

Abstract
Deficiencies in protein degradation and proteolytic function within neurons are linked to a number of neurodegenerative diseases and developmental disorders. Compartmentalized cultures of peripheral neurons were used to investigate the properties and relative abundance of the proteolytic machinery in the axons and cell bodies of sympathetic and sensory neurons. Immunoblotting of axonal proteins demonstrated that LAMP2, LC3 and PSMA2 were abundant in axons, suggesting that lysosomes, autophagosomes and proteasomes were located in axons. Interestingly, the expression of proteins associated with lysosomes and proteasomes were upregulated selectively in axons by NGF stimulation of the distal axons of sympathetic neurons, suggesting that axonal growth and maintenance requires local protein turnover. The regulation of the abundance of both proteasomes and lysosomes in axons by NGF provides a link between protein degradation and the trophic status of peripheral neurons. Inhibition of proteasomes located in axons resulted in an accumulation of ubiquitinated proteins in these axons. In contrast, lysosome inhibition in axons did not result in an accumulation of ubiquitinated proteins or the transferrin receptor, a transmembrane protein degraded by lysosomes. Interestingly, lysosomes were transported both retrogradely and anterogradely, therefore it is likely that ubiquitinated proteins that are normally destined for degradation by lysosomes in axons can be transported to the cell bodies for degradation. In summary, proteasomal degradation occurs locally, whereas proteins degraded by lysosomes can most likely either be degraded locally in axons, or be transported to cell bodies for degradation.
Keywords: Proteasome, Lysosome, NGF, Transport, Autophagy
Introduction
Protein degradation is a process common to all cell types and is important in both cell maintenance and disease (Klionsky and Emr 2000; Luzio et al. 2007; Tai and Schuman 2008). Protein degradation is involved in growth cone sprouting and cytoskeleton remodeling (Verma et al. 2005), receptor turnover (Arancibia-Carcamo et al. 2009; Colledge et al. 2003) and elimination of misfolded proteins (Goldberg 2003). Degradation occurs by three mechanisms: ubiquitin-proteasome degradation, endosomal-lysosomal degradation, and autophagy-lysosome degradation. The ubiquitin-proteasome system degrades cytosolic proteins that have been targeted for proteolysis by the addition of chains of four or more ubiquitin molecules (Thrower et al. 2000). Proteolysis occurs in the proteosome, a protein complex that identifies polyubiquitin chains and cleaves the target protein into small groups of amino acids (Voges et al. 1999). In contrast, monoubiquitination events target proteins to the endosomal-lysosomal degradation system. Transmembrane proteins typically are internalized after addition of monoubiquitin, often on several lysines, and are ultimately degraded by lysosomal enzymes after endosomes fuse with lysosomes (Chau et al. 1989; Hicke 2001; Luzio et al. 2007). The autophagy-lysosome system functions in large-scale degradation events, resulting in targeted degradation of protein aggregates or entire organelles that have been ubiquitinated and linked to p62/SQSTM1 (Kim et al. 2008; Mizushima et al. 2008; Pankiv et al. 2007; Yang and Klionsky 2009). Recruitment into autophagic vesicles is mediated by interaction between p62/SQSTM1 and the microtubule-associated protein LC3, a marker for autophagy that is involved in autophagic membrane formation (Kuma et al. 2007; Pankiv et al. 2007).
Defects in protein degradation have been implicated in a variety of neurodegenerative disorders including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease and amyotrophic lateral sclerosis (Rubinsztein 2006; Taylor et al. 2002). However, it is not fully understood how defects in protein degradation cause, or develop from, neurodegenerative diseases. Furthermore it is not explicitly known to what extent the protein degradation machinery is localized within axons of large, peripheral neurons and how they contribute to global cellular protein turnover. Although there is evidence for the localization of protein degradation machinery within cell somas and dendrites (Arancibia-Carcamo et al. 2009; Colledge et al. 2003; Lee et al. 2011), it is not clear how these machinery localize and carry out their functions within axons (Korhonen and Lindholm 2004; Overly et al. 1996; Song et al. 2008).
We utilized compartmentalized cultures of peripheral neurons to ascertain the abundance and regulation of the proteolytic machinery in axons as compared to cell bodies. Protein components of lysosomes, autophagosomes and proteasomes were all located in axons and lysosomes and proteasomes were upregulated in response to NGF stimulation. Inhibition of axonal proteasomes resulted in an accumulation of ubiquitinated proteins in axons. However, inhibition of axonal lysosomes did not result in an accumulation of ubiquitinated proteins, suggesting that proteasomal degradation occurs locally, whereas proteins degraded by lysosomes can be transported to cell bodies for degradation.
Materials and Methods
Sympathetic Neuron Cultures
Superior cervical ganglion (SCG) neurons were obtained from P0 Sprague-Dawley rats (Charles River, Portage, MI) (Goslin 1998; Tsui-Pierchala et al. 2002). Neurons were plated on gas-plasma treated substrates (Harrick Plasma, Ithaca NY) coated with type-I collagen (BD Biosciences, San Jose, CA). Neurons were plated in one of three formats: as mass cultures in the center of 35 mm2 dishes, as droplets in the center of 22×22 mm No 1.5 coverslips for fluorescence imaging, or as compartmentalized “Campenot” chambers (Tyler Research, Edmonton, Canada) (Campenot et al. 2009; Tsui-Pierchala and Ginty 1999). SCG neurons were maintained in minimal essential medium (MEM) containing 50 ng/mL NGF, 10% FBS, the antimitotic agents aphidicolin and flourodeoxyuridine (Sigma, St. Louis, MO), and penicillin-streptomycin-glutamine supplement (Invitrogen, Carlsbad, CA). Procedures involving animals were in accordance with the University Committee on Use and Care of Animals (UCUCA) and the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
NGF Withdrawal and Reintroduction
Sympathetic neurons were maintained for 21 days to allow an NGF-independent, adult phenotype to develop (Easton et al. 1997; Goedert et al. 1978; Orike et al. 2001). To test the effects of NGF on expression of proteins associated with the proteolytic machinery, neurons in compartmentalized cultures were deprived of NGF for 48 hours on both their cell bodies and axons (Fig 3A), a period of time sufficient to extinguish NGF signaling (unpublished observations). Following NGF withdrawal, cultures were either stimulated with NGF (50 ng/mL) or with medium alone for 4 or 48 hours. Cellular lysates were then collected from the cell body and terminal compartments.
Figure 3. NGF regulates lysosomes and proteasomes, but not autophagesomes.

(A) SCG neurons were cultured for 21 days, deprived of NGF for 48 hours, and then reintroduced to NGF before protein extracts were collected from both the cell bodies (CB) and distal axons (DA). (B) Representative immunoblots are shown for LAMP2, PSMA2 and LC3 in SCG neurons stimulated on the distal axons with medium without NGF for 4 hours (-NGF CB, DA), 50ng/mL NGF for 4 hours (NGF 4 CB, DA), and 50ng/mL NGF for 48 hours (NGF 48 CB, DA). (C–E) Measurements from NGF-treated samples were normalized to the -NGF controls and actin and statistically compared (one-way ANOVA on ranks). (C) LAMP2 expression in the distal axons was significantly higher after 48 hours of NGF treatment as compared to both the cell bodies at 48 hours and the terminals at 4 hours. (D) PSMA2 expression increased significantly in the distal axons as compared to the cell bodies after 48 hours of NGF treatment. (E) No significant differences were measured between NGF treatment conditions for LC3. P<0.05 was considered to be significant.
Monitoring Protein Degradation
Inhibition of protein degradation was achieved by selectively inhibiting proteasomes using 5 µM epoxomicin (Biomol Research Laboratories, Plymouth Meeting, PA), lysosomes using 1 µM concanamycinA (Biomol Research Laboratories), and autophagy using 10 mM 3-Methyladenine (Sigma). All inhibitors were used at doses similar to those suggested by the manufacturer and other published reports for inhibition of the respective pathways with minimal acute toxicity. After 24 hours of inhibition, lysates were collected separately from the cell body and terminal compartments and examined for the accumulation of ubiquitinated proteins. Mass cultures of SCG neurons were exposed to inhibitors for up to 96 hours to observe their effects on cell viability. Phase contrast images were collected using a Zeiss Axiovert inverted microscope equipped with a 10X objective (Zeiss, Jena, Germany).
Immunocytochemisty and Microscopy
SCG cultures were fixed for 10 minutes in 4% paraformaldehyde buffered with PBS, permeabilized for 5 min in 0.1% Triton X-100 in PBS, and blocked for 1 hour in 4% BSA with shaking. Samples were incubated overnight with a rabbit-anti-proteasome subunits antibody (1:500 dilution; Invitrogen) on a rotary shaker at 4°C. Samples were then incubated for 2 hours with Alexa-488 goat-anti-rabbit secondary antibody (1:1000 dilution; Invitrogen) with shaking, and counterstained for 30 minutes with Alexa-568 phalloidin (1:200 dilution; Invitrogen). Confocal imaging was performed using a SP5 inverted confocal microscope equipped with a resonance scanner and a 60X glycerol immersion objective (Leica, Wetzlar Germany). Lysosomes were labeled using the fluorescent dye LysoTracker Yellow HCK-123 (Invitrogen). Live SCG cultures were incubated for 45 minutes in culture medium containing LysoTracker and were then imaged in resonance scan mode (16000 HZ) within a humidified culture chamber maintained at 37°C, 5% CO2 (TokaiHit, Shizuoka-ken, Japan). Images were collected as single optical sections with fluorescence and phase contrast images acquired simultaneously every 2 seconds to track lysosome movement. Nocodazole was applied for 30 minutes after each initial imaging session and additional images were collected to demonstrate the importance of microtubules for lysosome transport.
Immunoblotting
It was not possible to obtain sufficient amounts of protein to assay the biochemical activity of protein degradation systems in axons. Therefore, immunoblots were used to assess the relative abundance of proteins involved in protein degradation. Protein extracts were loaded onto 4–20% Tris-glycine gradient minigels for SDS-PAGE and were transferred to a PVDF membrane (Millipore, Billerica, MA). Membranes were rinsed in TBST (10 mM Tris, pH 7.4, 100 mM sodium chloride, 0.1% Tween 20) and blocked for 1 hour in TBST containing 2% BSA. Membranes were then incubated for 3 hours in the primary antibody diluted in 2% BSA-TBST at room temperature or overnight at 4°C. HRP-conjugated secondary antibodies (Jackson ImmunoResearch Labs, West Grove, PA), diluted in 2% BSA-TBST, were used for detection along with a chemiluminescent substrate (Thermo Scientific, Waltham, MA). The following primary antibodies were used: rabbit anti-PSMA2 (1:1000 dilution; Cell Signaling, Danvers, MA), rabbit anti-LAMP2 (1:2000 dilution; Invitrogen), rabbit anti-LC3B (1:1000 dilution; Cell Signaling), mouse anti-transferrin receptor (1:500 dilution; Invitrogen), rabbit anti-total ubiquitin (1:200 dilution; Sigma), rabbit anti-beclin (1:1000 dilution; Cell Signaling), rabbit anti-atg5 (1:1000 dilution; Cell Signaling), goat anti-actin (1:1000 dilution; Santa Cruz Biotechnology), rabbit anti-MAP2a/b (1:1000 dilution, Abcam), rabbit anti-tau (1:1000 dilution, Cell Signaling), mouse anti-multiubiquitin (1:500 dilution; Stressgen, Victoria, Canada), and mouse anti-p62 (1:1000 dilution; Abnova, Taipie, Taiwan). Axon growth in compartmental cultures can be highly variable from chamber to chamber, and differences in overall protein levels between experiments often cannot be avoided. Thus, actin immunoblotting was used for protein normalization between compartments. Actin is known to be a faithful marker for the amount of axons and cell bodies in this culture system and, thus, actin normalization has been adopted by many investigators that perform immunoblotting on compartmentalized cultures. Furthermore, actin accurately reflects the response of neurons to NGF, a neurotrophic factor that increases ATP synthesis, protein synthesis and growth, and indicates the overall increases or decreases in cellular metabolism and axon growth upon manipulation of the trophic status of the neurons.
Image Analysis and Statistical Analyses
Scanned images of x-ray films were imported into ImageJ and processed using the gel analysis tool. Integrated density values obtained from western analysis were reported as mean values ± SEM. Values were normalized to both actin loading controls reprobed from the same blot and in-blot (vehicle treated) controls. In some cases, the immunoblot images do not exactly match the quantified data. This is due to inherent variations in the amount of neuronal axons and cell bodies between individual compartmented chambers, reflected in the actin controls. Lysosome movement was measured using the Image-J MTrackJ plugin (Erik Meijering, Erasmus MC - University Medical Center Rotterdam, Netherlands). Equal numbers of lysosomes moving either retrogradely or anterogradely were tracked for at least 3 frames and the average speed was calculated and statistically compared. Student’s unpaired t-test and one-way ANOVA on ranks were used as tests of statistical significance where appropriate. P-values of less than 0.05 were considered to be significant. Data analysis was performed using Sigmaplot with Sigmastat (Systat Software, Chicago, IL).
Results
Expression and localization of protein degradation machinery in peripheral neurons
Immunolabeling and immunoblotting with antibodies for MAP2a/b, which is selectively localized in dendrites, and Tau, which is specific for axons, indicated that the vast majority neurites produced by sympathetic neurons in vitro were axons (Fig. 1). Furthermore, MAP2 was absent from the distal axon compartments of compartmentalized cultures, suggesting that only axons extend into the side compartments (Fig. 1C,D).
Figure 1. Axons are exclusively present in the side compartments of compartmentalized cultures.

(A–B) Immunofluorescence revealed that dendrites (MAP2-positive structures) were less abundant than axons and extended only a short distance from somas. (C) Immunoblotting of lysates from cell bodies and distal axons revealed that MAP2 protein was not present the distal axon compartment, in contrast to Tau that was highly enriched in distal axons, relative to actin. (D) The relative abundance of MAP2 protein declined over time in culture.
Lysotracker labeling was used to determine the distribution of lysosomes in the cell bodies, axons and terminals of mass cultures of SCG neurons (Fig. 2A,C,E). Lysosomes were abundant in the cell bodies of SCG neurons and in large caliber axons extending away from the cell bodies (Fig 2A). Lysosomes were also present in the distal axon and terminal regions of SCG neurons (Fig. 2A,C,E). Acidic vesicles (lysosomes, late stage autophagic vesicles) were observed as either small (<1µm) single structures or larger (≥1µm) aggregates. It is possible that larger structures represented autophagic vesicles that fused with lysosomes.
Figure 2. Lysosomes, proteasomes and autophagesomes are expressed in both the cell bodies and axons of peripheral neurons.

Lysotracker was used to identify lysosomes (green punctuate structures) in (A) the cell bodies (dashed circles), (C) the axons and (E) the terminals/growth cones of SCG neurons (Scale = 10µm). Immunofluorescence was used to determine the localization of proteasomes (green) in (B) the cell bodies, (D) the axons and (F) the terminals/growth cones (Scale = 100µm). Phalloidin was used as a counterstain to label F-actin (red). (G) Immunoblots for protein extracts obtained from SCG mass cultures (SCG MC), compartmented SCG cell bodies (SCG CB) and compartmented SCG distal axons (SCG DA) confirmed that LAMP2, PSMA2 and three markers for autophagic vesicles (LC3, ATG5 and Beclin-1) were expressed in both peripheral nerve tissue and all parts of primary sympathetic neurons. (H) Differences in relative expression level of proteolytic machinery were observed between cell bodies and distal axons (p<0.01 by Student’s unpaired t-test). (I) Dorsal root ganglion (DRG) neurons and spinal motor (SM) neurons express PSMA2, LAMP2 and LC3 in both the cell bodies and distal axons. These data suggest that some results from SCG cultures may be generalized to other neuronal cell types that innervate peripheral targets.
Immunocytochemical labeling for proteasome subunits revealed that proteasomes were located in both the cell bodies and distal axons/terminals of SCG neurons (Fig. 2B,D,F). Proteasome labeling was diffuse and evenly distributed throughout the cell, as suggested by the large extent of overlap with phalloidin labeling of the actin cytoskeleton. Proteasome and actin signals did not overlap completely and proteasomes, for example, were not observed in growth cones (Fig. 2F, as indicated by arrows).
Immunoblotting of protein extracts collected from mass cultures of SCG neurons, and cell bodies and distal axons of compartmentalized sympathetic neurons confirmed the results from lysotracker labeling and provided additional insights into the localization of protein degradation machinery. Immunoblotting demonstrated that lysosome (LAMP2), proteasome (PSMA2) and autophagesome (LC3, ATG5 and Beclin-1) markers were all present in both the distal axons and cell bodies of SCG neurons (Fig 2G). LAMP2 was observed as a fully glycosylated species (~110 kDa) and in hypoglycosylated form (<110 kDa). The fully glycosylated (mature) species was enriched in SCG axons, suggesting that lysosomal constituents are post-translationally modified and assembled in the cell bodies before being transported to the axons in their mature form. Quantification of immunoblots revealed that only LAMP2 showed a significant difference in relative expression level between SCG cell bodies and terminals, but this was mostly due to an absence of hypoglycosylated LAMP2 in distal axons (Fig. 2H). Expression of proteasomes, lysosomes and autophagesomes was also observed both in the cell bodies and distal axons of sensory neurons from the dorsal root ganglion and spinal motor neurons, indicating that these data may be generalized to other neurons that innervate peripheral structures, not just sympathetic neurons (Fig. 2I). These results indicate that protein degradation pathways are present in the distal axons of neurons, although somewhat less abundant than they are in the cell bodies.
NGF regulates the expression of the proteolytic machinery
NGF is a target-derived neurotrophic factor that is required for axon growth and target-mediated survival during development as well as maintenance of adult neurons (Ernsberger 2009; Sofroniew et al. 2001). Because protein degradation facilitates these processes we hypothesized that NGF-dependant growth and maintenance are linked to NGF-dependent regulation of the proteolytic machinery. After 48 hours of NGF withdrawal, NGF was reintroduced to the distal axons of sympathetic neurons and protein extracts were collected either 4 or 48 hours later from both the cell bodies and distal axons (Fig. 3A,B). Immunoblots were performed using protein extracts collected from neurons given medium alone, stimulated with NGF for 4 hours or stimulated with NGF for 48 hr (Fig. 3). The expression levels of both LAMP2 and PSMA2 increased in the distal axons after NGF stimulation. LAMP2 expression was significantly increased in the distal axons as compared to the cell bodies after 48 hours of NGF treatment (Fig. 3C). After 48 hours of NGF stimulation LAMP2 expression was also significantly higher in the distal axons than after 4 hours of NGF stimulation. PSMA2 expression was not significantly different after 4 hours of stimulation, but increased significantly in the distal axons after 48 hours NGF application (Fig. 3D). PSMA2 and LAMP2 increased by more than 4-fold as compared to distal axons treated with medium alone. In contrast, no significant differences in LC3 expression were observed between any of the conditions (Figure 3E). LC3 was observed in two isoforms in SCG neurons: the larger, 16 kDa isoform represents the senescent LC3-I form and the smaller, 14 kDa isoform represents the cleaved (active) LC3-II isoform (Klionsky et al. 2008). LC3 cleavage indicates membrane fusion events that cause the formation of a fully-functional autophagic vesicle. Although no significant differences in LC3 expression were observed upon NGF stimulation, the ratio of LC3-II/LC3-I suggested that autophagy occurred more frequently in cell bodies than in the distal axons of SCG neurons (data not shown). In summary, these results suggest that NGF regulates the axonal expression of proteins associated with lysosomes and proteasomes, but did not regulate proteins required for autophagy.
Differential sensitivity of sympathetic neurons to inhibition of degradation pathways
Since NGF, a potent growth and survival factor for SCG neurons, upregulated the expression of proteins required for proteasomal and lysosomal degradation, we investigated how sensitive these neurons would be to inhibition of these protein degradation pathways. Neurons were exposed to one of three inhibitors of protein degradation, concanamycin, epoxomicin or 3-MeA, which inhibit lysosomes, proteasomes and autophagosomes, respectively. The toxic effects of these and other agents are typically obvious in SCG cultures, and are manifested as changes in structure of the cell soma and axonal network.
Sympathetic neurons exposed to the vehicle alone (DMSO) did not undergo degeneration or cell death over 96 hours (Fig. 4). When neurons were exposed to epoxomicin, no acute effects were observed. As expected, the sustained inhibition of proteasomes (96 hours) caused neurite degeneration and cell death, as evidenced by detachment of cell bodies and partial disintegration of processes that remained attached to the substrate (Fig. 4D–F). When neurons were given concanamycinA, cell death also occurred within 96 hours (Fig. 4G–I). However, we observed that neurons were capable of surviving short periods of inhibition (24–48 hours). Inhibition of autophagy using 3-MeA did not result in death of SCG neurons even after 96 hours of exposure (Fig. 4J–L). In fact, no discernable differences in cell morphology were observed between 3-MeA treated neurons and neurons given the vehicle alone, suggesting that autophagy was not necessary for the survival of sympathetic neurons maintained in nutrient-rich conditions over a period of 4 days. Based on these observations we chose treatment time points within the acute (24 hour, low toxicity) period to minimize any toxic effects in subsequent experiments.
Figure 4. Differential sensitivity of sympathetic neurons to proteasome, lysosome and autophagy inhibitors.

Mass cultures of SCG neurons treated with vehicle control (A–C), epoxomicin (D–F), conconamycinA (G–I) or 3-Methyl-Adenine (J–L) were observed over the course of 96 hours. SCG morphology was unaffected upon exposure to 3-Methyl-Adenine for over 96 hours. Application of proteasome or lysosome inhibitors (epoxomicin and concanamycinA respectively), in contrast, resulted in death of SCG neurons over the course of several days, as indicated by the detachment of cell somas and presence of granulated and degenerating neuronal processes. Scale bar = 100 µm.
Inhibition of proteasomes in axons causes the local accumulation of ubiquitinated proteins
It has been proposed that one cause of neurodegeneration is the accumulation of toxic or misfolded proteins in the axons of neurons (Taylor et al. 2002). We hypothesized that acute inhibition of the proteolytic pathways in either the cell bodies or distal axons of SCG neurons would result in the accumulation of ubiquitinated proteins in these respective compartments. A treatment period of 24 hours was chosen based on the observations that neuronal morphology was unaffected after the acute treatment with either epoxomicin or concanamycinA (Fig. 4 E,H).
Representative immunoblots of sympathetic neurons maintained in compartmentalized cultures that were given epoxomicin on either the cell bodies or distal axons are shown in Figure 5A–C. Levels of the proteasome component PSMA2 were measured to determine if perturbations in protein flux through the proteasome had an effect on the level of proteasome expression (Korolchuk et al. 2010). PSMA2 expression increased in the distal axons in response to proteasome inhibition on the terminals (Figure 5D). Ubiquitinated proteins increased in the SCG terminals after epoxomicin was applied to the terminals (Fig. 5E,F). An accumulation of multi-ubiquitinated proteins destined for the proteasome was also observed in the cell bodies after proteasomal degradation was selectively inhibited on the cell bodies. Interestingly, similar trends were observed for multi-ubiquitinated proteins and total-ubiquitin (both proteasome and lysosome), which accumulated in axons when proteasomes were inhibited in axons (Fig. 5F). These data suggest that proteasome inhibition results in the local accumulation of proteins targeted for proteasome degradation and that compensatory mechanisms, such as retrograde transport of ubiquitinated proteins to another cellular compartment, do not aid in degradation after acute proteasome inhibition.
Figure 5. Localized inhibition of proteasomes results in the accumulation of ubiquitinated proteins in compartmentalized neuronal cultures.
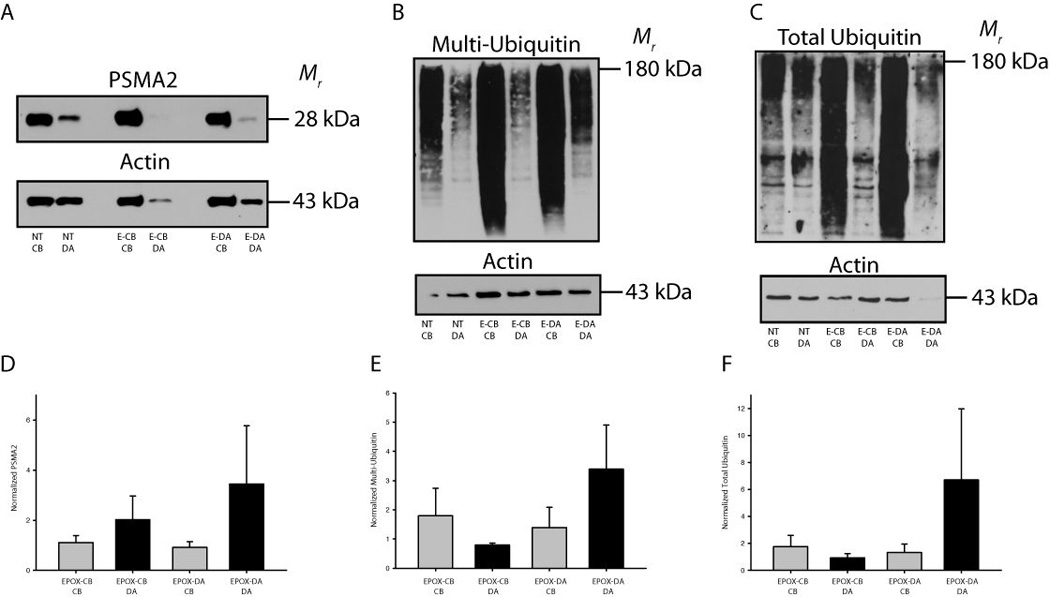
(A–C) Representative western blots for PSMA2 (proteasomes), multi-ubiquitin and total ubiquitin are shown. (D) Inhibition of proteasome function in distal axons (E-DA) or cell bodies (E-CB) resulted in increases in PSMA2 expression. (E–F) Ubiquitinated proteins accumulated in the axons/terminals following proteasome inhibition in the terminals. A modest accumulation of ubiquitinated proteins also occurred in the cell bodies after proteasome inhibition in the cell bodies. Values were normalized to actin and an in-blot vehicle treated control and displayed as mean ± SEM.
Inhibition of lysosomal degradation in axons does not cause local protein accumulation
We selectively inhibited lysosomes in either the cell bodies or the distal axons of SCG neurons to determine if deficiencies in lysosomal degradation in axons results in the accumulation of ubiquitinated proteins. LAMP2 expression was significantly higher in the cell bodies of SCG neurons as compared to the distal axons when lysosomes were inhibited in the cell body compartment. Likewise, LAMP2 expression was induced significantly in the distal axons as compared to the cell bodies when lysosomes were inhibited in the terminal compartments (Fig. 6A–B). These data suggest that either more LAMP2 protein is synthesized or that lysosomes are selectively transported in response to a decrease in lysosomal protein degradation to the cellular compartment where lysosomes are inhibited. Since fully glycosylated LAMP2 is enriched in axons it is likely that lysosomal constituents are synthesized in the cell bodies and trafficked to the axons after assembly.
Figure 6. Selective inhibition of lysosomes does not cause the accumulation of proteins in compartmentalized neuronal cultures.

ConcanamycinA was used to selectively inhibit lysosomes in either the cell body or distal axon compartments of SCG cultures. (A–B) LAMP2 protein expression was greater in the cell bodies after inhibition of lysosomes in the cell body compartment. LAMP2 expression was also significantly greater in the distal axons (DA) after inhibition of lysosomes in the terminal compartment. No significant accumulation of transferrin receptor (C–D) or total ubiquitin (E–F) was observed following selective lysosome inhibition on either the cell bodies or the distal axons. Bars represent mean ± SEM. P≤0.05 was considered significant by one way ANOVA on ranks. Values were normalized to actin and an in-blot vehicle treated control.
In contrast to proteasome inhibition, lysosome inhibition did not result in any trends in the local accumulation of proteins. Transferrin receptor, a model receptor for lysosomal degradation, did not accumulate in response to local lysosome inhibition (Fig. 6C–D). Importantly, no significant differences or trends between experiments were observed for the local accumulation of ubiquitinated proteins, suggesting that lysosome inhibition in subcellular regions of neurons does not lead to the local accumulation of proteins targeted for degradation (Fig. 6E–F). In addition, inhibition of autophagy did not result in local protein accumulation in either axons or cell bodies, as indicated by examination of the expression of the ubuiqitin binding adaptor protein, p62, and ubiquitinated proteins (Fig. 7). In summary, the local inhibition of lysosomes does not cause the regional accumulation of proteins targeted for lysosome degradation, such as ubiquitinated proteins and the transferrin receptor.
Figure 7. Inhibition of autophagy does not result in a significant accumulation of degradation products.

No significance differences between treatment conditions or trends among experiments were observed for (A) LC3, (B) the ubiquitin adaptor protein p62 or (C) total ubiquitin in response to treatment with the autophagy inhibitor 3-methyl adenine (3MA). Significance was tested using one-way ANOVA on ranks.
Lysosomes are transported between cell bodies and axons via microtubules
The lack of ubiquitinated protein accumulation following local lysosome inhibition in axons could be linked to the transport of lysosomes within axons. Although lysosomes are capable of moving short distances along actin filaments and microtubules in non-neuronal cells (Cordonnier et al. 2001), it is not known whether lysosomes can be transported over long distances in axons in either a retrograde or anterograde direction. To examine the possibility that lysosome transport prevented the accumulation of ubiquitinated proteins following lysosome inhibition, we examined the transport properties of lysosomes within axons.
Tracking lysosomes in living sympathetic neurons revealed that lysosomes moved considerable distances along the axons and terminals (Fig. 8A–F). Inspection of overlaid phase contrast and lysosome images using the fluorescent dye Lysotracker demonstrated that acidic vesicles moved retrogradely from the distal axons of SCG neurons (Griffiths et al. 1988). Two populations of lysosomes were observed in neurons: some lysosomes moved anterogradely towards the terminals, while other lysosomes moved retrogradely away from the terminals towards the cell bodies. Lysosomes segregated into two groups based on the direction and speed of their movement (Fig. 8G). Lysosomes moving retrogradely progressed shorter distances over a given time than those moving anterogradely. This data was used to compute the average instantaneous velocity of movement for lysosomes moving anterogradely and for those moving retrogradely. Lysosomes moving anterogradely within axons travelled at an average velocity of 1.2 µm/second (Fig. 8H). Based on their speed and relatively continuous rate of movement, it is likely that such lysosomes were transported along microtubules by kinesins. The population of lysosomes moving retrogradely travelled at a slower rate (0.46 µm/second) and moved discontinuously, indicating that they were transported along microtubules by dyneins (Fig. 8H).
Figure 8. Lysosomes move retrogradely and anterogradely within the axons of sympathetic neurons.

Lysosome movement was tracked in the axons of SCG neurons to assess the impact of lysosome translocation on axonal protein degradation. (A–F) Lysosomes were observed to move bidirectionally in the terminals of SCG axons. White arrows indicate the retrograde movement of a single lysosome over the course of 12 seconds. Scale bar = 10µm. (G) Movement profiles for individual lysosomes were color-coded based on the direction of movement (red for anterograde and cyan for retrograde). Lysosomes segregated into two populations based on their movement profiles. (H–I) Average velocity was plotted for all lysosomes, those moving anterogradely and those moving retrogradely. Average velocity was also measured in the presence of nocodazole. Thick dashed lines represent the mean. Solid lines within the boxes represent the median with first (top and bottom of box) and second (whisker bar) standard deviations. Grey circles represent individual data points. P≤0.05 was considered to be significant by Student’s unpaired T-test.
As expected from the data above, lysosome movement was abolished by destabilizing microtubules with nocodozole (Fig. 8I). In the absence of nocodazole, lysosomes moved at an average rate of 0.8 µm/second. After 30 minutes of nocodazole treatment the average velocity of lysosomes was reduced to 0.2 µm/second. Residual movement was due primarily to Brownian-type motion within the cell, thus demonstrating that lysosomes depend completely on microtubule transport to move between cell bodies and terminals.
Discussion
Our results indicate that protein degradation is pervasive within the axons of peripheral neurons. Proteins associated with all three protein degradation pathways (proteasomal, lysosomal and autophagic) were present in both the axons and cell bodies of sympathetic neurons. These proteins were also present in vitro within the axons of DRG sensory neurons and spinal motor neurons, suggesting that axonal protein degradation is a general property shared among peripheral neurons. The levels of proteins associated with proteasome and lysosome degradation were upregulated in response to NGF treatment, linking the processes of axon growth and maintenance with protein degradation. The inhibition of proteasome activity selectively in axons resulted in local accumulation of ubiquitinated proteins in axons. In contrast, inhibition of lysosome activity in axons did not result in accumulation of either transmembrane receptors typically degraded by lysosomes, such as the transferrin receptor, or ubiquitinated proteins, indicating a potential role of lysosome translocation in protein degradation. Interestingly, retrograde and anterograde lysosome movement were observed in the axons of sympathetic neurons, which were both disrupted upon the destabilization of microtubules with nocodazole. This raises the possibility that subcellular lysosomal inhibition does not cause the accumulation of ubiquitinated proteins because of the transport of lysosomes to other cellular regions.
Localization of protein degradation machinery in axons
The role of protein degradation in neuronal function and maintenance has been appreciated primarily in the context of protein turnover within cell somas and dendrites (Arancibia-Carcamo et al. 2009; Colledge et al. 2003; Goldberg 2003). Proteasome degradation has been linked to a number of dynamic processes that occur within dendrites, most notably dendrite morphogenesis, as well as activity dependant plasticity and long term potentiation (DiAntonio and Hicke 2004; Ehlers 2003). In dendrites proteasome function is closely linked to synaptic activity. This phenomenon has been illustrated by the recruitment and enrichment of proteasomes into dendritic spines (Bingol and Schuman 2006). Lysosomal degradation has also been appreciated in the contexts of neurite remodeling as well as turnover of receptor/complexes at postsynaptic structures (Luzio et al. 2007; Song et al. 2008). Alterations in protein degradation by lysosomes and proteasomes has been linked to protein accumulation disorders and degeneration/regeneration of axons (Rubinsztein 2006). While it is likely that some protein degradation occurs following retrograde translocation of endosomal vesicles to cell bodies, the abundance of lysosomes, proteasomes and autophagic proteins within distal axons suggests that local axonal protein degradation contributes to proteome homeostasis within axons. The accumulation of ubiquitinated proteins in axons upon local proteasome inhibition, along with the anterograde and retrograde transport of mature lysosomes, further supports this notion.
NGF and regulation of protein degradation
NGF supports the target-dependent survival, growth and maintenance of adult sympathetic and sensory neurons (Ernsberger 2009; Sofroniew et al. 2001). Our observation that proteasome- and lysosome-associated protein levels were upregulated by NGF selectively in axons suggests that axonal protein turnover is necessary for axonal growth and maintenance. Therefore, a loss of trophic factor support may contribute to degenerative disorders associated with protein accumulation in axons, such as Alzheimer’s and Parkinson’s diseases, by exacerbating the accumulation of misfolded and/or ubiquitinated proteins (Hennigan et al. 2007). It will be important in future studies to determine the precise function that the NGF-induced increase in protein degradation has in axons, especially since levels of NGF in target tissues often decline after development and during adulthood. Importantly, the increase in proteasomal and lysosomal proteins in axons in response to NGF was greater than the increase in actin, which is a faithful marker of the trophic effects of NGF on global protein levels.
The levels of both PSMA2 and LAMP2 increased selectively in the axons of NGF-stimulated neurons at 48 hours. The levels of LC3 did not increase in axons following NGF stimulation, and thus serves as an important control (in addition to actin) that suggests that NGF selectively upregulates proteins associated with proteasomes and lysosomes in axons. It is important to note that autophagy is active in NGF-maintained axons, as evidenced by the appearance of the active form of LC3 (LC3-II) in axonal lysates. This demonstrates that the lack of evidence for NGF regulation of autophagy is not due simply to an absence of active autophagy in these axons. Overall, these data suggest that an augmentation of protein degradation is necessary for some specific aspect of NGF-mediated axon growth or maintenance, rather than simply being necessary as a housekeeping function for axon homeostasis.
Transport properties of protein degradation machinery
The cell bodies and terminals of most neurons are often separated by large distances, requiring the transport of mRNA, protein components and complete protein complexes/organelles for the maintenance of functional axons and terminals. By inhibiting the function of proteolytic machinery within either cell bodies or axons it was possible to understand how transport properties can affect the flux of proteins through degradation machinery. Inhibition of proteasome function resulted in the accumulation of ubiquitinated proteins in the inhibited compartment, suggesting that the UPS system lacks mechanisms for transport of ubiquitinated proteins out of the inhibited compartment. This supports previous reports indicating that local degradation is particularly important in the UPS system (Bingol and Schuman 2006; DiAntonio and Hicke 2004). Indeed, proteasomes have been observed to aggregate in regions of high protein turnover. In contrast, protein accumulation was not observed following lysosome inhibition. However, an increase in lysosome-associated protein expression was observed in the inhibited compartments suggesting that either lysosome expression was upregulated or that lysosome translocation occurred in response to a decrease in local protein degradation. The lack of protein accumulation in concanamycinA-inhibited compartments is likely due to the transport of endosomes or lysosomes out of the inhibited compartment to a region of the neuron where degradation could occur.
Lysosomes were observed to move great distances both anterogradely and retrogradely within neurons, consistent with some previous studies (Overly et al., 1996). These data support a model in which lysosomal cargo can be either degraded in axons or transported between axons and cell bodies for degradation. During intracellular transport, lysosomes can move bidirectionally and undergo fusion events with endosomes and autophagesomes, thus providing a dynamic and locally tuned mechanism for protein degradation to accompany the stable local degradation of the ubiquitin-proteasome system. It is likely that autophagy is also occurring in axons given the presence of fully mature autophagosomes. Even so, a number of studies have demonstrated that lysosomes function independently of autophagosomes to degrade transmembrane proteins. For example, most plasma membrane-located receptors are trafficked to lysosomes for degradation and this process does not involve autophagy. Rather, autophagy is primarily involved in the degradation of organelles and large cellular structures, often as a response to nutrient deprivation. The observation that lysosomes can move within neurons to overcome deficiencies in local degradation has implications for neurodegenerative diseases, raising the possibility that diseases affecting axonal transport mechanisms may impede the adaptive ability of the neuron to remove ubiquitinated proteins that are accumulating in axons due to the disease process, thereby impacting the synaptic function of the neuron. It is thus possible that the apparent toxicity of some protein inclusion structures, such as those related to Aβ and tau, is caused in part by deficiencies in the transport of protein degradation machinery within axons.
Acknowledgments
Grant Information: This work was supported by an NIH R01 NS058510 (B.A.P.) and by the TEAM Tissue Engineering and Regeneration Training Grant NIDCR DE007057 (J.P.F.).
Literature Cited
- Arancibia-Carcamo IL, Yuen EY, Muir J, Lumb MJ, Michels G, Saliba RS, Smart TG, Yan Z, Kittler JT, Moss SJ. Ubiquitin-dependent lysosomal targeting of GABA(A) receptors regulates neuronal inhibition. Proc Natl Acad Sci U S A. 2009;106(41):17552–17557. doi: 10.1073/pnas.0905502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441(7097):1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Campenot RB, Lund K, Mok SA. Production of compartmented cultures of rat sympathetic neurons. Nat Protoc. 2009;4(12):1869–1887. doi: 10.1038/nprot.2009.210. [DOI] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243(4898):1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40(3):595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordonnier MN, Dauzonne D, Louvard D, Coudrier E. Actin filaments and myosin I alpha cooperate with microtubules for the movement of lysosomes. Mol Biol Cell. 2001;12(12):4013–4029. doi: 10.1091/mbc.12.12.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Hicke L. Ubiquitin-dependent regulation of the synapse. Annu Rev Neurosci. 2004;27:223–246. doi: 10.1146/annurev.neuro.27.070203.144317. [DOI] [PubMed] [Google Scholar]
- Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci. 1997;17(24):9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6(3):231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Ernsberger U. Role of neurotrophin signalling in the differentiation of neurons from dorsal root ganglia and sympathetic ganglia. Cell Tissue Res. 2009;336(3):349–384. doi: 10.1007/s00441-009-0784-z. [DOI] [PubMed] [Google Scholar]
- Goedert M, Otten U, Thoenen H. Biochemical effects of antibodies against nerve growth factor on developing and differentiated sympathetic ganglia. Brain Res. 1978;148(1):264–268. doi: 10.1016/0006-8993(78)90401-8. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G. Culturing Nerve Cells. Cambridge: MIT Press; 1998. [Google Scholar]
- Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell. 1988;52(3):329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- Hennigan A, O'Callaghan RM, Kelly AM. Neurotrophins and their receptors: roles in plasticity, neurodegeneration and neuroprotection. Biochem Soc Trans. 2007;35(Pt 2):424–427. doi: 10.1042/BST0350424. [DOI] [PubMed] [Google Scholar]
- Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2(3):195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105(52):20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen L, Lindholm D. The ubiquitin proteasome system in synaptic and axonal degeneration: a new twist to an old cycle. J Cell Biol. 2004;165(1):27–30. doi: 10.1083/jcb.200311091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584(7):1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3(4):323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011;31(21):7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8(8):622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orike N, Thrasivoulou C, Wrigley A, Cowen T. Differential regulation of survival and growth in adult sympathetic neurons: an in vitro study of neurotrophin responsiveness. J Neurobiol. 2001;47(4):295–305. doi: 10.1002/neu.1036. [DOI] [PubMed] [Google Scholar]
- Overly CC, Rieff HI, Hollenbeck PJ. Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J Cell Sci. 1996;109(Pt 5):971–980. doi: 10.1242/jcs.109.5.971. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Song JW, Misgeld T, Kang H, Knecht S, Lu J, Cao Y, Cotman SL, Bishop DL, Lichtman JW. Lysosomal activity associated with developmental axon pruning. J Neurosci. 2008;28(36):8993–9001. doi: 10.1523/JNEUROSCI.0720-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9(11):826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296(5575):1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19(1):94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ginty DD. Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J Neurosci. 1999;19(19):8207–8218. doi: 10.1523/JNEUROSCI.19-19-08207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Milbrandt J, Johnson EM., Jr NGF utilizes c-Ret via a novel GFL-independent, inter-RTK signaling mechanism to maintain the trophic status of mature sympathetic neurons. Neuron. 2002;33(2):261–273. doi: 10.1016/s0896-6273(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25(2):331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]