

Abstract
We compare the use of two-dimensional total internal reflection fluorescence microscopy with a rapid, simple-to-implement method for three-dimensional (3D) imaging using spinning-disk confocal microscopy suitable for reliable 3D tracking of clathrin-coated endocytic and endosomal carriers. These carriers contain about 20 EGFP (enhanced green fluorescent protein) equivalents of a chimeric fluorescent protein (either clathrin light chain or one of the clathrin adaptor subunits). Under tissue culture conditions, the clathrin-containing carriers correspond to a variable number of relatively sparse, diffraction-limited, fluorescent objects that can be identified with a spatial precision of ~30 nm or better and a temporal resolution of <1 s. The applicability of these approaches to mammalian cells in culture allows investigators detailed monitoring of the composition dynamics of the clathrin-containing carriers which can then be used to study in living cells the molecular mechanisms required for the formation and traffic of clathrin-coated pits and vesicles.
1. Introduction
Cells require ordered movement of proteins and lipids from one membrane-bound compartment to another, while maintaining the organization, function, and heterogeneity of the donor and acceptor membranes. A number of molecular assemblies (e.g., those based on clathrin, COPI, or COPII coatomers) have evolved to deform and invaginate membrane patches, which after pinching and scission become carriers of membrane traffic. Clathrin-coated pits and vesicles were the first membrane-traffic system to be recognized and analyzed in detail, because of the distinctive morphology of budding coated pits, the ease with which coated vesicles could be purified, and the importance of clathrin-coated structures for receptor-mediated endocytosis. Clathrin-coated vesicles are the most prominent form of traffic from the plasma membrane to endosomes (endocytosis), a pathway by which ligands such as hormones, transferrin, immunoglobulins, LDL (low-density lipoprotein), viruses, and their receptors enter cells. They are also important for traffic between endosomes and the trans-Golgi network (TGN; Bonifacino and Traub, 2003; Brodsky et al., 2001; Duncan and Payne, 2005; Hirst and Robinson, 1998; Kirchhausen, 2000; McMahon and Mills, 2004; Robinson, 2004; Traub, 2005; Ungewickell and Hinrichsen, 2007). They have become a paradigm for efforts to understand molecular mechanisms of other modes of vesicular transport (Harrison and Kirchhausen, 2010; Lee and Goldberg, 2010).
Cellular, biochemical, and high-resolution structural approaches have defined the molecular properties of clathrin and many of its associated proteins (Brett and Traub, 2006; Edeling et al., 2006; Gaidarov and Keen, 2005; Keyel et al., 2006; McMahon and Mills, 2004; Miwako and Schmid, 2006; Motley et al., 2006; Owen et al., 2004). In our own work, we have combined crystal structures with electron cryomicroscopy to image a clathrin coat at 8 Å resolution (Fotin et al., 2004b), a coat in association with a fragment of auxilin at 12 Å resolution (Fotin et al., 2004a), and a coat associated with both the auxilin fragment and a specifically bound Hsc70 (Xing et al., 2010). But biochemical and structural approaches, however powerful, can only provide snapshots or ensemble-averaged information about the properties of objects within a heterogeneous population. They are not sufficient to resolve important steps in coated vesicle formation and uncoating. To achieve temporal resolution in the context of a cell, many groups have turned to advanced imaging methods to study the localization of components during a given step, the order in which components are incorporated or released, and the way composition of an assembling vesicle affects its behavior (Ehrlich et al., 2004; Gaidarov et al., 1999; Kaksonen et al., 2005; Keyel et al., 2004; Le Clainche et al., 2007; Loerke et al., 2009; Merrifield et al., 2002, 2005; Mettlen et al., 2009; Newpher et al., 2005; Rappoport et al., 2005, 2006; Saffarian and Kirchhausen, 2008; Yarar et al., 2005; Zoncu et al., 2007). By analyzing assembly and disassembly of individual molecular complexes with fluorescence microscopy, one can link in vitro reconstitution studies, in which molecular concentrations and other external conditions can be fixed, thereby circumventing the hard-to-control complexities of an intact cell, with results from live-cell imaging, in which essentially identical detection schemes follow the same processes in their complete biological context.
This review highlights our use of total internal reflection fluorescence (TIRF) and spinning-disk confocal imaging of living cells to investigate the dynamics of clathrin coat formation. Depending on the acquisition mode (TIRF or spinning-disk confocal microscopy), the temporal resolution ranges between 10 and 100 ms. The required signal is typically emitted by 2–5 fluorescent molecules. Under carefully controlled TIRF conditions, it is also possible to record the signal from a single EGFP molecule. The spatial precision attained under these circumstances is 10–30 nm along the x-, y-, and z-axis (Kural et al., submitted; Saffarian and Kirchhausen, 2008). The signal-to-noise ratio (SNR) and resolution depend, in part, on such issues as detector response and particle tracking. We concentrate here primarily on the contributions of imaging modalities.
2. Dynamics of Clathrin Assembly
Our current picture of coated pit formation derives primarily from analysis by live-cell imaging of coated pits and vesicles at the plasma membrane (Fig. 4.1). The most important structural components of the assembly are clathrin and the AP-2 (α-β2-μ2-σ2) heterotetrameric adaptor complex. A number of accessory proteins associate with coated pits at specific stages of assembly/disassembly (Henne et al., 2010; Reider et al., 2009; Toshima et al., 2006; Traub, 2009). Eps15, epsin, FCHo1/2, and intersectin form an interacting complex that appears to be localized at the rim of a coated pit (Henne et al., 2010; Reider et al., 2009; Saffarian et al., 2009; Tebar et al., 1996; Traub and Wendland, 2010). This “rim complex” accumulates during early stages of coated pit assembly, but its components are excluded from a budded coated vesicle. Dynamin, the GTPase that drives membrane scission, accumulates both gradually during pit assembly and in a burst following clathrin lattice completion (Ehrlich et al., 2004; Loerke et al., 2009; Macia et al., 2006; Rappoport and Simon, 2003). Auxilin and Hsc70 arrive following scission, to direct uncoating (Lee et al., 2006; Massol et al., 2006). Hip1R, which binds clathrin light chains, recruits actin, required in some instances for coated vesicle maturation and budding (Ferguson et al., 2009; Merrifield et al., 2002, 2004; Saffarian et al., 2009).
Figure 4.1.

Coated pit formation proceeds by sequential addition of clathrin triskelions to an initial nucleus, generating a sharply curved coat; adaptor-mediated interactions with membrane-bound proteins (and lipids) deform the underlying membrane; dynamin mediates scission when the deformation has created a suitably narrow neck; auxilin, which arrives immediately following scission, recruits the uncoating ATPase, Hsc70. Under conditions of membrane tension (hyposmolarity, cell stretching, apical membranes of polarized cells, elongated cargo), coated pit maturation requires the formation of short-branched actin filaments; by contrast, actin polymerization does not generally accompany the assembly or budding of coated pits in membranes without tension. Continuous line, plasma membrane; dashed stripe, a clathrin coat (clathrin plus AP-2 adaptor); red rods, rim proteins (Eps15, epsin, FHCo1/2, SIG1a); gray lines, short-branched actin polymers plus the Arp2/3 complex, cortactin, N-Wasp; green bar dynamin; blue bar, F-BAR containing proteins; green dots, uncoating ATP Hsc70; red lines, auxilin. We use stable and transient expression of recombinant, fluorescently-tagged constructs to follow the dynamic behavior of plasma membrane structures containing different combinations of clathrin, AP-2, auxilin, Arp2/3 complex, cortactin, dynamin, etc. We mostly use TIRF and spinning disc confocal microscopy to obtain live-cell imaging data.
3. Limits of Fluorescence Microscopy
Even though fluorescence microscopy techniques enable real-time recordings from living cells and, hence, yield temporal information about cellular processes, spatial resolution is lost in most cases. Like most of the biological machinery, clathrin-coated structures (~100 nm in diameter) are smaller than the wavelength of the visible light (~500 nm), resulting in a convolution of a punctate image with the point-spread function (PSF) of the microscope (see below). Resolution of two identical fluorophores is limited by the Rayleigh criterion; two point sources that are closer than ~λ/2 (λ is the wavelength of the emitted light) cannot be resolved by conventional light microscopy. The accuracy of localizing a point source can be substantially better, however, depending on the SNR.
Other important limitations of fluorescence microscopy are photo-bleaching and phototoxicity. Organic fluorophores and fluorescent proteins can only emit a limited number of photons before ceasing to fluoresce. This phenomenon, also called photobleaching, results in a loss in signal as the sample is illuminated for long periods of time. Extensive illumination also creates toxic oxygen radicals. These factors necessitate using smart ways of illumination that can obtain high SNR without high levels of exposure. We start by outlining some of the microscopy techniques that enable cell biologists to obtain high-quality images with as little laser exposure as possible. Most of these methods are designed to confine the fluorescence excitation to a volume of interest, while leaving the rest of the specimen in the dark, to minimize background fluorescence and to avoid damage outside the illuminated region.
4. TIRF Microscopy
TIRF is based on the property of electromagnetic radiation that when it travels from a medium of high refractive index (n) to one of lower refractive index, it diverges from its original path toward the interface. This phenomenon, refraction, can result in a mirror effect (or total internal reflection) when the incidence angle exceeds a critical value (Fig. 4.2A). In this case, the reflected beam preserves most of its energy but a small portion of it is dissipated as an evanescent field at the interface. This quickly fading portion of light penetrates a few hundred nanometers (depending on wavelength, incidence angle, and the difference between the refractive indices) into the low-index medium, which becomes an ideal volume of illumination for fluorescence microscopy. In this geometry, fluorophores that are within a few hundred nanometers of the interface can be excited, while others that lie deeper within the sample are not illuminated (Axelrod, 1989). Because the plasma membranes of cells plated on glass coverslips are in contact with the glass–water interface (n = 1.51 and 1.33, respectively), TIRF microscopy is an excellent imaging technique for recording clathrin-coated pit formation at the adherent surface of the plasma membrane. In this configuration, TIRF illumination excites the fluorescently tagged components of the clathrin machinery that interact with the plasma membrane (high signal), but does not excite those that diffuse in the cytoplasm (low background; Fig. 4.3).
Figure 4.2.

TIRF illumination. (A) Schematic representation of ray paths at different angles of incidence as they reach the glass/sample interphase (panels a–d); total internal reflection conditions are achieved in panels c and d. The evanescence field depth depends on the incidence angle. (B) The images are of diffraction-limited fluorescent beads imaged using ~4 ms exposures with the same light source under total internal reflection (panels a and c) or widefield illumination (panels b) conditions. Panels b and c are color-coded representations of the SNR. Panel d highlights the substantial increase in SNR of the images acquired using TIRF microscopy.
Figure 4.3.

Schematic representation of the imaging strategies used to follow the dynamics of clathrin-coat assembly in different regions of the cell. The drawing indicates the location of various regions imaged in different cell types: apical surface of polarized cells (red box), leading edge of migrating cells (yellow box), dorsal surface of nonpolarized cells (blue box), and adherent surface (black box).
5. How to Optimize Your TIRF System
Objective-type and prism-type TIRF systems are the most widespread fluorescence microscopy techniques that use total internal reflection. Objective-type TIRF is generally preferred because water immersion objectives with lower numerical apertures used in prism type cannot collect as many photons. However, in both arrangements, it is possible to increase image quality by tweaking some of the parameters such as incidence angle and intensity of the excitation beam. Researchers generally use fluorescent bead samples for aligning their TIRF setups. The ideal bead sample prepared for TIRF imaging should be composed of a number of beads affixed to the coverslip and others that diffuse in the solution. In order to attain total internal reflection, the researcher should increase the incidence angle of the excitation beam to the point that the beads diffusing in the solution can no longer be imaged. Beads can be irreversibly immobilized to the coverslips by heating at 100 °C (Nugent-Glandorf and Perkins, 2004).
SNR of imaged fluorescent spots can be used as a measure to find the optimum TIRF configuration. SNR of a diffraction-limited spot can be calculated according to the formula where I is the signal collected at the brightest pixel and b is the standard deviation of the background. For instance, if the peak intensity of the PSF is 300 and the background fluctuations have a standard deviation of 10, then the SNR will be 300/(300 + 102)1/2 = 15. The first term in the noise (the denominator) is called the shot noise (also known as “photon” or “quantum” noise) which is inherent to systems following Poisson distribution and cannot be eliminated. Whereas the second term represents the noise stemming from the fluctuations of the background which is a combination of camera noise and out-of-focus signal coming from the diffusing fluorophores. The researcher can determine the optimum values of incidence angle and excitation intensity by searching for the highest SNR of immobilized beads (Fig. 4.2B).
In live-cell experiments, however, target molecules labeled with fluorescent proteins are not as bright as beads. In this low-signal regime, instead of using super-bright fluorescent bead samples, researchers may prefer single organic fluorophores immobilized on coverslip. In order to boost photo-stability of organic fluorophores, oxygen scavenging and reducing agents such as glucose oxidase (Yildiz et al., 2003) and trolox (Rasnik et al., 2006) can be used.
6. Spinning-Disk Confocal Microscopy
This modality, also called Nipkow type or Nipkow disk confocal fluorescence microscopy, uses a rapidly rotating (spinning) disk decorated with thousands of microlenses and pinholes, which focus the excitation beam onto a thin slab of the sample and filter out-of-focus fluorescence, respectively. In contrast to scanning confocal systems, spinning-disk microscopes record pulses of emission from many regions at once, scanning over and over to accumulate signal. The image can be recorded with a standard CCD camera to facilitate high rates of data acquisition. In cell biology, spinning-disk confocal microscopy is a more general technique than TIRF microscopy, because fluorescent objects that are microns away from the coverslip can be observed. (In TIRF microscopy, this distance is only a few hundred nanometers.) Thus, spinning-disk microscopy is well suited for imaging processes in the cytoplasm and at the nonadherent surface of the plasma membrane (Fig. 4.3). In some cases, spinning-disk imaging is preferable to TIRF, even at the ventral surface of cells, since it does not require any corrections for variations in intensity of illumination as a function of depth.
7. Spherical Aberration Correction Applied to Spinning-Disk Confocal Microscopy
Even though high NA (numerical aperture) objectives are necessary for obtaining better signal, they have their drawbacks as well. One of such problems occur due to the fact that light rays passing through the peripheral regions of the lens are refracted more than the ones closer to the optical axis (Fig. 4.4A). This results in longitudinally separated focal points which reduce spatial resolution. In optical microscopy, this phenomenon is called spherical aberration (SA). If there is a refractive index mismatch between the cover glass and media, the SA increases with the distance traversed the medium.
Figure 4.4.

Spherical aberration. (A) Schematic representation of ray paths in the absence and presence of spherical aberration. (B) Representation of the optical hardware used to correct spherical aberration. (C) Fluorescence image of two beads of different size acquired in three dimensions using the spinning-disk confocal microscope, in the absence (no correction) and presence (SAC) of spherical aberration correction. Note the increase in fluorescence intensity and decrease in the axial elongation of the diffraction-limited bead.
In order to circumvent this problem, commercially available spherical aberration correction (SAC) units can be installed to the emission path of the microscope. The working principle of SAC units is quite similar to correction collars attached to some objectives which are used to compensate the aberrations stemming from the variations in cover glass thickness. Just like the correction collar, the SAC unit contains a motile lens doublet. However, the position of the doublet is computer controlled; hence, the built-in calibration protocols of SAC units enable to find the best correction possible automatically. Another advantage of computer control is that SAC can dynamically correct for SA at varying penetration depths during a z-acquisition (Fig. 4.4B,C).
During the calibration process, the SAC will determine the correct setting for minimal SA at Z0, it will then determine the setting for minimal SA at the deepest penetration depth Zd. During the 3D acquisition, the SAC will dynamically move the correction lens relative to the z-position, therefore producing a 3D image that corrects SA at every plane in the series, as opposed to the correction collar that corrects for a single plane.
8. Getting Around the Diffraction Limit
Single light emitting fluorophores focused on a flat surface by using a lens with a circular aperture appear as an Airy function (generally referred as PSF). The Airy disk, the bright region at the center of Airy function, has a width of ~λ/2NA where NA is the numerical aperture of the lens (for oil-immersion objectives, NA is generally between 1.4 and 1.65). Determining the actual position of the fluorescent object in this extended “blob” of signal depends on localizing the center point of the Airy disk with high fidelity. A method called fluorescence imaging with one nanometer accuracy (FIONA), developed by the Selvin laboratory, has proved to be an effective way to locate a single fluorescent object with a precision of one nanometer in x and y (Kural et al., 2005; Yildiz et al., 2003). FIONA relies on the observation that the Airy disk can be represented to good approximation by a two-dimensional (2D) Gaussian function; if the SNR is high enough, then the centroid of the Gaussian can be determined to about 1 nm. The standard error of the mean (SEM) of the centroid can be used to represent the accuracy; it is given by (Thompson et al., 2002):
| (4.1) |
where s is the standard deviation of the Gaussian fit, N is the number of photons collected, a is the effective pixel size of the CCD camera (i.e., pixel size of the CCD chip divided by the total magnification), and b is the standard deviation of the background (which includes the camera noise together with the fluctuations of the background fluorescence). According to this equation, the most straightforward ways to increase the localization accuracy are (1) to maximize the total number of collected photons and (2) to minimize the standard deviation of the background.
There are multiple ways to increase the number of collected photons. The easiest is to increase the excitation power (or exposure time per imaging cycle). The experimenter must, however, consider that high power and long exposure increase the rate of photobleaching and phototoxicity. A more efficient but expensive way is to use a high NA lens. Especially in TIRF microscopy systems, numerical apertures up to 1.65 can be obtained by switching to oil-immersion lenses designed for sapphire coverslips (n ≈ 1.8). The background fluctuations can be minimized by filtering out the out-of-focus signal, for which both TIRF and spinning-disk systems are quite effective. In TIRF microscopy, the experimenter can increase the incidence angle of the excitation beam to reduce the penetration depth of the evanescent field and thus excite fewer fluorophores in the solution. For an objective type TIRF system, the back aperture of the objective sets a limit to the maximum incidence angle. Using advanced CCD cameras with low readout noise is another way to diminish fluctuations of the background. Binning CCD chips is an alternative approach to reduce the noise factor, as well as an efficient way to increase frame rate.
With the advent of FIONA and related techniques for localizing point sources (e.g., SHRIMP, SHREC, STORM, and PALM, some of which rely on specialized fluorophores and specialized excitation regimes), new possibilities have emerged for using fluorescence microscopy to obtain high temporal and spatial information about processes in living cells (Betzig et al., 2006; Churchman et al., 2005; Gordon et al., 2004; Rust et al., 2006). These techniques necessitate advanced computational and analytical tools to make use of the raw data. To illustrate these points, we will briefly describe some of the applications in which 2D imaging techniques have been used to study clathrin-mediated endocytosis at the ventral (adherent) and dorsal (free) surfaces of living cells (Fig. 4.3).
9. Use of 2D Spinning-Disk Confocal Microscope to Study Clathrin-Mediated Endocytosis at the Ventral and Dorsal Surfaces of a Cell
Clathrin interacts with lipid bilayers through its adaptor proteins. The major clathrin adaptor present solely at the plasma membrane is AP2. When imaged in the fluorescence microscope, labeled AP2 gives less-crowded images than does clathrin itself, because clathrin decorates endosomes and TGN compartments as well as the plasma membrane. For this reason, mammalian cells stably expressing AP2 adaptors tagged with fluorescent proteins are the systems of choice for studying clathrin-mediated endocytosis in real time. The high imaging rates and low background noise offered by spinning-disk confocal microscope enabled Ehrlich et al. (2004) to resolve the complete lifetime of endocytic clathrin coats at the adherent (ventral) surface BSC1 cells (Fig. 4.5). Their work revealed a nucleation-growth mechanism, which proceeds at a steady rate that is much slower than the uncoating reaction.
Figure 4.5.

Recruitment of clathrin and AP2 in endocytic coated pits. (A) Arrow heads point to an example of clathrin LCa-mRFP recruitment during the formation of a canonical endocytic clathrin-coated pit. (B) Plot of the fluorescence intensity normalized to the highest value before uncoating of a subset of 28 coated pits containing σ2-EGFP and LCa-mRFP, each with a lifetime of 42 s. The data were obtained using a spinning-disk confocal microscope from three BSC1 cells stably expressing σ2-EGFP and transiently expressing LCa-mRFP. (Reproduced from Fig. 4 of Ehrlich et al., 2004; from Fig. 4A and E in Ehrlich et al., 2004.)
Confocal imaging can penetrate several microns into the specimen, but at the expense of increased SA. Cureton et al. used a spinning-disk confocal microscope to characterize clathrin-mediated endocytosis of vesicular stomatitis virus (VSV) particles at the free (dorsal) surface of BSC1 cells. Their results showed that virus-containing clathrin coats are larger and longer lived than the virus-free clathrin coats at the same dorsal surface (Fig. 4.6; Cureton et al., 2009). The elongated virus particle stalls completion of the coat, which must be rescued by actin-driven membrane deformation.
Figure 4.6.

Live-cell imaging of clathrin-dependent endocytosis of single VSV particles. Plot of the kinetics of fluorescent AP-2 (tagged with σ2-EGFP) and clathrin (tagged with Tom-LCa) recruitment to endocytic clathrin-coated pits in BSC-1 cells associated or not associated with the internalization of VSV particles. (A) Fluorescence intensities were plotted relative to the time of clathrin detection and are expressed as a % of the average maximum clathrin observed in all pits lacking virus. (B) Plot of the average kinetics of AP-2 and clathrin recruitment to coated pits containing or not containing virus. Average fluorescence intensity and time are expressed as a % relative to clathrin-coated vesicles lacking virus observed in the same cells (from Fig. 1 in Cureton et al. 2009).
10. Use of TIRF Microscopy to Study Clathrin-Mediated Endocytosis
The evanescent field formed upon total internal reflection decays exponentially according to formula
| (4.2) |
where is the excitation intensity at the coverslip–medium interface, z is the distance from the interface, and d is the penetration depth. For an object that carries a fixed number of fluorescent particles, a similar equation would also work for fluorescence emission. That is, , where is the emission intensity at the interface. By using this relationship between z and emission intensity, it is in principle possible to map the changes in the z-position of an object which is moving within the evanescent field. In practice, clathrin-coated structures initiate, mature, and uncoat during the observation period, and in each one of these phases, the coat has different levels of fluorescent components. For objects, such as clathrin-coated structures, for which variation in fluorescence is part of the phenomenon being studied, it is essential, in order to map the z-position, to determine whether the variation in the emission signal stems from movement in z or changes in the number of fluorescent components. To overcome this ambiguity, Saffarian et al. coupled widefield illumination with TIRF microscopy on BSC1 cells stably expressing fluorescently labeled AP2 and clathrin (Saffarian and Kirchhausen, 2008). Making use of the uniform intensity of widefield illumination along the axial direction, they could quantify the total emitted signal, regardless of the position. In this case, the distance of the coat from the interface at time point t can be calculated as
| (4.3) |
where IWF is the intensity of the wide-field illumination, is the intensity of the TIRF illumination at the interface, FWF(t) is the fluorescence signal collected at the wide-field channel at time point t, and FTIRF(t) is the fluorescence signal collected at the TIRF channel at time point t.
Saffarian et al. extended this analysis to two different fluorophores recorded at the same time; they named the technique Differential Nanometry (DiNa). The DiNa analysis of coats carrying clathrin labeled with Tomato and AP2 adaptors labeled with EGFP showed that the center of fluorescence of adaptors is separated from that of clathrin, indicating an asymmetric distribution of adaptors within the clathrin coat (Saffarian and Kirchhausen, 2008; Fig. 4.7).
Figure 4.7.
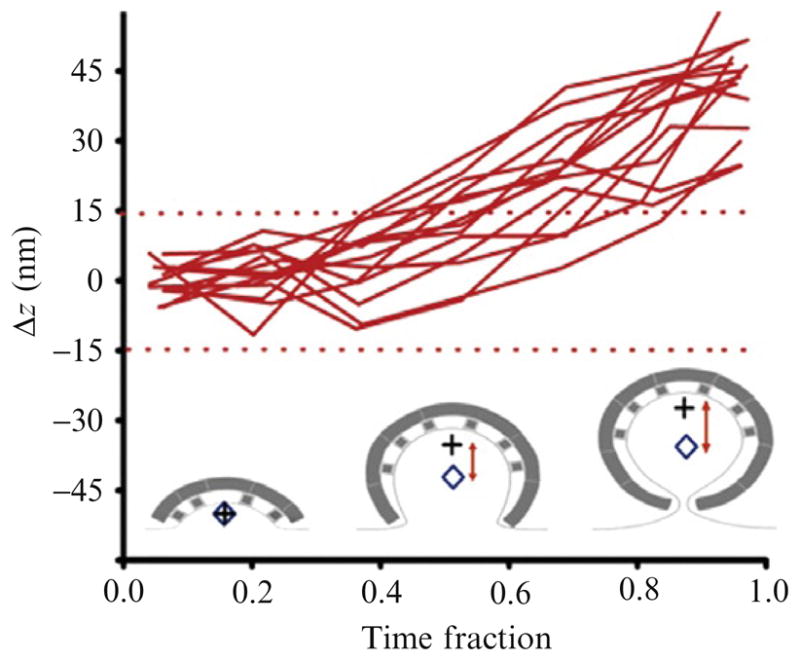
DiNa analysis applied to the recruitment of AP2 and clathrin during the formation of endocytic clathrin-coated pits. The experiment was done using the total internal reflection and widefield illumination signals of coated pits and vesicles forming on the ventral surface of BSC1 cells. AP2 was tagged by stable expression of s2-EGFP and clathrin by transient expression of Tom-LCa. Δz calculated for the difference in the average axial position between clathrin LCa-Tomato and AP-2 σ2-EGFP from the individual pits selected for DiNa analysis plotted as a function of the fraction of their respective lifetimes. Values outside the red dotted lines represent meaningful data in excess of 1.5 times the expected calculated resolution of Δz (from Fig. 5E in Saffarian and Kirchhausen, 2008).
11. The Third Dimension
2D time-lapse data acquisition has generally been the dominant high-resolution mode for fluorescence imaging experiments with living cells. This restriction has applied to a variety of illumination and recording modalities, for example, widefield, confocal, and TIRF microscopy. Although powerful for describing the localization of the fluorescent object and for resolving the time-dependence of the fluorescence intensity associated with a tagged protein, lipid, or organelle, 2D data fail to represent the intrinsically rich behavior of objects within the three-dimensional (3D) context of a cell. For instance, efforts to follow AP1-and AP3-containing clathrin carriers have not been successful because rapid movement, particularly along the direction of view (Z), has not permitted one to distinguish initiation of assembly or dissociation of the clathrin coat from passage into or out of the imaging plane. In order to circumvent this problem, we have developed a 3D analysis technique optimized for 3D time-lapse images taken with a spinning-disk confocal microscope. The method makes use of the fact that the 3D PSF of a diffraction-limited spot can be imaged at different planes in the axial dimension and its position can be estimated from the distribution of integrated intensities collected from each plane.
12. 3D Tracking in Spinning-Disk Imaging
A spinning-disk confocal microscope equipped with a piezo-driven stage can image up to 8 planes (in the axial dimension) per second. This rapid imaging makes it possible to determine the 3D trajectory of an intracellular object. The accuracy of trajectories depend on localizing each spot in x, y, and z with high precision at each time point. The FIONA procedure enables one to extract the x and y positions of fluorescent diffraction-limited objects from 2D movies, such as those taken with TIRF microscopy. In the case of 3D time-lapse movies acquired with a spinning-disk microscope, the projection of the brightest signals along the axial direction (maximum projection) can be used to project the 3D position onto the x–y plane for each z-stack and hence to obtain a 2D projected trajectory, to which FIONA can then be applied.
Determining the z-position needs some extra analytical tools that can make use of the information embedded within the z-stacks. If the distance between the consecutive planes is chosen appropriately, then the 3D PSF of fluorescent objects can be imaged on multiple planes in the z-stack. The intensity measured on each plane is related to distance from the actual position of the object in z. In other words, if an object is between planes n and n + 1, but closer to plane n, then the fluorescence intensity in plane n will be higher than the intensity in plane n + 1. Thus, one can determine the 3D PSF along z; by calculating the centroid of the distribution, the position of the object can be estimated.
The 3D tracking routine that we have programmed accepts data as a time-series of z-stacks, each plane in the stack being a recorded optical section. The required z-spacing is about one-fourth to one-third of the full height of the 3D PSF (in the examples described here, Δz ~ 250–450 nm). In order to perform automated tracking, single fluorescent spots are detected in the maximum projection image of a stack by using a local maximum-finding algorithm performed after 2D Gaussian and Laplacian filtering. x and y positions of the spots are then obtained in a window of 7 × 7 pixels by FIONA. For each spot, the 7 × 7 pixel window is then extended to each plane in the z-stack, and integrated intensities for each plane are calculated. The centroid of the intensity distribution in z is then calculated as , where n is the number of planes in the z-stack, zi is the coordinate of the plane on the z-axis, Ii is the integrated signal of that plane. The integrated intensity of the plane with the minimum signal is the threshold subtracted from all planes. By using this approach, biological molecules such as EGFP-tagged AP1 coats can be localized with ~20 nm accuracy in z. In living cells, we have followed AP1- and AP3-containing endosomal carriers from assembly to uncoating, despite quite rapid motion in all three dimensions, well displaced from either cell surface. These structures have lifetimes and sizes (the latter as estimated from maximum fluorescence intensity) comparable to those of endocytic, clathrin-coated carriers (Kural et al., submitted).
The analysis can be performed on hundreds of fluorescent objects simultaneously. It is therefore possible to classify objects in a single cell according to their position and movement. One such example is classification of the clathrin-coated structures forming at the ventral and dorsal surface of migrating U373 astrocytes (Fig. 4.3). By tracking AP2 puncta in 3D, we have found that AP2 carriers are absent from the ventral surface during migration and that they reappear when cellular migration ceases. The abundant AP2 structures that formed on the dorsal surface tended to initiate in the region close to the leading edge. The AP2 objects in this region moved with a characteristic centripetal retrograde flow that was inversely related to the migration rate of the cell (Fig. 4.8).
Figure 4.8.

Formation of endocytic clathrin and AP2-containing coated pits and vesicles on the dorsal surface of the leading lamellipodium of migrating U373 astrocytes. The image shows tracings color-coded for the z-position of AP2 fluorescent spots tagged with σ2-EGFP. The axial information was obtained by tracking in 3D the temporal position of individual AP2 spots imaged by spinning-disk confocal microscopy. Insets display representative tracings corresponding to the location along the z-axis of two AP2-containing objects.
13. Use of 3D Tracking to Monitor Clathrin-Mediated Entry of Reovirus Particles at the Apical Surface of Polarized Cells
Clathrin-mediated internalization of VSV particles can easily be traced by using spinning-disk confocal systems at the free surface of nonpolarized BSC-1 cells (Cureton et al., 2009, 2010). Unlike the dorsal membrane of nonpolarized cells, however, the apical membrane of polarized cells has a dome-shaped profile. The absence of a flat region at the top of a polarized cell means that one cannot monitor virus entry in a single optical plane. If the recording is restricted to 2D, many virus particles disappear from view because they move out of the confocal plane. To study virus entry using polarized MDCK (Madin-Darby canine kidney) cells, we therefore used rapid 3D imaging as outlined above to visualize a larger part of the apical plasma membrane (Fig. 4.3). In particular, we determined the 3D trajectory of reovirus particles. 3D movies were recorded from the apical surface of polarized cells, with z-stacks of 3–5 consecutive optical planes spaced at 0.35 μm. A 2D movie was then obtained by generating a maximum intensity z-projection for each time point. The colocalization of the viral particles and AP2-GFP was analyzed from the resulting 2D movies, and the position of the virus particles in the three dimensions was then extracted from the raw z-stack. As also found in nonpolarized BSC-1 cell, reovirus particles were internalized by clathrin-mediated endocytosis. Virions were captured by a coated pit and the intensity of the AP2-GFP signal increased steadily as the clathrin-coated pit formed. An abrupt disappearance of fluorescence was then observed. Our 3D imaging method allowed us to visualize the budding of the coated pit from the plasma membrane and the displacement of virus-containing vesicles as uncoating of AP2-GFP occurred (Fig. 4.9). Soon after the recruitment of AP2-GFP had reached a plateau, we detected a rapid displacement of the coated pits in the z-dimension, away from the plasma membrane. After budding of the clathrin-coated vesicles, uncoating occurred. We measured the average displacement of a virus-containing coated vesicles before uncoating and found that on average the vesicles moved ~500 nm away from the plasma membrane. Similar values were found for coated vesicles that did not contain virus particles.
Figure 4.9.

Real-time tracking of the internalization of a reovirus particle by an endocytic AP2-containing coated carrier. Top panel: schematic representation of the experimental outcome representing the internalization of a fluorescent reovirus particle at the apical surface of a polarized MDCK cell stably expressing σ2-EGFP. Bottom panel: plot of z-displacement of reovirus and the AP2-containing pit during coated pit formation. The z-position of the virion displays an abrupt displacement toward the cytoplasm at the onset of AP2 uncoating. The data was obtained using 3D spinning-disk confocal microscopy.
14. Using the Optimum Pixel and Step Sizes in z-Stacks
As shown in Eq. (4.1), SEM of the location in x and y is a function of effective pixel size. For larger pixel sizes (under low magnification), the PSF converges into a 2D delta function. In this case, the pixelation noise becomes dominant due to the fact that it is not possible to pinpoint the position where the photon arrived in the pixel. For that reason, using pixel sizes larger than the standard deviation of the PSF is not recommended. In the other extreme, for smaller size pixels (under high magnification), the maximum signal reduces dramatically (α magnification−2) and camera readout noise becomes stronger since the spot spreads over a larger number of pixels (Thompson et al., 2002). For systems with intrinsically high magnification, binning the CCD pixels (2 × 2) can be a good alternative to reduce the readout noise. Binning also increases CCD frame rate and by doing so enables faster imaging.
In localizing the center of 3D PSF in the axial dimension, using small steps to acquire z-stacks may increase spatial precision slightly. On the other hand, using larger steps to scan the same volume speeds up the acquisition and reduces exposure per imaging cycle which in return slows down photobleaching (Fig. 4.10). The researcher should consider these factors and find the optimum imaging parameters to be able to perform 3D tracking for his/her specific application. For tracking objects that move with slower pace, z-stacks can be acquired with longer time intervals to minimize photobleaching and elongate total time of imaging. To be able to track fast-moving objects, the researcher has to reduce the time interval between z-stacks to increase temporal resolution. Scanning the volume with larger step sizes can prevent excessive photobleaching.
Figure 4.10.

Monte Carlo simulation showing the effect of changing the size of the imaging z-step on SNR and precision determination along the z-axis. The calculation was carried out using 3D PSFs (width = 500 nm along the z-axis) and imaging steps of 200, 300, and 500 nm. (A) The SNR drop reflects the effect of photobleaching. (B) Step size has little effect on z-axis precision if the measurements are carried out before photobleaching ensues. The calculation was carried out using 50 PSFs, each simulated by using a Gaussian distribution of fluorescence intensity. The shot noise of each acquisition was obtained from the intensities corresponding to the values on each z-plane replaced with random values generated using the Poisson distribution of the same value. A Gaussian random number generator was added to simulate fluctuations in the background fluorescence. A uniform rate of photobleaching was used for each imaging step.
15. Conclusion
The strategies presented here have broad potential for investigating the dynamics of processes involving assembly, translocation, and disassembly of a small and variable number of diffraction-limited, fluorescent molecules at the ventral plasma membrane and throughout the volume of a cell.
Acknowledgments
We gratefully acknowledge David Cureton and Steeve Boulant for sharing their data shown in Figs. 4.2 and 4.9, respectively, and Eric Marino for maintaining the imaging resources used here. This work was supported in part by NIH grants GM 075252 (T. K.) and U54 AI057159 (New England Regional Center of Excellence in Biodefense and Emerging Infectious Diseases, Imaging Core).
References
- Axelrod D. Total internal reflection fluorescence microscopy. Methods Cell Biol. 1989;30:245–270. doi: 10.1016/s0091-679x(08)60982-6. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Brett TJ, Traub LM. Molecular structures of coat and coat-associated proteins: Function follows form. Curr Opin Cell Biol. 2006;18:395–406. doi: 10.1016/j.ceb.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Brodsky FM, Chen CY, Knuehl C, Towler MC, Wakeham DE. Biological basket weaving: Formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol. 2001;17:517–568. doi: 10.1146/annurev.cellbio.17.1.517. [DOI] [PubMed] [Google Scholar]
- Churchman LS, Okten Z, Rock RS, Dawson JF, Spudich JA. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc Natl Acad Sci USA. 2005;102:1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SP. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009;5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Whelan SP, Kirchhausen T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010;6(9):e1001127. doi: 10.1371/journal.ppat.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan MC, Payne GS. Cell biology: Protein choreography. Nature. 2005;438:571–573. doi: 10.1038/438571a. [DOI] [PubMed] [Google Scholar]
- Edeling MA, Smith C, Owen D. Life of a clathrin coat: Insights from clathrin and AP structures. Nat Rev Mol Cell Biol. 2006;7:32–44. doi: 10.1038/nrm1786. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Ferguson S, Raimondi A, Paradise S, Shen H, Mesaki K, Ferguson A, Destaing O, Ko G, Takasaki J, Cremona O, et al. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell. 2009;17:811–822. doi: 10.1016/j.devcel.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Grigorieff N, Walz T, Harrison SC, Kirchhausen T. Structure of an auxilin-bound clathrin coat and its implications for the mechanism of uncoating. Nature. 2004a;432:649–653. doi: 10.1038/nature03078. [DOI] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, Walz T. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004b;432:573–579. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- Gaidarov I, Keen JH. Membrane targeting of endocytic adaptors: Cargo and lipid do it together. Dev Cell. 2005;8:801–802. doi: 10.1016/j.devcel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- Gordon MP, Ha T, Selvin PR. Single-molecule high-resolution imaging with photobleaching. Proc Natl Acad Sci USA. 2004;101:6462–6465. doi: 10.1073/pnas.0401638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC, Kirchhausen T. Structural biology: Conservation in vesicle coats. Nature. 2010;466:1048–1049. doi: 10.1038/4661048a. [DOI] [PubMed] [Google Scholar]
- Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, Mittal R, McMahon HT. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science. 2010;328:1281–1284. doi: 10.1126/science.1188462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Robinson MS. Clathrin and adaptors. Biochem Biophys Acta. 1998;1404:173–193. doi: 10.1016/s0167-4889(98)00056-1. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG. A modular design for the clathrin-and actin-mediated endocytosis machinery. Cell. 2005;123:305–320. doi: 10.1016/j.cell.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Keyel PA, Watkins SC, Traub LM. Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J Biol Chem. 2004;279:13190–13204. doi: 10.1074/jbc.M312717200. [DOI] [PubMed] [Google Scholar]
- Keyel PA, Mishra SK, Roth R, Heuser JE, Watkins SC, Traub LM. A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol Biol Cell. 2006;17:4300–4317. doi: 10.1091/mbc.E06-05-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T. Three ways to make a vesicle [Review] Nat Rev Mol Cell Biol. 2000;1:187–198. doi: 10.1038/35043117. [DOI] [PubMed] [Google Scholar]
- Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: A tug-of-war or coordinated movement? Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- Le Clainche C, Pauly BS, Zhang CX, Engqvist-Goldstein AE, Cunningham K, Drubin DG. A Hip1R-cortactin complex negatively regulates actin assembly associated with endocytosis. EMBO J. 2007;26:1199–1210. doi: 10.1038/sj.emboj.7601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Goldberg J. Structure of coatomer cage proteins and the relationship among COPI, COPII, and clathrin vesicle coats. Cell. 2010;142:123–132. doi: 10.1016/j.cell.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Wu X, Eisenberg E, Greene LE. Recruitment dynamics of GAK and auxilin to clathrin-coated pits during endocytosis. J Cell Sci. 2006;119:3502–3512. doi: 10.1242/jcs.03092. [DOI] [PubMed] [Google Scholar]
- Loerke D, Mettlen M, Yarar D, Jaqaman K, Jaqaman H, Danuser G, Schmid SL. Cargo and dynamin regulate clathrin-coated pit maturation. PLoS Biol. 2009;7:e57. doi: 10.1371/journal.pbio.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Massol RH, Boll W, Griffin AM, Kirchhausen T. A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc Natl Acad Sci USA. 2006;103:10265–10270. doi: 10.1073/pnas.0603369103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Mills IG. COP and clathrin-coated vesicle budding: Different pathways, common approaches. Curr Opin Cell Biol. 2004;16:379–391. doi: 10.1016/j.ceb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Feldman ME, Wan L, Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Qualmann B, Kessels MM, Almers W. Neural Wiskott Aldrich syndrome protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur J Cell Biol. 2004;83:13–18. doi: 10.1078/0171-9335-00356. [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Perrais D, Zenisek D. Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells. Cell. 2005;121:593–606. doi: 10.1016/j.cell.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Mettlen M, Stoeber M, Loerke D, Antonescu CN, Danuser G, Schmid SL. Endocytic accessory proteins are functionally distinguished by their differential effects on the maturation of clathrin-coated pits. Mol Biol Cell. 2009;20:3251–3260. doi: 10.1091/mbc.E09-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwako I, Schmid SL. A cell-free biochemical complementation assay reveals complex and redundant cytosolic requirements for LRP endocytosis. Exp Cell Res. 2006;312:1335–1344. doi: 10.1016/j.yexcr.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Motley AM, Berg N, Taylor MJ, Sahlender DA, Hirst J, Owen DJ, Robinson MS. Functional analysis of AP-2 alpha and mu2 subunits. Mol Biol Cell. 2006;17:5298–5308. doi: 10.1091/mbc.E06-05-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher TM, Smith RP, Lemmon V, Lemmon SK. In vivo dynamics of clathrin and its adaptor-dependent recruitment to the actin-based endocytic machinery in yeast. Dev Cell. 2005;9:87–98. doi: 10.1016/j.devcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Nugent-Glandorf L, Perkins TT. Measuring 0.1-nm motion in 1 ms in an optical microscope with differential back-focal-plane detection. Opt Lett. 2004;29:2611–2613. doi: 10.1364/ol.29.002611. [DOI] [PubMed] [Google Scholar]
- Owen DJ, Collins BM, Evans PR. Adaptors for clathrin coats: Structure and function. Annu Rev Cell Dev Biol. 2004;20:153–191. doi: 10.1146/annurev.cellbio.20.010403.104543. [DOI] [PubMed] [Google Scholar]
- Rappoport JZ, Simon SM. Real-time analysis of clathrin-mediated endocytosis during cell migration. J Cell Sci. 2003;116:847–855. doi: 10.1242/jcs.00289. [DOI] [PubMed] [Google Scholar]
- Rappoport JZ, Benmerah A, Simon SM. Analysis of the AP-2 adaptor complex and cargo during clathrin-mediated endocytosis. Traffic. 2005;6:539–547. doi: 10.1111/j.1600-0854.2005.00280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappoport JZ, Kemal S, Benmerah A, Simon SM. Dynamics of clathrin and adaptor proteins during endocytosis. Am J Physiol Cell Physiol. 2006;291:C1072–C1081. doi: 10.1152/ajpcell.00160.2006. [DOI] [PubMed] [Google Scholar]
- Rasnik I, McKinney SA, Ha T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- Reider A, Barker SL, Mishra SK, Im YJ, Maldonado-Baez L, Hurley JH, Traub LM, Wendland B. Syp1 is a conserved endocytic adaptor that contains domains involved in cargo selection and membrane tubulation. EMBO J. 2009;28:3103–3116. doi: 10.1038/emboj.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarian S, Kirchhausen T. Differential evanescence nanometry: Live-cell fluorescence measurements with 10-nm axial resolution on the plasma membrane. Biophys J. 2008;94:2333–2342. doi: 10.1529/biophysj.107.117234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarian S, Cocucci E, Kirchhausen T. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009;7:e1000191. doi: 10.1371/journal.pbio.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebar F, Sorkina T, Sorkin A, Ericsson M, Kirchhausen T. Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J Biol Chem. 1996;271:28727–28730. doi: 10.1074/jbc.271.46.28727. [DOI] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima JY, Toshima J, Kaksonen M, Martin AC, King DS, Drubin DG. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc Natl Acad Sci USA. 2006;103:5793–5798. doi: 10.1073/pnas.0601042103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM. Common principles in clathrin-mediated sorting at the Golgi and the plasma membrane. Biochim Biophys Acta. 2005;1744:415–437. doi: 10.1016/j.bbamcr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Traub LM. Clathrin couture: Fashioning distinctive membrane coats at the cell surface. PLoS Biol. 2009;7:e1000192. doi: 10.1371/journal.pbio.1000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM, Wendland B. Cell biology: How to don a coat. Nature. 2010;465:556–557. doi: 10.1038/465556a. [DOI] [PubMed] [Google Scholar]
- Ungewickell EJ, Hinrichsen L. Endocytosis: Clathrin-mediated membrane budding. Curr Opin Cell Biol. 2007;19:417–425. doi: 10.1016/j.ceb.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Xing Y, Bocking T, Wolf M, Grigorieff N, Kirchhausen T, Harrison SC. Structure of clathrin coat with bound Hsc70 and auxilin: Mechanism of Hsc70-facilitated disassembly. EMBO J. 2010;29:655–665. doi: 10.1038/emboj.2009.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, Schmid SL. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:964–975. doi: 10.1091/mbc.E04-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: Single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Sebastian R, Nakatsu F, Chen H, Balla T, Ayala G, Toomre D, De Camilli PV. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci USA. 2007;104:3793–3798. doi: 10.1073/pnas.0611733104. [DOI] [PMC free article] [PubMed] [Google Scholar]