

Abstract
Nonalcoholic fatty liver disease (NAFLD) is closely related to insulin resistance, metabolic syndrome, obesity, type 2 diabetes, and dyslipidaemia. Obesity and metabolic syndrome are associated with an increased cancer risk, and recent evidence demonstrated an association between NAFLD and colorectal cancer (CRC). The mechanism of how NAFLD can be associated with increased risk of CRC is not fully understood; however, NAFLD represents a condition of profound insulin resistance and a proinflammatory state. Insulin and insulin-like growth factors may promote the development of CRC through their proliferative and antiapoptotic effects. Patients with NAFLD have reduced expression of adiponectin, an adipokine with anti-inflammatory effects. Importantly, hypoadiponectinemia is associated with an increased risk of CRC. Decreased levels of adiponectin lead to increased insulin levels due to marked insulin resistance and in turn increased insulin growth factor-1 (IGF-1). Insulin binds to IGF-1 receptors and plays an important role in cell proliferation, apoptosis, and increased production of vascular endothelial growth factor, an angiogenic factor that supports cancer growth. Further studies are needed to establish (i) the pathophysiology of NAFLD with colorectal cancer, (ii) the benefit of early screening of CRC in NAFLD patients, and (iii) the impact of treatment of NAFLD in the modulation of the risk of colorectal cancer.
1. Introduction
Colorectal cancer (CRC) remains as one of the commonest cancers worldwide, with a million new cases diagnosed each year [1]. In the UK, there are 35,000 new cases of colorectal cancer diagnosed each year [2]. It is also the second leading cause of cancer deaths worldwide [3, 4]. Established risk factors include increased age, black race, smoking, and low fibre diet. Suggestions from data now point at obesity, insulin resistance, and the metabolic syndrome also as potential risk factors [5]. The incidence of the metabolic syndrome is increasing worldwide in overweight populations. It includes at least three of the following: increased waist circumference, hypertriglyceridaemia, low high-density lipoprotein (HDL), hypertension, and hyperglycaemia [5]. It is also stipulated that one of its hepatic manifestations, the nonalcoholic fatty liver disease (NAFLD), may be an independent risk factor for developing colorectal cancer [6, 7]. This paper highlights the association between colorectal cancer and NAFLD, the metabolic syndrome, diabetes, and hyperlipidaemia.
2. NAFLD
NAFLD is now the commonest chronic liver disease in Western populations. It is an umbrella term describing a spectrum of diseases associated with excessive fat accumulation in the liver (steatosis), in the absence of excessive alcohol consumption. The spectrum of the condition ranges from simple steatosis, to steatohepatitis (NASH), through to advanced fibrosis/cirrhosis and even hepatocellular carcinoma. The characteristic histological features of NAFLD are indistinguishable from those of alcohol-induced fatty liver disease but are also found in people who consume minimal alcohol [8]. It is regarded as the commonest cause of deranged liver enzymes in the USA and is associated with type 2 diabetes, obesity, and hyperlipidaemia [8, 9]. NAFLD and colon cancer (Figure 1).
Figure 1.
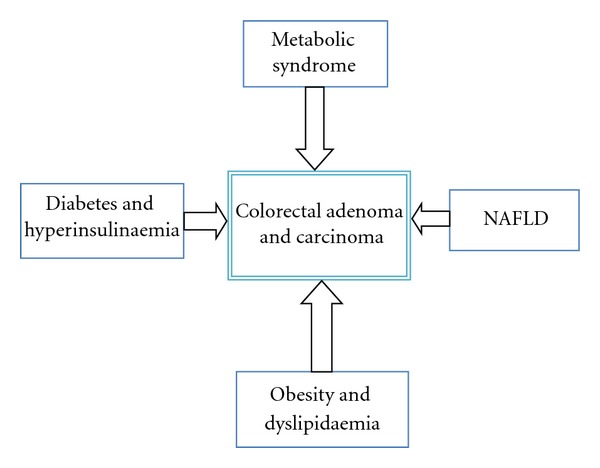
Relationship between metabolic syndrome, NAFLD, and development of colorectal adenoma and carcinoma.
It is defined as the accumulation of liver fat by >5% per liver weight, in the presence of <10 g of daily alcohol consumption [10–12]. It is very common and occurs in individuals of all ages and ethnic groups [13]. The estimated prevalence of NAFLD in USA is thought to be around 34%, and interestingly 90% of these cases are attributed largely to obesity and diabetes [12]. The Dallas Heart Study which was based on a large, multiethnic probability-based population reported that the prevalence of NAFLD is around 33.6% [14]. Additionally, the use of ultrasound in the Dionysos nutrition and liver disease study in Italy showed that the prevalence of steatosis was increased in heavy drinkers and obese persons. This study concluded that steatosis is frequently encountered in healthy people and is usually present in the obese who drink more than 60 g of alcohol per day. Steatosis is more strongly associated with obesity than with heavy drinking, suggesting a greater role of obesity than alcohol consumption in the accumulation of fat in the liver [15].
NAFLD is associated with morbid obesity, type 2 diabetes, and hyperlipidaemia; however, excess alcohol intake is still the main cause of fatty liver disease. The increase in prevalence of the epidemic of type 2 diabetes and obesity is likely to increase the prevalence of NAFLD in the near future with serious financial implications for health care providers [16, 17]. A failure in diagnosing NAFLD may lead to an insidious progression of the condition which may result in cirrhosis, portal hypertension, and liver-related death in early adulthood. Importantly, NAFLD is associated with an increased risk of all-cause death and predicts future CVD events, independently of age, sex, LDL cholesterol, smoking, and the cluster of features of the metabolic syndrome [12]. NAFLD is regarded as the hepatic manifestation of the metabolic syndrome, an entity associated with colorectal carcinogenesis [16]. The commonest risk factors for acquiring NAFLD are obesity, diabetes, and hyperlipidaemia [8]. The reported prevalence of obesity with NAFLD varies between 30% and 100%, whereas the prevalence of NAFLD with type 2 diabetes varies between 10% and 75% [16].
The lack of sensitive and specific biochemical markers have attracted a lot research interest, as it is predicted that mass screening for significant liver injury in patients with NAFLD may play an important role in the years to come because of the increase in obesity and diabetes [18].
3. NAFLD and Colorectal Cancer
Much work has gone into the relationship between NAFLD and colorectal cancer. One of these studies was carried out by Adams et al., who looked at the impact of NAFLD on mortality among 337 patients with type 2 diabetes followed for 9 years [19]. Among these, 116 were diagnosed with NAFLD 0.9 ± 4.6 years after diabetes was diagnosed. Those found to have developed NAFLD were younger and more likely to be female and obese. In multivariate analysis, they showed that beside increased risk of IHD and diabetes, NAFLD is associated with an increased risk of dying from malignancy (HR 2.3; 95% CI 0.9, 5.9; P = 0.09) [20]. In context, a diagnosis of NAFLD should be taken seriously, and steps to exclude malignancy should be taken. Treatment for all its other associated complications should be also addressed.
Hwang et al. presented the first evidence that demonstrated an association between NAFLD and an increased rate of colorectal adenomatous polyps. In their study, a population of 2917 participants were investigated via colonoscopy, abdominal ultrasonography, and liver tests. The prevalence of NAFLD was 41.5% in the adenomatous polyp group versus 30.2% in the control group providing the first evidence that an association may exist. By multiple logistic regression analysis, NAFLD was found to be associated with an increased risk of colorectal adenomatous polyps (odds ratio, 1.28; 95% confidence interval, 1.03–1.60). Interestingly, an increased risk for NAFLD was evident in patients with a greater number of adenomatous polyps. If left untreated, these polyps develop into colorectal cancer (adenoma-carcinoma sequence) [7].
Furthermore, Wong et al. showed that in a cross-sectional study of 40–70 year olds (N = 199), NAFLD patients (N = 199) had a higher prevalence of colorectal adenomas (34.7% versus 21.5%; P = 0.043) and advanced neoplasms (18.6% versus 5.5%; P = 0.002). Among patients with biopsy-proven NAFLD, patients with NASH (N = 49) had a higher prevalence of adenomas (51.0% versus 25.6%; P = 0.005) and advanced neoplasms (34.7% versus 14.0%; P = 0.011) than those with simple steatosis (N = 86). After adjusting for demographic and metabolic factors, NASH remained associated with adenomas (adjusted OR 4.89, 95% CI 2.04 to 11.70) and advanced neoplasms (OR 5.34, 95% CI 1.92 to 14.84) [21]. This may suggest that NASH is associated with higher risk of colorectal adenoma, demonstrating a correlation between histological severity with colorectal cancer, adding further fuel to the relationship between NAFLD and colorectal cancer, and suggesting the potential benefit of colorectal cancer screening in this group.
In a large retrospective cohort study of Korean women, Lee et al. found a significant relationship between NAFLD and colorectal neoplasia. The study included data from 5517 women, aged 35–80 years, who underwent life insurance health examination between 2002 and 2006 at their institution. Of these subjects, 15.1% (833 women) had NAFLD. During followup, 65 women were found to have adenomatous polyps, and 15 women were found to have colorectal cancer. Furthermore, the risk of adenomatous polyps in patients with NAFLD was twice that of patients without NAFLD, and adjusted RRs (95% CI) for adenomatous polyps by age, low HDL-cholesterol, and NAFLD were 1.12 (95% 1.09–1.15), 2.56 (95% CI 1.53–4.28), and 1.94 (95% CI 1.11–3.40). There was a threefold increase in the risk of colorectal cancer in patients with NAFLD compared to the healthy subjects, and adjusted RRs (95% CI) for colorectal cancer by age and NAFLD were 1.23 (95% CI 1.17–1.29) and 3.08 (95% CI 1.02–9.34). However, the presence of NAFLD had no influence on the prognosis of CRC and in particular with disease recurrence during followup [22].
In another study, 1200 patients (603 males and 608 females) underwent screening colonoscopy. Stadlmayr et al. showed that the patients with NAFLD had significantly more colorectal adenomas and early colorectal cancers compared to those without NAFLD [23]. The mean age of the patients in the study was approximately 60 years. NAFLD was diagnosed in 367 (60.8%) in the male group and in 265 (43.5%) in the female group. The rate of colorectal adenoma was significantly higher in patients with NAFLD. In the male group with NAFLD, the rate of adenomas was 243/367 versus 107/236, P = 0.010. In the female group with NAFLD, the rate of adenomas was 96/265 versus 78/343 in women without NAFLD, P = 0.014. However, significantly more colorectal cancers were observed in the males with NAFLD, 6/367 versus 1/236, P < 0.001, and not in the female group with NAFLD. Multivariate regression analyses demonstrated an independent association of colorectal adenomas with NAFLD after adjustment of age, sex, body mass index (BMI), and glucose intolerance (OR 1.47; 95% CI 1.079–2.003, P = 0.015). Overall, the males with NAFLD had more tubular adenomas, rectal adenomas, and more cancers, whilst the females with NAFLD were shown to have more tubular adenomas and adenomas of the proximal colon.
In contrast, Touzin et al. have failed to demonstrate an increase in incidence of colorectal adenomas in patients with NAFLD compared to those without [1]. In this retrospective observational study of smaller number of patients of 233 who underwent screening colonoscopy, they compared patients with sonographic imaging and biopsy-proven simple steatosis (n = 65), or non-alcoholic steatohepatitis (NASH), (n = 29), to a control group of 139 patients without NALFD on sonographic imaging [1]. The mean age of the patients was 55 years of whom 48% were women. The prevalence of adenomas was 35/139 (25.1%) in the control group compared to 23/94 (24.4%) in patients with NAFLD, P = 1.00. After adjustment of the known cofounder to include race, BMI, and family history, no significant difference was found in the two groups, P = 0.33. Interestingly, the mean number of adenomas was significantly higher in patients with NAFLD compared to the control group, P = 0.016. Compared to the previous study, this study has a smaller number of patients, 233 versus 1200 patients. The patients in this group were younger, 55 years compared to 65 years in the study by Stadlmayr et al. [23].
The proposed mechanism for how NAFLD influences carcinogenesis is not completely understood (Figure 2). It is known, however, that NAFLD represents a condition of profound insulin resistance and a proinflammatory state [24]. Insulin and insulin-like growth factors may promote the development of colorectal neoplasm through their proliferative and antiapoptotic effects. Patients with NAFLD have reduced expression of adiponectin, an adipokine with anti-inflammatory effects [25, 26]. This is also the case in the obese, diabetic, and those with insulin resistance [27].
Figure 2.
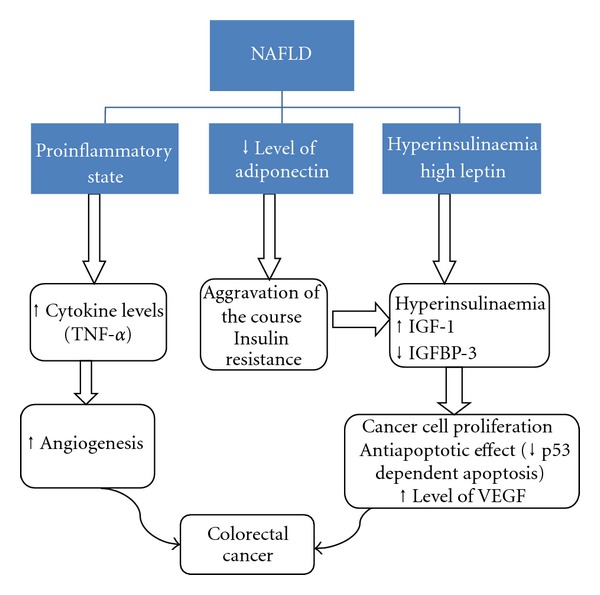
A proposed mechanism of how metabolic syndrome and NAFLD influence carcinogenesis in colorectal cancer. MS: metabolic syndrome; ILGF-1: insulin-like growth factor-1; IGFBP-3: insulin growth factor binding protein-3; VEGF: vascular endothelial growth factor; TNF: tumour necrosis factor.
Decreased levels of adiponectin lead to increased insulin levels due to marked insulin resistance and in turn increased insulin growth factor-1 (IGF-1). Insulin binds to IGF-1 receptors and plays an important role in cell proliferation, apoptosis, and increased production of vascular endothelial growth factor, an angiogenic factor that supports cancer growth [1]. The new anticancer antibody Bevacizumab is human monoclonal antibody that inhibits vascular endothelial growth factor A and is licensed for use in metastatic colorectal cancer.
Studies in humans have shown an association between hypoadiponectinemia and an increased risk of colorectal adenomas [28]. Adiponectin inhibits colorectal cancer cell growth in vitro [29], and mice with hypoadiponectinaemia had increased risk of colonic polyps when exposed to a high-fat diet [30]. Mediators derived mainly from adipose tissue such as adiponectin or leptin could be critically involved in such processes, and, therefore, these so-called adipocytokines might reflect attractive candidates linking obesity-related disorders with tumour development both intra- and extrahepatically [6]. It is well established that those with NAFLD, especially NASH, have significantly lower levels of serum adiponectin compared to healthy controls [26].
Indeed, an association of obesity and hypoadiponectinaemia with colorectal adenomas and cancer has been shown in men [31]. Another study demonstrated an inverse correlation of total and high molecular weight adiponectin serum levels with development of colorectal adenomas [28]. It demonstrates anticarcinogenic effects in vitro through the inhibition of colon cancer cell growth via the stimulation of AMP-activated protein kinase activity [32]. This has been further supported by mice tumour model which also demonstrated that adiponectin significantly stopped the growth of the primary tumour in a caspase-dependent manner resulting in endothelial cell apoptosis [33].
According to Ferroni et al. and Rose et al., adiponectin also directly inhibits tumour necrosis factor alpha (TNF-α), a mediator known to play a role in tumour cell proliferation and angiogenesis [34, 35]. Low adiponectin levels were shown to be inversely related to colonic tumour stage and independently predicted cancer recurrence [36].
Inflammatory cytokines may also play a role in colorectal cancer and are related to the metabolic syndrome via adipocyte secretion of various cytokines, including TNF-α [37, 38]. Through hypoxia, adipocytes would then secrete cytokines which stimulate angiogenesis into the adipose cells, and these cytokines in turn also promote insulin resistance and increase circulating triglycerides. This pro-inflammatory state influences growth, apoptosis and tumour-cell proliferation in many cancers [39–41].
Many studies have also investigated the effects of leptin, another adipocytokine dysregulated in obesity and NAFLD, in experimental cellular and animal models [42]. These suggest that leptin can induce carcinogenesis, whilst adiponectin seems to decrease cell proliferation. It is purported that through its interference with leptin, adiponectin may have an anti-carcinogenic effect, while leptin would exert a carcinogenic state under low adiponectin level [6].
The relationship between NAFLD and colonic carcinogenesis emphasises the importance of a healthy lifestyle to prevent and treat the metabolic syndrome and its hepatic manifestation. Since NAFLD and colorectal neoplasms share many common risk factors, Wong et al. postulated that NAFLD patients have an increased risk of colorectal neoplasms and should be a target group for screening. Colorectal cancer is a leading cause of cancer-related deaths worldwide, and screening would reduce the incidence and mortality of colorectal cancer [21].
They strongly advocated screening in NASH patients as proximal colonic lesions are common in these patients, and colonoscopy would be considered as the preferred screening method. Furthermore, patients with metabolic syndrome are more likely to develop cancer in the proximal colon and may only be detected by full colonoscopy [21, 43]. It has recently been shown that in average-risk individuals, 40–49 years of age, men with abdominal obesity or metabolic syndrome might benefit from screening colonoscopy starting at 45 years of age to detect colonic neoplasms. Gut presented data which would favour early screening in NASH populations. The presence of NASH might be an important prognostic factor in liver disease and additionally in extra hepatic prognosis, that is, cancer development [6].
4. The Metabolic Syndrome and Colorectal Cancer
The metabolic syndrome and visceral obesity have an increasing prevalence and incidence in the general population, with an estimated 24.6% and 30.9% in Europe [44]. Novel studies have shown link between metabolic syndrome and the development of colorectal cancer. Several clinical features of the metabolic syndrome, including obesity, dyslipidaemia, and impaired glucose tolerance, have been linked to an increased risk for colorectal cancer in several recent epidemiological studies [44].
The mechanism by how it does this is mainly related to abdominal obesity and insulin resistance, with studies demonstrating that carcinogenesis is linked to hyperinsulinaemia, increased C-peptide, increased BMI, high levels of IGF-1, low levels of insulin growth factor binding protein-3, high leptin levels, and low adiponectin levels [44]. In a nested control study that included 1093 individuals, among individual components of the metabolic syndrome, abdominal obesity (RR = 1.51; 95% CI: 1.16–1.96) was associated with colon cancer, whereas abnormal glucose metabolism was associated with both colon (RR = 2.05; 95% CI: 1.57–2.68) and rectal cancers (RR = 2.07; 95% CI: 1.45–2.96) [45]. In another study in the USA of 900,000 individuals who had undergone 16-year follow up, they found 57,145 cancer-related deaths [46]. They found that for all cancers, there was a trend of increasing death rate with increased BMI, and the relative risk of colorectal cancer showed a dose response to an increasing BMI as well. A further study by Ahmed et al. of 14,109 individuals identified 194 colorectal cancers and found that after adjusting for age and gender, they found that baseline metabolic syndrome (>3 components versus 0 components) had a positive association with colorectal cancer incidence [47]. After multivariate adjustment, they found a dose response association between colorectal cancer incidence and the number of metabolic syndrome components. This was reenforced in a study which suggested that the effects of the individual components of the metabolic syndrome may be additive, as they demonstrated that the relative risk of death from colorectal cancer was increased in cluster analysis compared to glucose level alone [48]. Out of the individual components, only “glucose level” was associated with an increased risk of colorectal cancer death, and only if both genders were combined. The relationship between individual components of metabolic syndrome and the risk of colorectal cancer has been analysed in other studies. One such study was carried out by Colangelo et al. [49] who found that there was a 35% increased risk of colorectal cancer associated with high blood pressure and also found that clustering of features of the metabolic syndrome significantly increased the risk of associated colorectal cancer. Furthermore, Kabat et al. showed that an increase in glucose level and systolic blood pressure is also associated with high risk of colon cancer [50].
5. Diabetes and Insulin Resistance and Colorectal Cancer
There is increasing evidence that abnormal glucose metabolism is associated with an increased risk of colorectal cancer [51, 52]. There is an extensive body of research examining the relationship between diabetes and colorectal cancer, and findings have so far varied, with relative risks of colorectal cancer ranging from 1.2 to 2.8 in prospective cohort studies [48]. Studies have positively correlated colorectal cancer with C-peptide levels (insulin-release marker), 2-hour postchallenge insulin and glucose concentrations, and haemoglobin A1C levels [48]. In one study, greater median fasting insulin values (14–400 IU/mL) were associated with a 1.6-fold increased risk of colorectal cancer compared to values of 3–13 IU/mL [53]. In another study, there was also a weak positive association between incidental colorectal cancer and fasting insulin level [48].
Khaw et al. examined the relationship between known diabetes and glycated haemoglobin (HbA1c) concentrations measured in 9,605 men and women and subsequent incident colorectal cancer after 6 years follow up in the European Prospective Investigation into Cancer-Norfolk Study [51, 52, 54]. Their main finding is that the risk of colorectal cancer increased after HbA1c concentrations above 7%. Additionally, they found that an absolute increase of 1% in HbA1c was associated with a 33% increase in risk of colorectal cancer. Interestingly, when both HbA1c and known diabetes were included in the same multivariate model, glycated HbA1c remained a significant risk predictor, but the effect of diabetes status was no longer significant. They suggested that the increased risk of colorectal cancer in diabetics was mediated via the increased HbA1c levels. This relationship between colorectal cancer and HbA1c was also present in the CLUE II cohort [52, 54].
Several studies have reported stronger associations between impaired glucose tolerance and cancer than diabetes with cancer [51]. Saydah et al. reported a significant risk of colorectal cancer mortality with impaired glucose tolerance, but not diabetes (RR 1.87 versus 1.13) [53]. Overall findings of diabetes association with colorectal cancer have not been entirely consistent [51]. Some have suggested that this may be due to the alternating course of diabetes, with insulin resistance initially associated with hyperinsulinaemia, and in latter stages it leads to hypoinsulinaemia secondary to B-cell failure. This variability in severity and duration may explain the inconsistencies observed in studies associating diabetes with cancer [51].
There are also studies which have refuted this association. A metaanalysis by Mulholland et al. looked at the association between diets with a high glycaemic index (GI) and high glycaemic load (GL), with the risk of digestive tract cancers [55]. They reported that pooled cohort studies failed to demonstrate an association between colorectal cancer risk and high glycaemic intake. They did report, however, that two studies observed positive associations between GI and GL intake with colorectal cancer risk in those with an above-normal BMI. The impact of BMI has been shown to be inconsistent, with other studies failing to show such an association.
Insulin has been shown to affect the growth of both normal and neoplastic epithelial cells and to have the capability of promoting mitosis in vitro, directly or indirectly via IGF-1. It has been proposed that insulin and insulin-like growth factors may be involved in colon carcinogenesis [45, 48]. Insulin acts as a growth factor, and insulin receptors are present in normal and malignant colorectal cells. This action would be potentiated by hyperglycaemia which would induce secretion of insulin [48].
Hyperinsulinaemia may also affect the development of colorectal cancer, indirectly by downregulating insulin-like growth factor binding proteins (IGFBP-1, 2, 3), thus enhancing IGF-1 bioavailability [56]. This also happens via insulin binding to IGF-1 receptors (IGF1Rs) [55]. IGF-1 which is an important mitogen for progression through the cell cycle having autocrine, paracrine, and endocrine actions on cell proliferation and apoptosis, leads to increased cell proliferation, inhibition of apoptosis and promotion of tumour growth [57]. IGF-1 also increases the production of vascular endothelial growth factor (VEGF), an angiogenic factor that may support cancer growth [58].
The association of IGF-1 and IGFBP-3 levels with their effects on increased risk of colorectal adenoma/cancer initially came into knowledge through acromegalics, which have chronically increased growth hormone (GH) levels. Excess GH leads to hepatic and peripheral insulin resistance leading to excess insulin, causing IGF-1 hypersecretion and low IGFBP-3 levels [59].
Giovannucci et al. studied the relationships between IGF-1 and IGFBP-3 levels and colorectal cancer in a study of 32,826 women [60]. They showed that those with higher levels of IGF-1 were at increased risk of intermediate/late-stage colorectal neoplastic adenoma and cancer. The women with higher levels of IGFBP-3 were at lower risk of intermediate/late-stage colorectal adenoma and cancer, indicating opposing effects of IGF-1 and IGFBP-3 to colorectal cancer.
William et al. analysed the independent role of IGFBP-3 in colorectal cancer [61]. IGFBP-3 has been shown to enhance p53-dependent apoptosis after DNA damage. Hence, loss of IGFBP-3 could contribute to the development of colorectal adenomas that retain wild-type p53 function through the suppression of p53-dependent apoptotic signals, promoting survival of abnormal cells and tumourigenesis. These studies suggest that IGFBP-3 may have an important role in the regulation of differentiation and apoptosis in human colonic epithelium.
6. Obesity and Dyslipidaemia and Colorectal Cancer
A high body mass index (BMI) has been shown to be associated with an increased mortality not only from atheromatous disease, but also from some cancers [62]. There is a plethora of evidence to suggest an increased risk of colorectal cancer in the obese, and obesity has been included as an important risk factor in the recent guidelines from the American College of Gastroenterology for screening colorectal cancer. It has a direct and independent relationship with colorectal cancer, though the association seems to be weaker than previously reported [63]. A study suggests that obesity and weight gain may induce early colorectal carcinogenesis in women especially those arising in the distal colon [64]. Obesity is the usual cause for the development of NAFLD, an entity that has been linked to both extrahepatic and colorectal cancers [6].
In a study of 600 patients with a median age of 56, 185 patients were classified as obese, and of these there was a 44.3% prevalence of “any” adenomas and a 13% prevalence for advanced neoplasia after undergoing colonoscopy. After multivariate analysis, obesity (BMI ≥ 30) was significantly associated with an increased risk of advanced neoplasia (odds ratio (OR) = 3.83; 95% CI 1.94–7.55) [65]. Furthermore, the increase in waist circumference and BMI was associated with an increase in risk of colorectal cancer [66]. Wang et al. showed that in both men and women, waist circumference was associated with increased colorectal incidence. High BMI levels were also associated with an increased risk. However, after adjustment for BMI, waist circumference remained associated with a nonstatistically significant increase in colorectal cancer incidence in both genders. The conclusion was that increased incidence of colorectal cancer due to an increased waist circumference may be partially independent of BMI [66]. For colorectal cancer, the RR of death varied from 1.34 (95% CI: 0.94–1.34), for a BMI of 25–29.9, to 1.90 (95% CI: 1.46–2.47) and 4.52 (95% CI: 2.94–6.94), for a BMI between 30.0–34.9 and 35.0–39.9, respectively. The relationship between BMI and colon cancer was also studied in the EPIC study, which was based on 984 cases of colon cancer [67]. In a retrospective study of 7566 subjects by Moore et al., 306 cases of incident colorectal cancer were found. They showed a twofold increased risk of colorectal cancer for waist circumference of >99 cm in women and >101 cm in men, and this risk increased linearly with increased waist-circumference [67]. Another suggestion is that visceral adipose tissue rather than whole body adipose tissue correlates better with the risk of colorectal adenoma [67].
Furthermore, in a large prospective study, high circulating triglycerides were associated with a nonsignificant twofold elevation in risk of colorectal cancer in men, but no clear association was observed in women [48]. In addition, hypertriglyceridemia and hyperglycaemia were associated with colorectal carcinoma [68, 69]. However, Liu et al. showed that central obesity and dyslipidaemia are independent risk factors for colorectal cancer [70]. Importantly, meta-analysis of 18 studies involving more than 1.5 million patients showed that statins treatment is not associated with significant decrease in risk of colorectal cancer [71]. Therefore, we recommend that further research is urgently needed to establish the role of hyperlipidaemia as risk factor for colorectal cancer.
7. Conclusion
NAFLD is hepatic component of the metabolic syndrome. NAFLD is associated with an increase in risk of colorectal cancer. Additional prospective studies are needed to better define a true association between NAFLD and adenomatous colon polyps. Therefore, it is good clinical practice to exclude the presence of colon cancer in individuals with long-standing NAFLD. Furthermore, several studies suggest that metabolic syndrome and its components are associated with increased risk for colonic neoplasm. This relationship has an important impact on society and further emphasizes the importance of a healthy lifestyle to prevent and treat metabolic syndrome and its hepatic manifestation, NAFLD. It is not yet clear what an impact of the treatment of NAFLD on modulation of the risk of colon cancer is.
References
- 1.Touzin NT, Bush KNV, Williams CD, Harrison SA. Prevalence of colonic adenomas in patients with nonalcoholic fatty liver disease. Therapeutic Advances in Gastroenterology. 2011;4(3):169–176. doi: 10.1177/1756283X11402118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thorne AL, Mercer SJ, Harris GJC, Simson JNL. Reduction in late diagnosis of colorectal cancer following introduction of a specialist colorectal surgery service. Annals of the Royal College of Surgeons of England. 2006;88(6):562–565. doi: 10.1308/003588406X130688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards BK, Brown ML, Wingo PA, et al. Annual report to the nation on the status of cancer, 1975-2002, featuring population-based trends in cancer treatment. Journal of the National Cancer Institute. 2005;97(19):1407–1427. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- 4.Byeon JS, Yang SK, Kim TI, et al. Colorectal neoplasm in asymptomatic Asians: a prospective multinational multicenter colonoscopy survey. Gastrointestinal Endoscopy. 2007;65(7):1015–1022. doi: 10.1016/j.gie.2006.12.065. [DOI] [PubMed] [Google Scholar]
- 5.Reilly MP, Rader DJ. The metabolic syndrome: more than the sum of its parts? Circulation. 2003;108(13):1546–1551. doi: 10.1161/01.CIR.0000088846.10655.E0. [DOI] [PubMed] [Google Scholar]
- 6.Tilg H, Diehl AM. NAFLD and extrahepatic cancers: have a look at the colon. Gut. 2011;60(6):745–746. doi: 10.1136/gut.2011.239392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang ST, Cho YK, Park JH, et al. Relationship of non-alcoholic fatty liver disease to colorectal adenomatous polyps. Journal of Gastroenterology and Hepatology. 2010;25(3):562–567. doi: 10.1111/j.1440-1746.2009.06117.x. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed MH, Byrne CD. The Metabolic Syndrome. Wiley-Blackwell; 2011. Non-alcoholic fatty liver disease; pp. 245–277. [Google Scholar]
- 9.Ahmed MH, Byrne CD. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD) Drug Discovery Today. 2007;12(17-18):740–747. doi: 10.1016/j.drudis.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed MH, Abu EO, Byrne CD. Non-Alcoholic Fatty Liver Disease (NAFLD): new challenge for general practitioners and important burden for health authorities? Primary Care Diabetes. 2010;4(3):129–137. doi: 10.1016/j.pcd.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed MH, Byrne CD. Obstructive sleep apnea syndrome and fatty liver: association or causal link? World Journal of Gastroenterology. 2010;16(34):4243–4252. doi: 10.3748/wjg.v16.i34.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrne CD, Olufad R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clinical Science. 2009;116(7):539–564. doi: 10.1042/CS20080253. [DOI] [PubMed] [Google Scholar]
- 13.Giovannucci E. Metabolic syndrome, hyperinsulinemia, and colon cancer: a review. The American Journal of Clinical Nutrition. 2007;86(3):s836–842. doi: 10.1093/ajcn/86.3.836S. [DOI] [PubMed] [Google Scholar]
- 14.Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. American Journal of Physiology. 2005;288(2):E462–E468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 15.Bellentani S, Saccoccio G, Masutti F, et al. Prevalence of and risk factors for hepatic steatosis in northern Italy. Annals of Internal Medicine. 2000;132(2):112–117. doi: 10.7326/0003-4819-132-2-200001180-00004. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed MH, Byrne CD. Metabolic syndrome, diabetes & CHD risk. In: Packard CJ, editor. The Year in Lipid Disorders. Oxford, UK: Clinical Publishing; 2007. pp. 3–26. [Google Scholar]
- 17.Ahmed MH, Byrne CD. Current treatment of non-alcoholic fatty liver disease. Diabetes, Obesity and Metabolism. 2009;11(3):188–195. doi: 10.1111/j.1463-1326.2008.00926.x. [DOI] [PubMed] [Google Scholar]
- 18.Ahmed MH. Biochemical markers: the road map for the diagnosis of nonalcoholic fatty liver disease. American Journal of Clinical Pathology. 2007;127(1):20–22. doi: 10.1309/JXWUM661T8VT1ETX. [DOI] [PubMed] [Google Scholar]
- 19.Adams LA, Lymp JF, Sauver JS, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 20.Adams LA, Harmsen S, Sauver JLS, et al. Nonalcoholic fatty liver disease increases risk of death among patients with diabetes: a community-based cohort study. American Journal of Gastroenterology. 2010;105(7):1567–1573. doi: 10.1038/ajg.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong VWS, Wong GLH, Tsang SWC, et al. High prevalence of colorectal neoplasm in patients with non-alcoholic steatohepatitis. Gut. 2011;60(6):829–836. doi: 10.1136/gut.2011.237974. [DOI] [PubMed] [Google Scholar]
- 22.Lee YI, Lim Y-S, Park HS. Colorectal neoplasms in relation to non-alcoholic fatty liver disease in Korean women: a retrospective cohort study. Journal of Gastroenterology and Hepatology. 2012;27(1):91–95. doi: 10.1111/j.1440-1746.2011.06816.x. [DOI] [PubMed] [Google Scholar]
- 23.Stadlmayr A, Aigner E, Steger B, et al. Nonalcoholic fatty liver disease: an independent risk factor for colorectal neoplasia. Journal of Internal Medicine. 2011;270(1):41–49. doi: 10.1111/j.1365-2796.2011.02377.x. [DOI] [PubMed] [Google Scholar]
- 24.Wong VW, Hui AY, Tsang SW, et al. Metabolic and Adipokine Profile of Chinese Patients With Nonalcoholic Fatty Liver Disease. Clinical Gastroenterology and Hepatology. 2006;4(9):1154–1161. doi: 10.1016/j.cgh.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 25.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology. 2004;40(1):46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 26.Wong VWS, Wong GLH, Tsang SWC, et al. Genetic polymorphisms of adiponectin and tumor necrosis factor-alpha and nonalcoholic fatty liver disease in Chinese people. Journal of Gastroenterology and Hepatology. 2008;23(6):914–921. doi: 10.1111/j.1440-1746.2008.05344.x. [DOI] [PubMed] [Google Scholar]
- 27.Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends in Endocrinology and Metabolism. 2010;21(6):345–352. doi: 10.1016/j.tem.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaji T, Iwasaki M, Sasazuki S, Tsugane S. Interaction between adiponectin and leptin influences the risk of colorectal adenoma. Cancer Research. 2010;70(13):5430–5437. doi: 10.1158/0008-5472.CAN-10-0178. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama M, Takahashi H, Hosono K, et al. Adiponectin inhibits colorectal cancer cell growth through the AMPK/mTOR pathway. International Journal of Oncology. 2009;34(2):339–344. [PubMed] [Google Scholar]
- 30.Fujisawa T, Endo H, Tomimoto A, et al. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut. 2008;57(11):1531–1538. doi: 10.1136/gut.2008.159293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS. Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. Journal of the National Cancer Institute. 2005;97(22):1688–1694. doi: 10.1093/jnci/dji376. [DOI] [PubMed] [Google Scholar]
- 32.Kim AY, Lee YS, Kim KH, et al. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Molecular Endocrinology. 2010;24(7):1441–1452. doi: 10.1210/me.2009-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bråkenhielm E, Veitonmäki N, Cao R, et al. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(8):2476–2481. doi: 10.1073/pnas.0308671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferroni P, Palmirotta R, Spila A, et al. Prognostic significance of adiponectin levels in non-metastatic colorectal cancer. Anticancer Research. 2007;27(1 B):483–489. [PubMed] [Google Scholar]
- 35.Rose DP, Komninou D, Stephenson GD. Obesity, adipocytokines, and insulin resistance in breast cancer. Obesity Reviews. 2004;5(3):153–165. doi: 10.1111/j.1467-789X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 36.Stattin P, Lukanova A, Biessy C, et al. Obesity and colon cancer: does leptin provide a link? International Journal of Cancer. 2004;109(1):149–152. doi: 10.1002/ijc.11668. [DOI] [PubMed] [Google Scholar]
- 37.Cowey S, Hardy RW. The metabolic syndrome: a high-risk state for cancer? American Journal of Pathology. 2006;169(5):1505–1522. doi: 10.2353/ajpath.2006.051090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. British Journal of Nutrition. 2004;92(3):347–355. doi: 10.1079/bjn20041213. [DOI] [PubMed] [Google Scholar]
- 39.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pittas AG, Joseph NA, Greenberg AS. Adipocytokines and insulin resistance. Journal of Clinical Endocrinology and Metabolism. 2004;89(2):447–452. doi: 10.1210/jc.2003-031005. [DOI] [PubMed] [Google Scholar]
- 41.Sonnenberg GE, Krakower GR, Kissebah AH. A novel pathway to the manifestations of metabolic syndrome. Obesity Research. 2004;12(2):180–186. doi: 10.1038/oby.2004.24. [DOI] [PubMed] [Google Scholar]
- 42.Procaccini C, Galgani M, de Rosa V, et al. Leptin: the prototypic adipocytokine and its role in nafld. Current Pharmaceutical Design. 2010;16(17):1902–1912. doi: 10.2174/138161210791208884. [DOI] [PubMed] [Google Scholar]
- 43.Chiu HM, Lin JT, Shun CT, et al. Association of metabolic syndrome with proximal and synchronous colorectal neoplasm. Clinical Gastroenterology and Hepatology. 2007;5(2):221–229. doi: 10.1016/j.cgh.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Pais R, Silaghi H, Silaghi AC, Rusu ML, Dumitrascu DL. Metabolic syndrome and risk of subsequent colorectal cancer. World Journal of Gastroenterology. 2009;15(41):5141–5148. doi: 10.3748/wjg.15.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aleksandrova K, Boeing H, Jenab M, et al. Metabolic syndrome and risks of colon and rectal cancer: the european prospective investigation into cancer and nutrition study. Cancer Prevention Research. 2011;4(11):1873–1883. doi: 10.1158/1940-6207.CAPR-11-0218. [DOI] [PubMed] [Google Scholar]
- 46.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. Adults. New England Journal of Medicine. 2003;348(17):1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed RL, Schmitz KH, Anderson KE, Rosamond WD, Folsom AR. The metabolic syndrome and risk of incident colorectal cancer. Cancer. 2006;107(1):28–36. doi: 10.1002/cncr.21950. [DOI] [PubMed] [Google Scholar]
- 48.Trevisan M, Liu J, Muti P, Misciagna G, Menotti A, Fucci F. Markers of insulin resistance and colorectal cancer mortality. Cancer Epidemiology Biomarkers and Prevention. 2001;10(9):937–941. [PubMed] [Google Scholar]
- 49.Colangelo LA, Gapstur SM, Gann PH, Dyer AR, Liu K. Colorectal cancer mortality and factors related to the insulin resistance syndrome. Cancer Epidemiology Biomarkers and Prevention. 2002;11(4):385–391. [PubMed] [Google Scholar]
- 50.Kabat GC, Kim MY, Peters U, et al. A longitudinal study of the metabolic syndrome and risk of colorectal cancer in postmenopausal women. European Journal of Cancer Prevention. 2012;21(4):326–332. doi: 10.1097/CEJ.0b013e32834dbc81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu Y-C, Chiu H-M, Liou J-M, et al. Glycated hemoglobin A1c is superior to fasting plasma glucose as an independent risk factor for colorectal neoplasia. Cancer Causes and Control. 2012;23(2):321–328. doi: 10.1007/s10552-011-9880-y. [DOI] [PubMed] [Google Scholar]
- 52.Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N. Preliminary communication: glycated hemoglobin, diabetes, and incident colorectal cancer in men and women: a prospective analysis from the European Prospective Investigation into Cancer-Norfolk Study. Cancer Epidemiology Biomarkers and Prevention. 2004;13(6):915–919. [PubMed] [Google Scholar]
- 53.Schoen RE, Tangen CM, Kuller LH, et al. Increased blood glucose and insulin, body size, and incident colorectal cancer. Journal of the National Cancer Institute. 1999;91(13):1147–1154. doi: 10.1093/jnci/91.13.1147. [DOI] [PubMed] [Google Scholar]
- 54.Saydah SH, Platz EA, Rifai N, Pollak MN, Brancati FL, Helzlsouer KJ. Association of markers of insulin and glucose control with subsequent colorectal cancer risk. Cancer Epidemiology Biomarkers and Prevention. 2003;12(5):412–418. [PubMed] [Google Scholar]
- 55.Mulholland HG, Murray LJ, Cardwell CR, Cantwell MM. Glycemic index, glycemic load, and risk of digestive tract neoplasms: a systematic review and meta-analysis. American Journal of Clinical Nutrition. 2009;89(2):568–576. doi: 10.3945/ajcn.2008.26823. [DOI] [PubMed] [Google Scholar]
- 56.Sandhu MS, Dunger DB, Giovannucci EL. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. Journal of the National Cancer Institute. 2002;94(13):972–980. doi: 10.1093/jnci/94.13.972. [DOI] [PubMed] [Google Scholar]
- 57.Grimberg A, Cohen P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. Journal of Cellular Physiology. 2000;183(1):1–9. doi: 10.1002/(SICI)1097-4652(200004)183:1<1::AID-JCP1>3.0.CO;2-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warren RS, Yuan H, Matli MR, Ferrara N, Donner DB. Induction of vascular endothelial growth factor by insulin-like growth factor 1 in colorectal carcinoma. Journal of Biological Chemistry. 1996;271(46):29483–29488. doi: 10.1074/jbc.271.46.29483. [DOI] [PubMed] [Google Scholar]
- 59.Jenkins PJ. Acromegaly and cancer. Hormone Research. 2004;62(supplement 1):108–115. doi: 10.1159/000080768. [DOI] [PubMed] [Google Scholar]
- 60.Giovannucci E, Pollak MN, Platz EA, et al. A prospective study of plasma insulin-like growth factor-1 and binding protein-3 and risk of colorectal neoplasia in women. Cancer Epidemiology Biomarkers and Prevention. 2000;9(4):345–349. [PubMed] [Google Scholar]
- 61.Williams AC, Collard TJ, Perks CM, et al. Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Research. 2000;60(1):22–27. [PubMed] [Google Scholar]
- 62.De Gonzalez AB, Hartge P, Cerhan JR, et al. Body-mass index and mortality among 1.46 million white adults. New England Journal of Medicine. 2010;363(23):2211–2219. doi: 10.1056/NEJMoa1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moghaddam AA, Woodward M, Huxley R. Obesity and risk of colorectal cancer: a meta-analysis of 31 studies with 70,000 events. Cancer Epidemiology Biomarkers and Prevention. 2007;16(12):2533–2547. doi: 10.1158/1055-9965.EPI-07-0708. [DOI] [PubMed] [Google Scholar]
- 64.Morois S, Mesrine S, Josset M, Clavel-Chapelon F, Boutron-Ruault MC. Anthropometric factors in adulthood and risk of colorectal adenomas. American Journal of Epidemiology. 2010;172(10):1166–1180. doi: 10.1093/aje/kwq258. [DOI] [PubMed] [Google Scholar]
- 65.Stein B, Anderson JC, Rajapakse R, Alpern ZA, Messina CR, Walker G. Body mass index as a predictor of colorectal Neoplasia in ethnically diverse screening population. Digestive Diseases and Sciences. 2010;55(10):2945–2952. doi: 10.1007/s10620-009-1113-9. [DOI] [PubMed] [Google Scholar]
- 66.Wang Y, Jacobs EJ, Patel AV, et al. A prospective study of waist circumference and body mass index in relation to colorectal cancer incidence. Cancer Causes and Control. 2008;19(7):783–792. doi: 10.1007/s10552-008-9141-x. [DOI] [PubMed] [Google Scholar]
- 67.Pischon T, Lahmann PH, Boeing H, et al. Body size and risk of colon and rectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) Journal of the National Cancer Institute. 2006;98(13):920–931. doi: 10.1093/jnci/djj246. [DOI] [PubMed] [Google Scholar]
- 68.Tabuchi M, Kitayama J, Nagawa H. Hyperglycemia and hypertriglyceridemia may associate with the adenoma-carcinoma transition in colorectal epithelial cells. Journal of Gastroenterology and Hepatology. 2008;23(6):985–987. doi: 10.1111/j.1440-1746.2007.05072.x. [DOI] [PubMed] [Google Scholar]
- 69.Tabuchi M, Kitayama J, Nagawa H. Hypertriglyceridemia is positively correlated with the development of colorectal tubular adenoma in Japanese men. World Journal of Gastroenterology. 2006;12(8):1261–1264. doi: 10.3748/wjg.v12.i8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu CS, Hsu HS, Li CI, et al. Central obesity and atherogenic dyslipidemia in metabolic syndrome are associated with increased risk for colorectal adenoma in a Chinese population. BMC Gastroenterology. 2010;10, article 51 doi: 10.1186/1471-230X-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonovas S, Filioussi K, Flordellis CS, Sitaras NM. Statins and the risk of colorectal cancer: a meta-analysis of 18 studies involving more than 1.5 million patients. Journal of Clinical Oncology. 2007;25(23):3462–3468. doi: 10.1200/JCO.2007.10.8936. [DOI] [PubMed] [Google Scholar]