 5′-end with a terminal anion group and covalently bound to carboxylated fluorescent microbeads using ethylene dichloride (EDC), following the manufacturer's instructions. Oligonucleotide-labeled microbeads (oligobeads) were mixed together to make a 5000/5 μL oligobead mixture for hybridization. The 5′-biotin-labeled PCR amplicons were hybridized to the oligobeads in a total volume of 50 μL per well in a 96-well plate by adding 5 μL of the PCR amplicon to 40 μL of hybridization buffer (3.75 M TMAC, 62.5 mM TB [pH 8.0], 0.5 mM EDTA, and 0.125% N-lauroylsarcosine). This reaction mixture was denatured at 95°C for 2 min and then hybridized at 55°C for 30 min using the Eppendorf Mastercycler gradient.
After hybridization, the oligobeads were washed with 1× PBS (pH 7.5, containing 0.01% Tween 20) and centrifuged at 2200 ×g for 2 min. The supernatant was discarded (washed twice), and the pelleted oligobeads were reacted with 70 μL aliquot of a 1000× diluted solution of SA-PE (Prozyme). The hybridized amplicons were labeled with SA-PE at 52°C for 15 min using the Eppendorf Mastercycler gradient. Then the reactions were measured by the Luminex100 flow cytometer.
2.10. Determination of mtDNA Full-Length Sequence
2.10.1. First PCR
The entire mitochondrial genome was amplified as six fragments, each approximately 3.0 kb in length, by a symmetric PCR method with primer pairs as stated previously [28]. The PCR amplification was conducted in a final reaction volume of 20 μL, containing 50 ng human genomic DNA, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.2 mM concentration of each dNTP, 1.0 μM concentration of each primer, and 1 unit of pfu DNA polymerase (Takara Bio Inc.). The PCR conditions used were as follows: an initial denaturation step at 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 15 s, annealing at 58°C for 35 s, and extension at 72°C for 3 min, with a final extension of 10 min at 72°C. The amplified fragments were analyzed by electrophoresis on a 1% agarose gel and visualized by ethidium bromide staining.
2.10.2. Second PCR
The first PCR DNA templates for sequence analysis of the entire mitochondrial genome were amplified as 32 overlapping segments, each approximately 500–1100 bp as shown previously [28]. The PCR amplifications were conducted in a final reaction volume of 20 μL, containing 200 ng of the first PCR product, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.2 mM concentration of each dNTP, 1.0 μM concentration of each primer, and 0.5 units of pfu DNA polymerase (Takara Bio Inc.). The PCR conditions used were the same as those for the first PCR except that the annealing temperature used was 60°C. The second PCR fragments were analyzed by electrophoresis on a 1.5% agarose gel and visualized by ethidium bromide staining.
2.11. Sequence Analysis and mtSNP Identification
The second PCR template and primers were sent to the Genome Research Center, Sequencing Center of National Yang-Ming University, which performed the sequence using Applied Biosystems. The primers used for sequencing were as described previously [28].
3. Results
3.1. Production of ρ 0 Cells from 143B Osteosarcoma Cells
EtBr was bound to the double-helix DNA and inhibited replication. MtDNA was more susceptible to EtBr than nuclear DNA in an indicated concentration of EtBr. Low dosages of EtBr were used for long-term exposure to fully deplete mtDNA while maintaining cellular growth. The reversion of mtDNA in ρ 0 cell in the absence of EtBr was reported to be minimized by prolonged culturing [37]. Thus, instead of shorter durations of EtBr exposure [29, 38], 143B cells were grown in EtBr for 8 weeks to reduce the possibility of mtDNA reversion. In order to obtain stable clones, individual clones were further isolated. The reversion of mtDNA was examined in the absence of EtBr.
The mtDNA of 143B cells decreased to 0.93 copy/cell after 8 weeks (Table 2). However, mtDNA reversed to the level of the parental 143B cells after EtBr removal for 14 days. The possibility of mtDNA reversion was eliminated by isolating single clones, and six of 16 isolated clones maintained mtDNA; a level of less than one copy per cell after EtBr withdrawal for 50 days. Clone no. 38 was selected for another 60 days of culture without EtBr to confirm the irreversibility of 143B-derived ρ 0 cells (Table 2). The ρ 0 cells died within 4 weeks without pyruvate and uridine (data not shown).
Table 2.
mtDNA copy number of 143B osteosarcoma following EtBr exposure.
| Cell type | Days without EtBr | mtDNA (copy/cell) |
|---|---|---|
| 143B | — | 1060.11 |
| ρ 08w | 0 | 0.93 |
| ρ 08w revertant | 14 | 1009.9 |
| Clone #23 | 50 | 0.98 |
| Clone #24 | 50 | 0.86 |
| Clone #31 | 50 | 0.98 |
| Clone #35 | 50 | 0.85 |
| Clone #36 | 50 | 0.97 |
| Clone #38 | 50 | 0.97 |
| Clone #38 | 110 | 0.69 |
143B: parental osteosarcoma; ρ 08w: 143B following 8 weeks of EtBr exposure; ρ 08w revertant: recovered mtDNA level in the absence of EtBr for 14 days.
Clones 23, 24, 31, 35, 36, and 38 were selected clones through limiting dilution.
3.2. Loss of Mitochondrial Oxygen Consumption and Defects in Respiratory Enzyme Complexes
Since subunit 2 of cytochrome c oxidase (COX2) was mtDNA encoded, COX2 was examined for ρ 0 cells. Immunoblotting demonstrated undetectable COX2 of ρ 0 cells (Figure 1). In terms of oxygen consumption ability of the ρ 0 cells, mitochondrial complex I- and II-linked respiration monitored in the presence of glutamate/malate and succinate, respectively, were deficient in ρ 0 cells (Figure 3).
Figure 1.
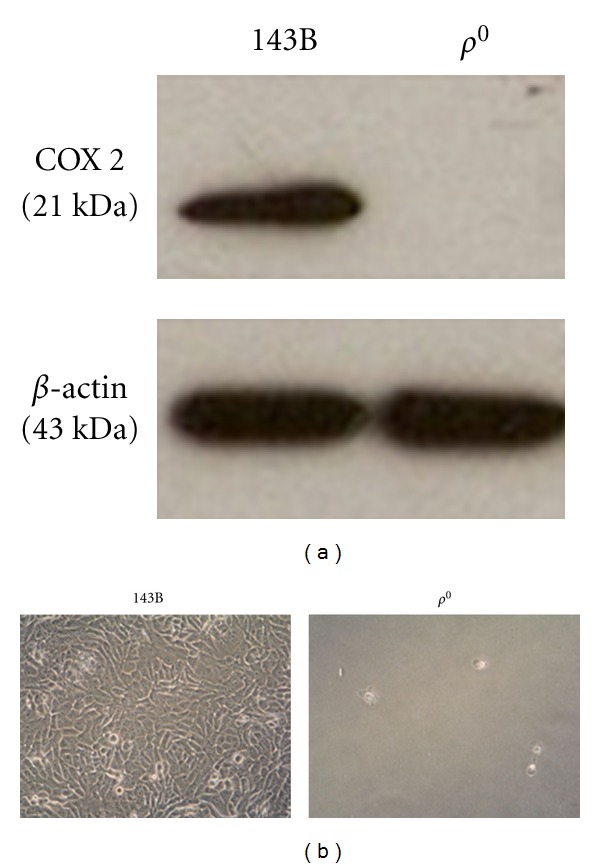
(a) The absence of mtDNA-encoded protein in ρ 0 cells. The expression of mtDNA-encoded COX2 and nuclear-encoded β-actin in the parental 143B osteosarcoma and ρ 0 cell was analyzed by Western blotting. (b) The ρ 0 cells failed to survive in the galactose medium over 48 h, in contrast to the normal growth of the parental 143B cells.
Figure 3.
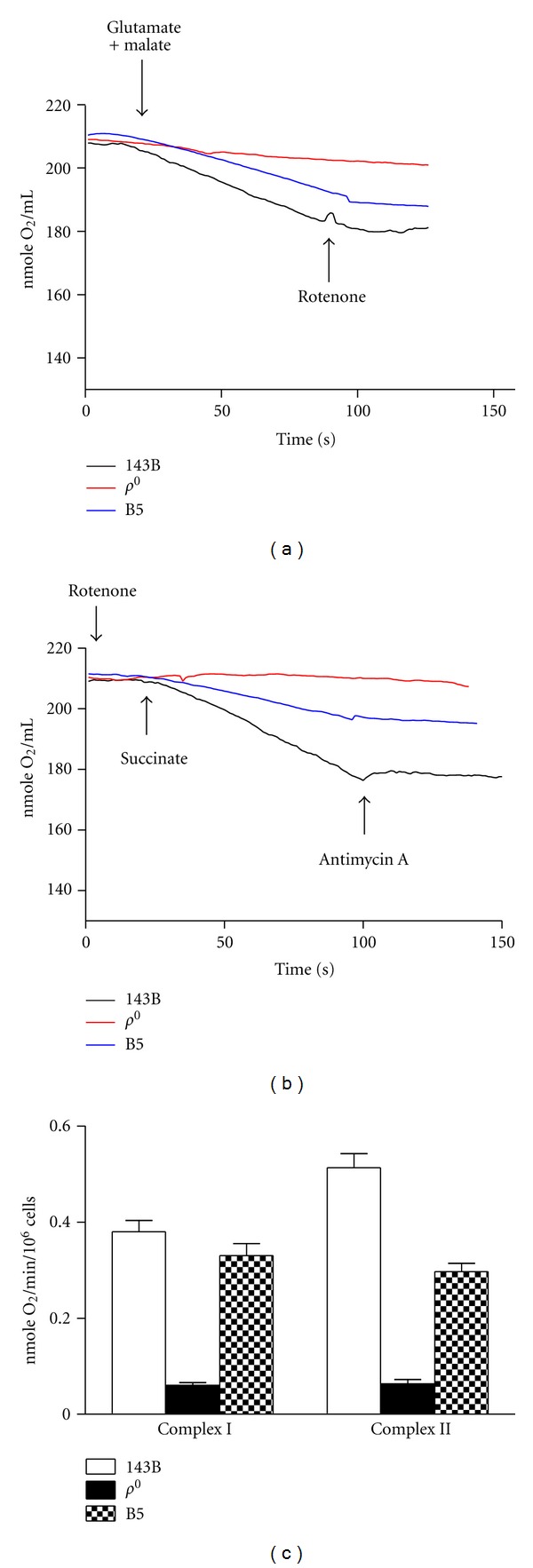
Oxygen consumption of 143B osteosarcoma, ρ 0 cell, and the cybrid harboring the mtDNA haplogroup of B5. (a) Complex I-linked respiration was performed by adding 10 mM glutamate and 10 mM malate in the presence of 0.1 mM ADP. The complex I inhibitor, rotenone, terminated the respiration. (b) Complex II-linked respiration preincubated with rotenone was performed by adding succinate in the presence of 0.1 mM ADP. The complex III inhibitor, antimycin A, terminated the respiration. (c) Quantitative data of oxygen consumption expressed in slope were shown as mean ± SEM for three independent experiments.
3.3. Production of Cybrids Harboring Different Haplogroups from Human Platelet Mitochondria
Sixteen different haplogroups were selected from healthy volunteers for mitochondrial genotyping. Platelets from the 16 known mtDNA haplogroups were fused with ρ 0 cells, followed by selection. All of the transmitochondrial experiments contained negative control groups in which ρ 0 cells underwent the same fusion process in the absence of human platelets. All of the negative control cells died after selection, while cells fused with platelets survived (data not shown), suggesting that fusion was successful, and the mtDNA of ρ 0 cells were unable to revert.
3.4. Mitochondrial Respiratory Function and Mitochondrial DNA Content Reversed after Transmitochondrial Fusion
The mtDNA copy number of parental 143B, ρ 0 cells and cybrids harboring human mtDNA suggested that the depleted mtDNA content in ρ 0 cells was reversed following transmitochondrial fusion (Figure 2). Because most cellular metabolisms relied on normal mitochondria, the respiratory function of the cybrids was examined. Parental 143B cells, ρ 0 cells, and cybrids harboring the B5 haplogroup assayed for oxygen consumption rate indicated that ρ 0 cells with depleted mtDNA demonstrated loss of respiratory function, whereas transmitochondrial fusion successfully restored the lost respiration (Figure 3).
Figure 2.
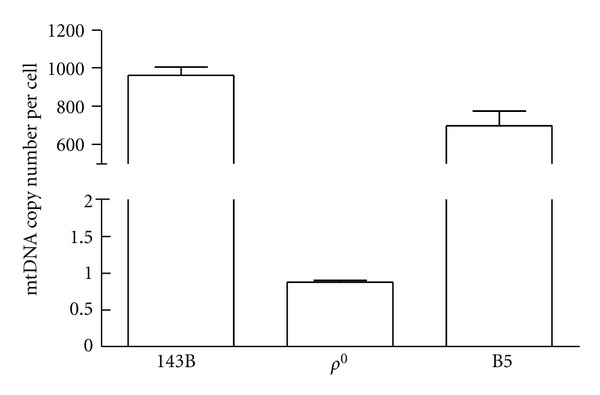
The mtDNA content of 143B osteosarcoma, ρ 0 cell, and the cybrid harboring the B5 haplogroup was analyzed by real-time PCR and shown as mtDNA copy number. Data was expressed as mean ± SEM for three independent experiments.
3.5. Mitochondrial Polymorphisms Generated Cybrids Harboring Identical Mitochondrial Genomarkers as the Original Platelet Donors
To verify the mtDNA haplogroup of the 16 cybrids, polymorphism sites were reexamined by Luminex 1000. All 16 cybrids had similar mtDNA haplogroups as their original donors (data not shown). To further verify the mtDNA haplogroup of these constructed cybrids, full-length sequencing for mtDNA was performed. Nonsynonymous sites in mtDNA genome caused amino acid substitution in the 16 cybrids (Tables 3 and 4), showing characterizations of the genetic aspect and corresponding features of the amino acid.
Table 3.
Nonsynonymous substitution of 16 cybrid cell lines.
| Gene | Substitution | AA change | B4a | B4b | B4c | B4d | B5 | R | F1a | F2 | D4a | D4e | D5a | D5b | E | M8 | C4 | N9a | 143B |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G4491A | V→I | — | — | — | — | — | — | — | — | — | — | — | — | A | — | — | — | — | |
| G4596A | V→I | — | — | — | — | — | — | — | — | — | — | — | — | — | — | A | — | — | |
| ND2 | C5178A | L→M | — | — | — | — | — | — | — | — | A | A | A | A | — | — | — | — | — |
| A5301G | I→V | — | — | — | — | — | — | — | — | — | — | G | G | — | — | — | — | — | |
| A5466G | T→A | — | — | — | — | — | — | — | — | G | — | — | — | — | — | — | — | — | |
|
| |||||||||||||||||||
| T6253C | M→T | — | — | — | — | — | — | — | — | — | — | — | C | — | — | — | — | — | |
| COI | G6267A | A→T | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | A |
| T7266G | S→A | G | G | G | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
|
| |||||||||||||||||||
| COII | G7598A | A→T | — | — | — | — | — | — | — | — | — | — | — | — | A | — | — | — | — |
| A7934G | I→V | — | — | — | — | — | — | — | — | — | — | — | — | G | — | — | — | — | |
|
| |||||||||||||||||||
| ATPase8 | C8414T | L→I | — | — | — | — | — | — | — | — | T | T | — | — | — | — | — | — | — |
|
| |||||||||||||||||||
| G8584A | A→T | — | — | — | — | A | — | — | — | — | — | — | — | — | A | A | — | — | |
| C8684T | T→I | — | — | — | — | — | — | — | — | — | — | — | — | — | T | — | — | — | |
| A8701G | T→A | — | — | — | — | — | — | — | — | G | G | G | G | G | G | G | — | — | |
| ATPase6 | A8860G | T→A | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G |
| G9053A | S→N | — | — | A | — | — | — | A | — | — | — | — | — | — | — | — | — | — | |
| C9099T | I→M | — | — | — | — | — | — | — | T | — | — | — | — | — | — | — | — | — | |
| T9128C | I→T | — | — | — | — | — | — | — | — | — | — | — | C | — | — | — | — | — | |
|
| |||||||||||||||||||
| T10084C | I→T | — | C | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| ND3 | A10398G | T→A | — | — | — | — | G | — | — | — | G | G | G | G | G | G | G | — | — |
| C10400T | T→A | — | — | — | — | — | — | — | — | T | T | T | T | T | T | T | — | — | |
|
| |||||||||||||||||||
| ND4L | T10609C | M→T | — | — | — | — | — | — | C | — | — | — | — | — | — | — | — | — | — |
|
| |||||||||||||||||||
| 11138T | (Insertion) | T | T | ||||||||||||||||
| ND4 | G11969A | A→T | — | — | — | — | — | — | — | — | — | — | — | — | — | — | A | — | — |
| A12026G | I→V | — | — | — | — | — | — | — | — | — | — | G | — | — | — | — | — | — | |
|
| |||||||||||||||||||
| T12338C | M→T | — | — | — | — | — | — | — | C | — | — | — | — | — | — | — | — | — | |
| A12358G | T→A | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | G | — | |
| G12406A | V→I | — | — | — | — | — | — | A | — | — | — | — | — | — | — | — | — | — | |
| G13135A | A→T | — | — | — | — | — | — | — | — | — | A | — | — | — | — | — | — | A | |
| G13708A | A→T | — | — | — | A | — | — | — | A | — | — | — | — | — | — | — | — | — | |
| ND5 | G13759A | A→T | — | — | — | — | — | — | A | — | — | — | — | — | — | — | — | — | — |
| A13834G | T→A | — | — | — | — | — | — | — | — | G | — | — | — | — | — | — | — | — | |
| G13928C | S→T | — | — | — | — | — | C | C | C | — | — | — | — | — | — | — | — | — | |
| A13942G | T→A | — | — | — | G | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| 13944C | (Insertion) | C | |||||||||||||||||
| A13966G | T→A | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | G | |
|
| |||||||||||||||||||
| ND6 | T14318C | N→S | — | — | — | — | — | — | — | — | — | — | — | — | — | — | C | — | — |
| T14577C | I→V | — | — | — | — | — | — | — | — | — | — | — | — | C | — | — | — | — | |
|
| |||||||||||||||||||
| C14751T | T→I | T | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
| C14766T | I→T | — | — | — | — | — | — | — | — | — | — | — | T | — | — | — | — | — | |
| Cytb | T14979C | I→T | — | — | — | — | — | — | — | — | C | — | — | — | — | — | — | — | — |
| T15204C | I→T | — | — | — | — | — | — | — | — | — | — | — | — | — | — | C | — | — | |
| A15326G | T→A | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | |
| A15758G | I→V | — | G | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | |
Nucleotide change, corresponding amino acid change, and the reference mitochondrial sequence were based on MITOMAP (http://www.mitomap.org/MITOMAP).
AA: amino acid; ND2: NADH dehydrogenase subunit 2; COI: cytochrome c oxidase subunit I; COII: cytochrome c oxidase subunit II; ATPase8: ATP synthase F0 subunit 8; ATPase6: ATP synthase F0 subunit 6; ND3: NADH dehydrogenase subunit 3; ND4L: NADH dehydrogenase subunit 4L; ND4: NADH dehydrogenase subunit 4; ND5: NADH dehydrogenase subunit 5; ND6: NADH dehydrogenase subunit 6; Cytb: cytochrome b.
Table 4.
Summary of changes in each gene in each haplogroup.
| B4a | B4b | B4c | B4d | B5 | R | F1a | F2 | D4a | D4e | D5a | D5b | E | M8 | C4 | N9a | 143B | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ND2 | C5178A, | C5178A | C5178A, | C5178A, | G4491A | G4596A | |||||||||||
| A5466G | A5301G | A5301G | |||||||||||||||
|
| |||||||||||||||||
| ND3 | T10084C | A10398G | A10398G, | A10398G, | A10398G, | A10398G, | A10398G, | A10398G, | A10398G, | ||||||||
| C10400T | C10400T | C10400T | C10400T | C10400T | C10400T | C10400T | |||||||||||
|
| |||||||||||||||||
| ND4 | A12026G | ins11138T | ins11138T, | ||||||||||||||
| G11969A | |||||||||||||||||
|
| |||||||||||||||||
| ND4L | T10609C | ||||||||||||||||
|
| |||||||||||||||||
| G13708A, | 13928, | G12406A, | T12338C, | A13834G | G13135A | A12358G | G13135A, | ||||||||||
| ND5 | A13942G | insl3944C | G13759A, | G13708A, | A13966G | ||||||||||||
| G13928C | G13928C, | ||||||||||||||||
|
| |||||||||||||||||
| ND6 | T14577C | T14318C | |||||||||||||||
|
| |||||||||||||||||
| Cytb | T14751C, | A15326G, | A15326G | A15326G | A15326G | A15326G | A15326G | A15326G | T14979C, | A15326G | A15326G | C14766T, | A15326G | A15326G | T15204C, | A15326G | A15326G |
| A15326G | A15758G | A15326G | A15326G | A15326G | |||||||||||||
|
| |||||||||||||||||
| COI | T7266G | T7266G | T7266G | T6253C | G6267A | ||||||||||||
|
| |||||||||||||||||
| COII | G7598A, | ||||||||||||||||
| A7934G | |||||||||||||||||
|
| |||||||||||||||||
| ATPase6 | A8860G | A8860G | A8860G, | A8860G | G8584A, | A8860G | A8860G, | A8860G, | A8701G, | A8701G, | A8701G, | A8701G, | A8701G, | G8584A, | G8584A, | A8860G | A8860G |
| G9053A | A8860G | G9053A | C9099T | A8860G | A8860G | A8860G | A8860G, | A8860G | C8684T, | A8701G, | |||||||
| T9128C | A8701G, | A8860G | |||||||||||||||
| A8860G | |||||||||||||||||
|
| |||||||||||||||||
| ATPase8 | C8414T | C8414T | |||||||||||||||
ND2: NADH dehydrogenase subunit 2; COI: cytochrome c oxidase subunit I; COII: cytochrome c oxidase subunit II; ATPase8: ATP synthase F0 subunit 8; ATPase6: ATP synthase F0 subunit 6; ND3: NADH dehydrogenase subunit 3; ND4L: NADH dehydrogenase subunit 4L; ND4: NADH dehydrogenase subunit 4; ND5: NADH dehydrogenase subunit 5; ND6: NADH dehydrogenase subunit 6; cytb: cytochrome b; ins: insertion.
3.6. Cybrids with Various mtDNA Haplogroups Exhibited Differences in Oxygen Consumption Capacity and Mitochondrial Membrane Potential
Haplogroups N9a and D4a were reported to be protective against diabetes [24, 25] and promoted longevity [14], respectively. Haplogroup B4 was diabetes susceptible, while haplogroup D4 was diabetes-resistant [28]. To clarify if mtDNA genetic variations among these haplogroups influenced mitochondrial respiratory capacity, cellular oxygen consumption rates were examined in cell lines created from these haplogroups. Cybrid-harboring mtDNA haplogroups N9a and D4a had significantly higher oxygen consumption rates compared to cybrid-harboring mtDNA haplogroups B4b, F1a, B5, and D5a in complex I- and II-linked respiration (Figure 4(a)). Moreover, compared to cybrid-harboring mtDNA haplogroup B4b, haplogroups F1a, B5, and D5a also exhibited higher O2 consumption rates in complex I-linked respiration.
Figure 4.
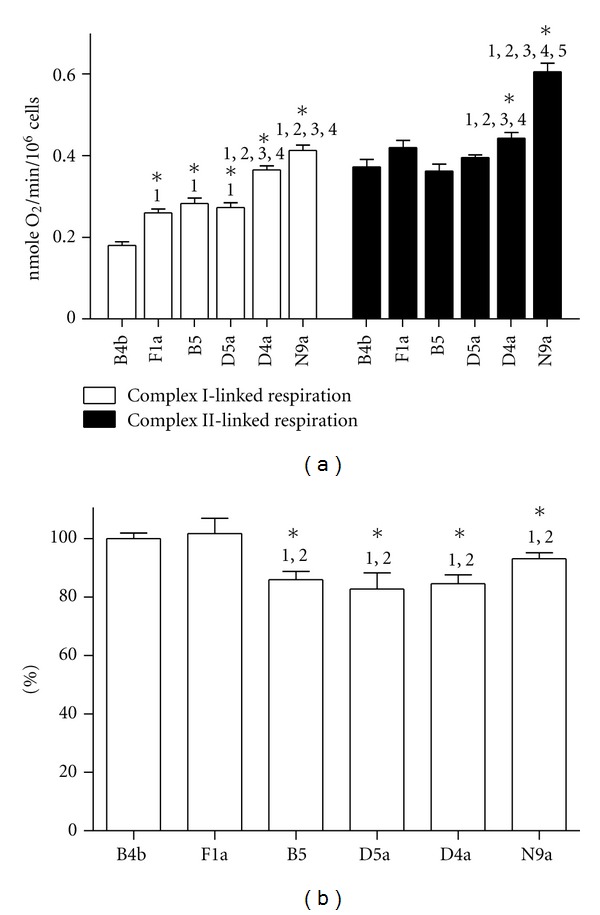
Oxygen consumption rate and mitochondrial membrane potential of cybrids harboring B4b, F1a, B5, D5a, D4a and N9a. (a) Complex I-linked respiration was performed by adding 10 mM glutamate and 10 mM malate in the presence of 0.1 mM ADP. Complex II-linked respiration pre-incubated with rotenone was performed by adding succinate in the presence of 0.1 mM ADP. (b) The mean TMRE fluorescence value of B4b was set to 100%. Data were expressed as mean ± SEM for four independent experiments. The statistical significance of differences between groups was determined by one-way analysis of variance followed by Bonferroni post hoc test. *P < 0.05 compared to (1) B4b, (2) F1a, (3) B5, (4) D5a, and (5) D4a.
Measuring mitochondrial membrane potential (ΔΨm) among these cybrid lines, the ΔΨm of cybrid-harboring mtDNA haplogroups B4b and F1a was significantly higher compared to those of B5, D5a, D4a, and N9a (Figure 4(b)). This was consistent with a previous report that a cybrid with higher oxygen consumption rate had lower mitochondrial membrane potential [39].
3.7. Cybrids Harboring Different mtDNA Haplogroups Exhibited Discriminating Tolerance against Oxidative Stress
To assess whether differences in OXPHOS capacity affected cellular tolerance to oxidative stress, the viability of cybrid cells exposed to various doses of hydrogen peroxide (H2O2) was examined. Under higher H2O2 concentration, cybrids-harboring mtDNA haplogroups F1a, D4a, and N9a had significant resistance and higher cellular viability compared to B4b (Figure 5), suggesting that cybrid B4b was much more sensitive to oxidative stress induced by H2O2. Notably, cybrid-harboring mtDNA haplogroup N9a demonstrated superior viability under oxidative stress compared to those with B4b, F1a, B5, D5a, and D4a.
Figure 5.
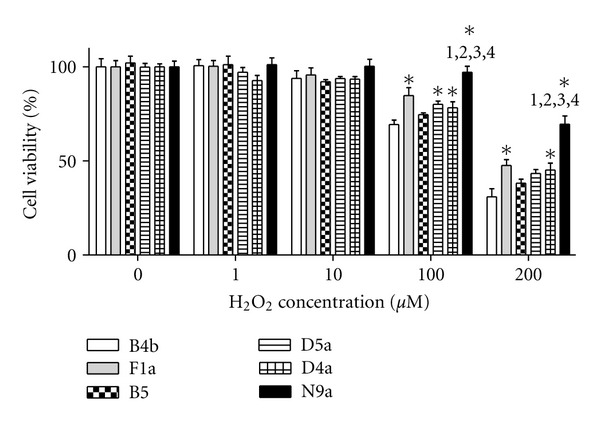
Cellular viability of cybrids under various concentration of H2O2 for 4 h. WST-8 assay for viability was determined by spectrophotometry at 450 nm. Data (mean ± SEM, n = 8) expressed as percentage relative to B4b in the untreated control was determined by one-way analysis of variance followed by Bonferroni post hoc test. *P < 0.05 compared to B4b and to (1) F1a, (2) B5, (3) D5a, and (4) D4a.
4. Discussion
The present study illustrates the process behind the construction of cybrid lines using samples from a representative sample of the Taiwanese population harboring different mtDNA haplogroups. The study further demonstrates that the mitochondria of recomposed cells are functionally intact. The ρ 0 cells defective in mtDNA and lacking in O2 consumption ability have been successfully constructed from 143B osteosarcoma cells. Their defective mitochondrial function is restored after transmitochondrial fusion with mitochondria donated from volunteer platelet donors of different mtDNA haplogroups. The recomposed cybrid lines generated and tested systemically harbor the sixteen most commonly found mtDNA haplogroups identified in the Taiwanese population.
Associations between degenerative diseases and mtDNA haplogroups have previously been documented. Epidemiologic studies reveal that certain mtDNA haplogroups are prone to disease generation, whereas other haplogroups are resistant. While the relationship between mtDNA variants and disease generation is generally accepted, some studies on the association of clinical diseases and haplogroup variations remain controversial. In terms of the 16189 T to C transition variant of mtDNA and its association with type II diabetes, both positive and negative reports have been published [27]. Thus, reliable and reproducible material is essential for studies concerning the impact of mtDNA haplogroups on the development of these diseases.
Approximately two decades ago, Attardi's group demonstrated the construction of transmitochondrial cybrids and provided a platform for studying mitochondrial function in cells carrying mtDNA variations in the same nuclear background [29]. Since then, studies using cybrids have shown the importance of mitochondrial functions and genetics for normal physiology. An unpublished finding has previously suggested that Taiwanese harboring the haplogroup B4 have higher prevalence of diabetes mellitus, whereas those harboring haplogroup B5 are resistant to Parkinson's disease. Other studies involving Eastern Asian subjects also reveal that certain haplogroups exhibit dominant physiologic performance, including resistance to Parkinson's disease generation [18]. However, to date, there is still no complete set of cybrid cells carrying different mtDNA haplogroups from the general Taiwanese population. This study, therefore, has created cybrid lines carrying the most commonly occurring haplotypes found among ethnic Chinese population for future functional and mechanical studies.
One of the problems with constructing cybrid cell lines is the reversion of the ρ 0 cells' mtDNA, causing contamination of the parental mtDNA. EtBr chelation may distort the double-strand DNA structure and is widely used in producing mtDNA-less or mtDNA-depleted cells. Appropriate EtBr dosage chelating mtDNA with minimal damage to the nucleus is considered ideal for interfering with the replication process of mtDNA without affecting cell survival. Very long-term exposure to EtBr can generate stable ρ 0 cells with permanent loss of mtDNA. As such, to harvest stable ρ 0 cells, a modest period of exposure followed by isolating single-cell clones has been used in the present study. The data suggest that these ρ 0 cells must be stable to prevent mtDNA reversion and hence ensure the mtDNA consistency of subsequent cybrids. Characteristics were examined by genetic testing, translational protein assessment, and functional respiration testing to verify the validity of transmitochondrial fusion. In addition, the identity of mtDNA haplogroups of original donors and cybrids has been reconfirmed by multiple probes, while full-length sequencing has further demonstrated the solidity of cybrids.
Physiologic differences in common European mtDNA haplogroups using the cybrid model have been reported in the works of Pello et al. [40] and of Gómez-Durán et al. [39]. Thus, it is feasible to test the mitochondrial respiratory function of the cybrids generated in this study. A previous work has identified the diabetes-susceptible group (haplogroup B4) to consist of 10398A and diabetes-resistant group (haplogroup D4) to consist of C5178A, A10398G, and T152C [28]. As was expected, the cybrid harboring the mtDNA haplogroup B4b that carries the 10398A variant is more susceptible to developing diabetes and has a significantly lower oxygen consumption capacity compared to other commonly seen haplogroups in Asian populations of ethnic Chinese background (Figure 4(a)). Consistent with a previous clinical genetic study, cybrids harboring the specific mtDNA haplogroup carrying A10398G (B5) or in combination with C5178A and A10398G (D4a and D5a) display superior oxygen consumption capacity compared to cybrids with B4b (Figure 4(a)). Furthermore, the superior oxygen consumption capacity of cybrids with N9a and D4a is also compatible with findings of Tanaka and colleagues, who reported that D4a is related to a longer lifespan, and haplogroups N9a and D5 are resistant against metabolic syndromes [14, 25].
Closely related to oxygen consumption and cellular energy need, the value of ΔΨm is a key indicator of cellular metabolic activity. Compatible with a previous report [39], ΔΨm is inversely related to the decrease in oxygen consumption rate in the cybrid related to disease conditions (B4b) (Figure 4(b)). This suggests that efficient energy production through the OXPHOS machinery is associated with less ΔΨm and related to cellular resistance to stressful environment. Testing cellular response of the cybrid cell lines to H2O2-induced oxidative stress, cybrid cells carrying various mitochondrial genomic backgrounds have significant differences in survival rates in response to ROS exposure (Figure 5). Consistent with oxygen consumption data, the cybrid harboring the haplogroup N9a is the most resistant to H2O2-induced damage.
In conclusion, the present study has generated cybrid lines harboring the most commonly occurring haplogroups in the Taiwanese population of ethnic Chinese background. Mitochondrial respiratory function, membrane potential, and cellular survival in response to oxidative stress are also different among these common cybrid lines. This set of cybrid cell lines may be useful for future studies regarding the impact of common human mtDNA variances on disease generation and progression.
Authors' Contribution
Drs. T.-K. Lin and H.-Y. Lin contributed equally to this work.
Acknowledgments
The work was supported by Grants from Chang Gung Memorial Hospital (CMRPG870881-3; CMRPG891101-2; CMRPG891081-2) and the National Science Council, Taiwan, (NMRPG896131-2). The authors thank the Chang Gung Medical Foundation Kaohsiung Chang Gung Memorial Hospital Tissue Bank (CLRPG8B0031) for the technical support, and James Waddell of the Han Mei Institute and ATS Medical Editing and Review Solutions for the English editing and proofreading services.
References
- 1.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283(5407):1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Bankier AT, Barrell BG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 3.Poyton RO, McEwen JE. Crosstalk between nuclear and mitochondrial genomes. Annual Review of Biochemistry. 1996;65:563–607. doi: 10.1146/annurev.bi.65.070196.003023. [DOI] [PubMed] [Google Scholar]
- 4.Wei YH. Oxidative stress and mitochondrial DNA mutations in human aging. Proceedings of the Society for Experimental Biology and Medicine. 1998;217(1):53–63. doi: 10.3181/00379727-217-44205. [DOI] [PubMed] [Google Scholar]
- 5.Johns DR. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. New England Journal of Medicine. 1995;333(10):638–644. doi: 10.1056/NEJM199509073331007. [DOI] [PubMed] [Google Scholar]
- 6.Scheffler IE. Mitochondria make a comeback. Advanced Drug Delivery Reviews. 2001;49(1-2):3–26. doi: 10.1016/s0169-409x(01)00123-5. [DOI] [PubMed] [Google Scholar]
- 7.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283(5407):1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 8.Chinnery PF, Turnbull DM. Clinical features, investigation, and management of patients with defects of mitochondrial DNA. Journal of Neurology Neurosurgery and Psychiatry. 1997;63(5):559–563. doi: 10.1136/jnnp.63.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martínez-Fernández E, Gil-Peralta A, García-Lozano R, et al. Mitochondrial disease and stroke. Stroke. 2001;32(11):2507–2510. doi: 10.1161/hs1101.098328. [DOI] [PubMed] [Google Scholar]
- 10.Rogounovitch TI, Saenko VA, Shimizu-Yoshida Y, et al. Large deletions in mitochondrial DNA in radiation-associated human thyroid tumors. Cancer Research. 2002;62(23):7031–7041. [PubMed] [Google Scholar]
- 11.Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325(6099):31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 12.Merriwether DA, Clark AG, Ballinger SW, et al. The structure of human mitochondrial DNA variation. Journal of Molecular Evolution. 1991;33(6):543–555. doi: 10.1007/BF02102807. [DOI] [PubMed] [Google Scholar]
- 13.Macaulay V, Richards M, Hickey E, et al. The emerging tree of west Eurasian mtDNAs: a synthesis of control-region sequences and RFLPs. American Journal of Human Genetics. 1999;64(1):232–249. doi: 10.1086/302204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilal E, Rabadan R, Alexe G, et al. Mitochondrial DNA haplogroup D4a is a marker for extreme longevity in Japan. PLoS ONE. 2008;3(6) doi: 10.1371/journal.pone.0002421.e2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren WH, Li XH, Zhang HG. Mitochondrial DNA haplogroups in a Chinese Uygur population and their potential association with longevity. Clinical and Experimental Pharmacology and Physiology. 2008;35(12):1477–1481. doi: 10.1111/j.1440-1681.2008.05028.x. [DOI] [PubMed] [Google Scholar]
- 16.Takasaki S. Mitochondrial SNPs associated with Japanese centenarians, Alzheimer’s patients, and Parkinson’s patients. Computational Biology and Chemistry. 2008;32(5):332–337. doi: 10.1016/j.compbiolchem.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 17.Chinnery PF, Taylor GA, Howell N, et al. Mitochondrial DNA haplogroups and susceptibility to AD and dementia with Lewy bodies. Neurology. 2000;55(2):302–304. doi: 10.1212/wnl.55.2.302. [DOI] [PubMed] [Google Scholar]
- 18.van der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. American Journal of Human Genetics. 72(4):804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ban M, Elson J, Walton A, et al. Investigation of the role of mitochondrial DNA in multiple sclerosis susceptibility. PLoS ONE. 2008;3(8) doi: 10.1371/journal.pone.0002891.e2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magri C, Gardella R, Barlati SD, Valsecchi P, Sacchetti E, Barlati S. Mitochondrial DNA haplogroups and age at onset of schizophrenia. American Journal of Medical Genetics, Part B. 2007;144(4):496–501. doi: 10.1002/ajmg.b.30496. [DOI] [PubMed] [Google Scholar]
- 21.Rollins B, Martin MV, Sequeira PA, et al. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS ONE. 2009;4(3) doi: 10.1371/journal.pone.0004913.e4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishigaki Y, Yamada Y, Fuku N, et al. Mitochondrial haplogroup A is a genetic risk factor for atherothrombotic cerebral infarction in Japanese females. Mitochondrion. 2007;7(1-2):72–79. doi: 10.1016/j.mito.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Nishigaki Y, Yamada Y, Fuku N, et al. Mitochondrial haplogroup N9b is protective against myocardial infarction in Japanese males. Human Genetics. 2007;120(6):827–836. doi: 10.1007/s00439-006-0269-z. [DOI] [PubMed] [Google Scholar]
- 24.Fuku N, Park KS, Yamada Y, et al. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. American Journal of Human Genetics. 2007;80(3):407–415. doi: 10.1086/512202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka M, Fuku N, Nishigaki Y, et al. Women with mitochondrial haplogroup N9a are protected against metabolic syndrome. Diabetes. 2007;56(2):518–521. doi: 10.2337/db06-1105. [DOI] [PubMed] [Google Scholar]
- 26.Wang PW, Lin TK, Weng SW, Liou CW. Mitochondrial DNA variants in the pathogenesis of type 2 diabetes—relevance of asian population studies. Review of Diabetic Studies. 2009;6(4):237–246. doi: 10.1900/RDS.2009.6.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liou CW, Lin TK, Chen JB, et al. Association between a common mitochondrial DNA D-loop polycytosine variant and alteration of mitochondrial copy number in human peripheral blood cells. Journal of Medical Genetics. 2010;47(11):723–728. doi: 10.1136/jmg.2010.077552. [DOI] [PubMed] [Google Scholar]
- 28.Liou CW, Chen JB, Tiao MM. Mitochondrial DNA coding and control region variants as genetic risk factors for type 2 diabetes mellitus. Diabetes. 2012;61(10):2642–2651. doi: 10.2337/db11-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 30.Chomyn A. Platelet-mediated transformation of human mitochondrial DNA-less cells. Methods in Enzymology. 1996;264:334–338. doi: 10.1016/s0076-6879(96)64031-2. [DOI] [PubMed] [Google Scholar]
- 31.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. Journal of Biological Chemistry. 1979;254(8):2669–2676. [PubMed] [Google Scholar]
- 32.Gohil VM, Sheth SA, Nilsson R, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nature Biotechnology. 2010;28(3):249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Djafarzadeh S, Vuda M, Takala J, Ochs M, Jakob SM. Toll-like receptor-3-induced mitochondrial dysfunction in cultured human hepatocytes. Mitochondrion. 2011;11(1):83–88. doi: 10.1016/j.mito.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Granger DL, Lehninger AL. Sites of inhibition of mitochondrial electron transport in macrophage-injured neoplastic cells. Journal of Cell Biology. 1982;95(2):527–535. doi: 10.1083/jcb.95.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vrbacký M, Krijt J, Drahota Z, Mělková Z. Inhibitory effects of Bcl-2 on mitochondrial respiration. Physiological Research. 2003;52(5):545–554. [PubMed] [Google Scholar]
- 36.Kong QP, Bandelt HJ, Sun C, et al. Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Human Molecular Genetics. 2006;15(13):2076–2086. doi: 10.1093/hmg/ddl130. [DOI] [PubMed] [Google Scholar]
- 37.Miller SW, Trimmer PA, Parker WD, Jr., Davis RE. Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. Journal of Neurochemistry. 1996;67(5):1897–1907. doi: 10.1046/j.1471-4159.1996.67051897.x. [DOI] [PubMed] [Google Scholar]
- 38.Trounce I, Neill S, Wallace DC. Cytoplasmic transfer of the mtDNA nt 8993 T → G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(18):8334–8338. doi: 10.1073/pnas.91.18.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gómez-Durán A, Pacheu-Grau D, López-Gallardo E, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Human Molecular Genetics. 2010;19(17):3343–3353. doi: 10.1093/hmg/ddq246. [DOI] [PubMed] [Google Scholar]
- 40.Pello R, Martín MA, Carelli V, et al. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Human Molecular Genetics. 2008;17(24):4001–4011. doi: 10.1093/hmg/ddn303. [DOI] [PubMed] [Google Scholar]