

Abstract
Activation of the dynorphin/κ-opioid receptor (KOR) system by repeated stress exposure or agonist treatment produces place aversion, social avoidance, and reinstatement of extinguished cocaine place preference behaviors by stimulation of p38α MAPK, which subsequently causes the translocation of the serotonin transporter (SERT, SLC6A4) to the synaptic terminals of serotonergic neurons. In the present study we extend those findings by showing that stress-induced potentiation of cocaine conditioned place preference occurred by a similar mechanism. In addition, SERT knock-out mice did not show KOR-mediated aversion, and selective reexpression of SERT by lentiviral injection into the dorsal raphe restored the prodepressive effects of KOR activation. Kinetic analysis of several neurotransporters demonstrated that repeated swim stress exposure selectively increased the Vmax but not Km of SERT without affecting dopamine transport or the high-capacity, low-affinity transporters. Although the serotonergic neurons in the dorsal raphe project throughout the forebrain, a significant stress-induced increase in cell-surface SERT expression was only evident in the ventral striatum, and not in the dorsal striatum, hippocampus, prefrontal cortex, amygdala, or dorsal raphe. Stereotaxic microinjections of the long-lasting KOR antagonist norbinaltorphimine demonstrated that local KOR activation in the nucleus accumbens, but not dorsal raphe, mediated this stress-induced increase in ventral striatal surface SERT expression. Together, these results support the hypothesis that stress-induced activation of the dynorphin/KOR system produces a transient increase in serotonin transport locally in the ventral striatum that may underlie some of the adverse consequences of stress exposure, including the potentiation of the rewarding effects of cocaine.
Introduction
Although acute stress exposure can produce proadaptive responses, prolonged stress exposure can evoke maladaptive responses including increased risk of mood disorders and drug addiction (Koob, 2008; Krishnan and Nestler, 2008). Prior studies have identified the endogenous dynorphin/κ-opioid receptor (KOR) system as a key regulator of this stress response in animal models of depression-like and addiction-like behaviors (Bruchas et al., 2010; Knoll and Carlezon, 2010). However, the neuronal sites of action and signaling mechanisms responsible for these behaviors are not yet understood.
Sustained KOR activation by stress-induced release of endogenous dynorphins leads to G-protein receptor kinase 3 (GRK3)-dependent, p38α mitogen-activated protein kinase (MAPK) activation, which has been implicated previously in mediating both aversive effects of stress and stress-induced reinstatement of drug seeking (Bruchas et al., 2007a, 2011; Land et al., 2009). In addition, a possible role for KOR-mediated regulation of serotonin transporter (SERT, SLC6A4) by p38α MAPK has been suggested (Bruchas et al., 2011), although the underlying kinetic mechanisms, brain region(s) involved, and transporter selectivity of this effect remains unknown. Altered SERT and dopamine transporter (DAT; SLC6A3) functions have been linked to stress, prodepressive, and proaddictive behaviors (Kuhar, 1992; Lesch et al., 1996; Heinz et al., 1998; Malison et al., 1998; Laasonen-Balk et al., 1999; Sora et al., 2001; Lira et al., 2003; Wellman et al., 2007). Interestingly, previous reports demonstrated a role for p38 MAPK in the modulation of SERT and DAT function in vitro (Zhu et al., 2004, 2005; Samuvel et al., 2005), further supporting the suggested role for this MAPK in monoamine transport regulation. Monoamines can also be sequestered by low-affinity, high-capacity transporters such as the organic cation transporters (Octs) and the plasmalemmal monoamine transporters (Daws, 2009; Hagan et al., 2011). Stress exposure has been shown to decrease the function of Oct3 at micromolar concentrations of serotonin (5-HT) (Baganz et al., 2010), although a role for KOR was not assessed.
In the present study, we used rotating disk electrode voltammetry (RDEV) to measure neurotransmitter uptake kinetics in synaptosomal preparations (Earles and Schenk, 1998; Schenk et al., 2005; Hagan et al., 2010). To determine whether stress exposure can regulate these transporters, we measured uptake of 5-HT by SERT, dopamine (DA) by DAT, and 5-HT and DA by low-affinity, high-capacity transporters following acute or repeated stress exposure, direct KOR activation, or during nicotine withdrawal. To determine whether stress exposure can regulate the surface expression of SERT, we used a biotinylation approach to label cell surface proteins. In this study, we found that rather than having a global effect on serotonergic tone, repeated stress exposure selectively regulated SERT function via p38 MAPK in the synaptic terminals of dorsal raphe (DR) neurons projecting to the ventral striatum (VS). We posit that this stress-induced alteration in ventral striatum surface SERT expression results in a transient and localized hyposerotonergic state in the nucleus accumbens, an effect that may underlie prodepressive and proaddictive behaviors.
Materials and Methods
Animals and housing
Male C57BL/6 mice (Charles River Laboratories) or transgenic mice on a C57BL/6 genetic background weighing 18–30 g were used in these experiments. Homozygous GRK3 knock-out (−/−) (Peppel et al., 1997), SERT knock-out (−/−) (Jackson Laboratories), KOR (−/−) (Hough et al., 2000), and respective wild-type (WT) (+/+) littermate control mice were prepared by heterozygous crosses and genotyped as described previously (Xu et al., 2004; Hagan et al., 2010). Mice were group housed, two to four per cage, and the housing rooms were maintained on a 12 h light/dark cycle (lights on at 07:00) with food pellets and water available ad libitum. Animal procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Generation of p38α conditional knock-out (p38αCKOePet) transgenic mice
Breeding.
A floxed p38α MAPK mouse line (Nishida et al., 2004) with loxP sites flanking the third exon of p38α was obtained from the RIKEN Bioresearch Center (Tsukuba, Japan). p38αlox/+ heterozygotes were crossed to mice broadly expressing Cre recombinase under the Mox2 promoter (Tallquist and Soriano, 2000) to generate p38αΔ/+; Mox2Cre/+ heterozygotes bearing a null p38α allele no longer susceptible to Cre recombination, thereby guarding against ectopic germ-line excision of the floxed p38α allele during later generations of breeding. The null p38αΔ allele was segregated away from the Mox2-Cre allele by backcrossing with C57BL/6 wild-type mice.
The p38αΔ/+ mice globally heterozygous for p38α were then crossed to the ePet1-Cre line (Scott et al., 2005) to yield p38αΔ/+ mice, also heterozygous for ePet1-Cre. ePet1-Cre is a transgene driven by an enhancer element for a transcription factor (Pet1) that is uniquely expressed in serotonergic neurons (Scott et al., 2005). These mice were then mated with p38αlox/lox mice to give p38αΔ/lox; ePet1Cre/+ conditional knock-out (CKO) progeny (p38αCKOePet) as well as p38αΔ/lox (p38αΔ/lox) and p38αlox/+ (p38α+/+) mice, which can be regarded as littermate controls carrying one or two functional p38α alleles, respectively. Conditional knock-out mice showed no apparent differences in growth, lifespan, or overt health from either their p38αΔ/lox (heterozygote) or functionally wild-type littermates, and were produced in expected Mendelian frequency (Bruchas et al., 2011).
Genotyping.
Mice weaned at 28 d of age were briefly anesthetized with isoflurane (Hospira), and a 0.5 cm tail biopsy was obtained. Tail tissue was digested by proteinase K overnight and genomic DNA purified using Qiagen DNEasy columns according to manufacturer's instructions. Tail DNA was then used as a template for PCR using Promega GoTaq Flexi polymerase with one of two buffers (5× Green GoTaq Flexi, Promega catalog #M8911, or TaqDNA polymerase 10× PCR reaction buffer, Promega catalog #M1902) used depending on the reaction. PCR products were then resolved on a 1.5% agarose electrophoresis gel and photographed under UV illumination for analysis.
PCR screening was performed using the following primers. A3 (5′-ATGAGATGCAGTACCCTTGGAGACCAGAAG-3′) and A4 (5′-AGCCAGGGCTATACAGAGAAAAACCCTGTG-3′) were used for the floxed and wild-type p38α alleles, giving bands of 230 and 180 bp, respectively. Primers A1 (5′-CCACAGAAGAGATGGAGCTATATGGATCTC-3′) and A4 were used to detect the null p38αΔ allele as a 420-bp PCR product. The Mox2-Cre and ePet1-Cre transgenes were detected using 5′-AGCGTTCGAACGCACTGATTTCG-3′ and 5′-CGCCGTAAATCAATCGATGAGTTG-3′, yielding a 330 bp band.
Drugs and chemicals
Cocaine-HCl, norbinaltorphimine (norBNI)-HCl, and (±)U50,488(2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide) were provided by the National Institute of Drug Abuse Drug Supply Program (Bethesda, MD) and were dissolved in 0.9% saline. (−)-Nicotine hydrogen tartrate salt [(−)-1-methyl-2-(3-pyr-idyl)pyrrolidine (+)-bitartrate salt (nicotine)] was purchased from Sigma and was dissolved in 0.9% saline. Citalopram hydrobromide was from Tocris Bioscience and was dissolved in 0.9% saline. KCl, MgSO4, paraformaldahyde, and Tween-20 were from Fisher Scientific. CaCl2, KH2PO4, NaCl, and NaHCO3 were from JT Baker. Bovine serum albumin (BSA), CHAPS, EDTA, glucose, HEPES, Na-deoxycholate, Ponceau-S reagent, sodium dodecyl sulfate, sucrose, Tris buffer, and Triton X-100 were from Sigma-Aldrich. GBR 12935 (1-(2-(diphenylmethoxy)ethyl)-4-(3-phenylpropyl)piperazine; DAT inhibitor), nisoxetine hydrochloride [norepinephrine transporter (NET) inhibitor], and paroxetine hydrochloride (SERT inhibitor) were from Sigma-Aldrich and were dissolved in physiological buffer as described previously (Hagan et al., 2010). Dopamine hydrochloride and serotonin hydrochloride were from Sigma-Aldrich and were dissolved in pH 7.2 PBS as described previously (Hagan et al., 2010). Glycine and powdered milk were from Bio-Rad. EZ-link Sulfo-NHS-SS-Biotin was from Pierce.
Forced swim stress
Mice were exposed to a modified Porsolt forced swim stress as described previously (Porsolt et al., 1977; McLaughlin et al., 2003a). All swim sessions were performed in 30 ± 1°C water. After each swim session, mice were removed, towel dried, and returned to their home cage for at least 6 min before further testing. For acute stress exposure (A-FSS), mice were exposed to one 15 min swim 10 min before decapitation and synaptosome generation. For repeated forced swim stress (R-FSS), mice were exposed to one 15 min swim, and 24 h later were exposed to four 6 min swims, each separated by a 6 min break in the home cage, 10 min before decapitation and synaptosome generation or final cocaine-conditioned place preference (CPP) (see below). For repeated stress exposure recovery (R-Recovery), mice were exposed to one 15 min swim, and 24 h later were exposed to four 6 min swims, each separated by a 6 min break in the home cage, and then 24 h later were decapitated and synaptosomes were prepared. For acute stress exposure recovery (A-Recovery), mice were exposed to one 15 min swim, and then 24 h later were decapitated and synaptosomes were prepared.
Cocaine conditioned place preference
Mice were used in a balanced three-compartment place-conditioning apparatus as described previously (Schindler et al., 2010). Noldus Ethovision software (version 3.0; Norldus) was used to analyze movement through each compartment recorded previously using video capture (Canon ZR90) from above. Briefly, on Day 1, mice were tested for initial place preference bias; mice spending >720 s in the middle chamber or spending triple the amount of time in one of the outer chambers over the other outer chamber were excluded from the study. An unbiased design was used; approximately half the animals received cocaine in their nonpreferred box (n = 99) and half in their preferred box (n = 90), and pretest time spent in the subsequently drug paired box was equivalent to pretest time spent in the subsequently saline paired box (drug paired box mean, 674 ± 9.6 s, n = 189; saline paired box mean, 701 ± 10.6 s, n = 189; unpaired, two-sample t tests, p > 0.05). On Days 2 and 3, mice were administered saline (10 ml/kg of body weight, s.c.) and confined to their assigned saline-paired compartment for 30 min in the morning, and then 4 h later were administered cocaine (15 mg/kg, s.c.) and confined to their assigned drug-paired compartment for 30 min in the afternoon. On Day 4 of testing, mice were assessed for postconditioning place preference. Some mice were exposed to one 15 min FSS 2 h following completion of cocaine training on Day 3 and then on Day 4 were exposed to four 6 min swims before postconditioning preference testing. On Day 4, some mice were pretreated with either saline (10 ml/kg of body weight, i.p.) or citalopram (15 mg/kg, i.p.) 30 min before administration of either saline (10 ml/kg of body weight, i.p.) or U50,488 (5 mg/kg, i.p.) 60 min before postconditioning preference testing. Cocaine CPP scores were calculated as time spent in the drug-paired compartment before training subtracted from time spent in the drug-paired compartment after conditioning.
Conditioned place aversion
Methods are the same as those used for cocaine CPP except that on Days 2 and 3, mice were administered saline (10 ml/kg of body weight, i.p.) and confined to their assigned saline-paired compartment for 30 min in the morning, and then 4 h later were given U50,488 (2.5 mg/kg, i.p.) and confined to their assigned drug-paired compartment for 30 min in the afternoon. U50,488 conditioned place aversion (CPA) scores were calculated as time spent in the drug-paired compartment before training subtracted from time spent in the drug-paired compartment after training.
Chronic nicotine exposure
Mice were administered saline (four injections of 10 ml/kg of body weight, s.c.; 2 h apart) or nicotine (20 mg/kg/d, s.c.; four injections of 5 mg/kg; 2 h apart) for 6 d. RDEV was carried out on Day 8. The mice showed tolerance to the hypolocomotor effects of nicotine after repeated dosing (data not shown), but specific behavioral signs of nicotine withdrawal were not further documented.
Preparation of brain synaptosomes
Mice were decapitated, whole-brain or specific brain regions were dissected, and synaptosomes prepared as described previously (Hagan et al., 2010). The synaptosomal preparations were resuspended in 5 ml preoxygenated Krebs–Ringer–HEPES (KRH) buffer [containing (in mm) 124 NaCl, 1.8 KCl, 1.3 MgSO4, 1.24 KH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 glucose] and maintained blanketed with 95% O2/5% CO2 gas in a 50 ml conical tube on ice.
Rotating disk electrode voltammetry
RDEV is an electrochemical technique used to measure neurotransmitter uptake kinetics (Earles and Schenk, 1998; Schenk et al., 2005; Hagan et al., 2010). A voltage potential sufficient to oxidize an electroactive neurotransmitter is applied to the synaptosome suspension. When 5-HT or DA is added, a small proportion of substrate transfers electrons to the electrode surface, producing a temporally resolved detection current proportional to the concentration of extrasynaptosomal 5-HT or DA. RDEV can be used to measure uptake kinetics for DAT, NET, SERT, and low-affinity, high-capacity transporter systems (Burnette et al., 1996; Earles and Schenk, 1998; Schenk et al., 2005; Hagan et al., 2010, 2011).
As described previously (Hagan et al., 2010), a Pine Instruments AFMD03 glassy carbon electrode was used. The electrode was lowered into an electrochemical well and rotated at 3000 rpm for 5-HT uptake studies and 2000 rpm for DA uptake studies. A constant potential of +550 mV was applied for 5-HT uptake studies and +450 mV for DA uptake studies relative to a Ag/AgCl reference electrode. After background subtraction, the initial velocities of 5-HT or DA uptake by synaptosomal preparations were calculated from the linear slope of the initial zero order portion of the plot of [5-HT] versus time or [DA] versus time. Data were normalized to synaptosomal protein concentration using a bicinchoninic acid colorimetric based assay (Pierce) using BSA as the standard. Uptake rates were expressed as femtomoles of 5-HT or DA per second per milligram protein.
5-HT studies
SERT-specific uptake.
For control experiments, synaptosomal preparations were allowed to stabilize for 10 min in the presence of 100 nm nisoxetine and 1 μm GBR 12935 (selective NET and DAT inhibitors, respectively) (see Fig. 2a, control trace). To measure nonspecific 5-HT uptake, synaptosomal preparations were allowed to stabilize for 10 min in the presence of 100 nm nisoxetine, 1 μm GBR 12935, and 1 μm paroxetine (see Fig. 2a, paroxetine trace). Once a stable baseline was reached, 5-HT was added (10, 30, 100, 300, or 1000 nm final concentration), and uptake was recorded for 3 min. The slope of uptake by the paroxetine-treated synaptosomes (paroxetine) was subtracted from the slope of uptake by the non-paroxetine-treated synaptosomes (control) to obtain SERT-specific uptake velocities (see Fig. 2a). For all studies except the SERT kinetic analysis experiments, 100 nm 5-HT (final concentration) was used, as 100 nm was determined previously to be the SERT Km using RDEV and a synaptosomal preparation (Hagan et al., 2010).
Figure 2.
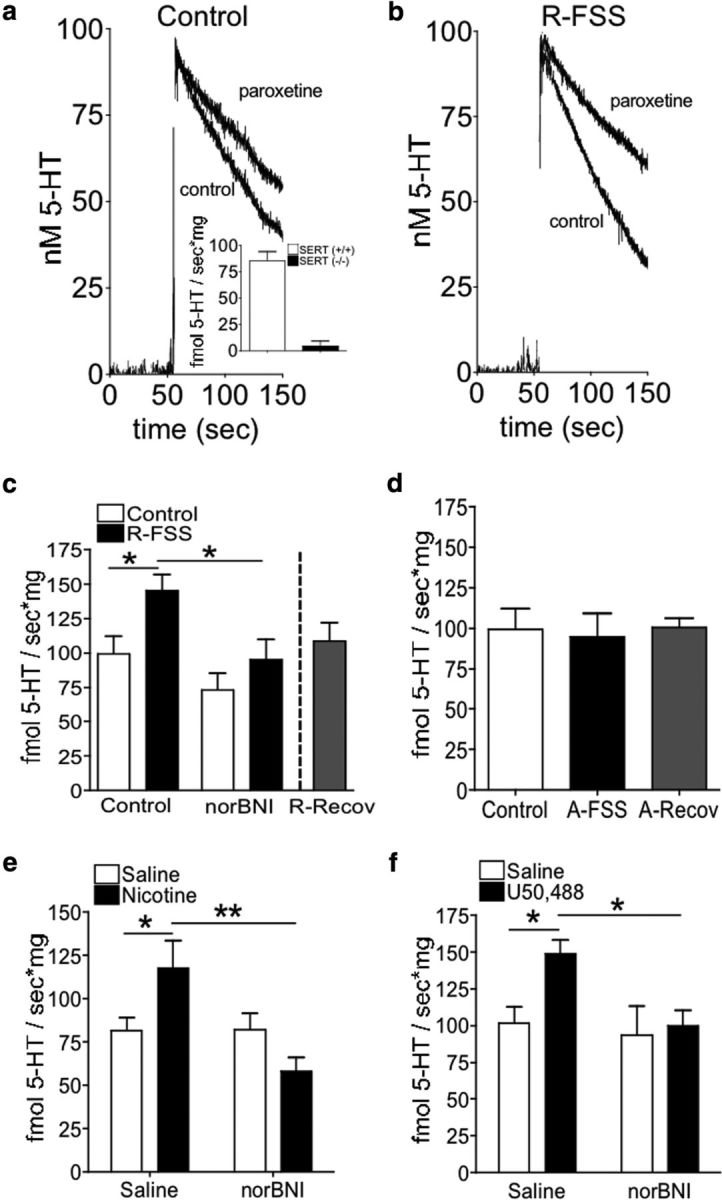
R-FSS, direct KOR activation, and withdrawal from repeated nicotine, but not A-FSS, increase the rate of 5-HT uptake by SERT in an norBNI-dependent manner. a, Representative traces from control and paroxetine-treated synaptosomes prepared from control mice. Inset, 5-HT uptake by SERT was present in SERT (+/+) littermates, but absent in (−/−) mice, demonstrating isolation of SERT-specific uptake. b, Representative traces from control and paroxetine-treated synaptosomes prepared from mice exposed to R-FSS. c, RDEV data demonstrating that R-FSS increased the rate of 5-HT uptake by SERT that was blocked by pretreatment with norBNI and recovered 24 h post final stress (two-way ANOVA followed by Bonferroni's post hoc test; n = 6–9). d, RDEV data demonstrating that A-FSS did not increase the rate of 100 nm 5-HT uptake by SERT, and that 24 h incubation period was not required for a FSS effect. e, RDEV data demonstrating that withdrawal from repeated nicotine increases the rate of 5-HT uptake by SERT and was blocked by pretreatment with norBNI (two-way ANOVA followed by Bonferroni's post hoc test; n = 7–8). f, RDEV data demonstrating that direct KOR activation by U50,488 increased 100 nm 5-HT uptake by SERT and was blocked by pretreatment with norBNI (two-way ANOVA followed by Bonferroni's post hoc test; n = 9–11). Data are shown as mean ± SEM. *p < 0.05; **p < 0.01.
Low-affinity uptake.
The slope of uptake by the paroxetine-treated synaptosomes (paroxetine) was used to obtain 5-HT uptake by low-affinity, high-capacity transporters (see Fig. 2a) (see the aforementioned experimental conditions).
DA studies
DAT-specific uptake.
For control experiments, synaptosomal preparations were allowed to stabilize for 10 min in the presence of 100 nm nisoxetine and 1 μm paroxetine. To measure nonspecific DA uptake, synaptosomal preparations were allowed to stabilize for 10 min in the presence of 100 nm nisoxetine, 1 μm paroxetine, and 1 μm GBR 12935. Once a stable baseline was reached, DA was added (30, 100, 300, 1000, or 3000 nm final concentration), and uptake was recorded for 3 min. The slope of uptake by the GBR 12935-treated synaptosomes (GBR 12935) was subtracted from the slope of uptake by the non-GBR 12935-treated synaptosomes (control) to obtain DAT-specific uptake velocities.
Low-affinity uptake.
The slope of uptake by the GBR 12935-treated synaptosomes (GBR 12935) was used to obtain DA uptake by low-affinity, high-capacity transporters.
Purification, biotinylation, and Western blotting of synaptosomes
Whole-brain and brain region-specific synaptosomes were prepared as described previously (Hagan et al., 2010), except protease and phosphatase inhibitors (Calbiochem) were added to each buffer. For brain region studies, tissue from two to four mice were pooled for each independent measure (n). Synaptosomes were purified by layering over a sucrose gradient consisting of 2.6 ml each of 0.85, 1.0, and 1.2 m sucrose (top to bottom) and were centrifuged at 85,000 × g for 2 h at 4°C. Purified synaptosomes were collected and appeared as a creamy colored band at the interface of the 1.0 and 1.2 m sucrose fractions. The purified synaptosomes were washed once with 0.32 m sucrose and then once with KRH buffer. After protein concentration determination, the purified synaptosomes were biotinylated using the EZ-link Sulfo-NHS-SS-Biotin to label cell surface proteins, according to manufacturer's instructions. Excess biotin was quenched with 100 mm glycine, and synaptosomes were lysed in RIPA lysis buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 1% CHAPS, and protein and phosphatase inhibitors). The lysates were rocked at 4°C for 45 min and then centrifuged at 15,000 rpm for 45 min. Supernatants were incubated with neutravidin beads (Pierce Biotechnology) overnight at 4°C to capture the cell surface biotinylated proteins. Beads were then pelleted, an aliquot of each supernatant (nonbiotinylated proteins) was saved, and the remaining supernatant was aspirated. Beads were then washed with RIPA buffer, and bound proteins were extracted with Laemmli's buffer. To obtain total SERT protein levels, before incubation with neutravidin beads, an aliquot of each biotinylated lysate was extracted in Laemmli's buffer. Surface (biotinylated), intracellular (nonbiotinylated), and total protein samples were then electrophoresed using 10% tris-glycine gels and processed for Western blotting. The blots were first stained with Ponceau-S to visualize total proteins. Blots were then washed three times with water and 1× Tris buffered saline with Tween-20 (TBS-T), and then blocked for 1 h in block buffer (2.5% milk, 2.5% BSA, 1× TBS-T). Blots were then probed overnight at room temperature in 2.5% BSA, 2.5% milk, 1× TBS-Tween 20 with the anti-SERT antibody (1:500; Santa Cruz Biotechnology), which recognizes predominantly the 75 kDa SERT species and the anti-calnexin antibody (1:4000; Enzo Life Sciences) to asses the extent of biotinylation of nominally intracellular proteins. Blots were washed three times with 1× TBS-T and then incubated for 1 h at room temperature with 680 donkey anti-goat or 800 donkey anti-rabbit (1:10,000; LI-COR) in Li-Cor blocking buffer and 2.5% BSA, 2.5% milk, and 1× TBS-T (1:1 dilution). Blots were then washed three times with 1× TBS-T and imaged as described previously (Bruchas et al., 2007a). The band densities for surface and total SERT samples were quantified and normalized by the densities of calnexin in the corresponding total SERT sample as described previously (Samuvel et al., 2005).
Cannulation and intracerebroventricular injections
As described previously (Land et al., 2008), isoflurane-anesthetized mice were mounted on a stereotaxic alignment system. Guide cannulas were placed in the lateral ventricle at 1.0 mm lateral, 0.4 mm posterior from bregma, and 3.0 mm below the skull, and were anchored with screws using dental cement. Mice were allowed to recover from surgery in their home cage for 6–8 d. Injection volumes (1 μl) of saline or corticotropin releasing factor (CRF; 1 μg; to induce dynorphin release and subsequent KOR activation) were performed during a 2 min period. Internal cannulas were removed 1 min after injection.
Immunohistochemistry
Cannulated mice were injected as described above, and 30 min later were anesthetized with pentobarbital and intracardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (PB). Brains were dissected, postfixed with 4% paraformaldehyde for 2 h at 4°C, cryoprotected with a 30% (w/v) sucrose solution at 4°C for 3 d, cut into 40 μm sections, and processed for immunohistochemistry. Sections containing the nucleus accumbens were washed three times in PBS and blocked in PBS containing 0.3% Triton X-100 and 5% normal goat serum. Sections were then incubated with a mixture of mouse anti-glutamic acid decarboxylase 67 (GAD67) (Millipore Bioscience Research Reagents) and rabbit anti-phospho-KOR Ab as described previously (McLaughlin et al., 2004; Bruchas et al., 2007a). Sections were then washed six times in PBS and incubated for 2 h at room temperature in Alexa Fluor 488 goat anti-mouse IgG (1:500; Invitrogen) and Alexa Fluor 555 goat anti-rabbit IgG (1:500; Invitrogen). Sections were washed six times in PBS, rinsed threes times for 10 min in PB, and then mounted on glass slides with Vectashield (Vector Labs) and sealed with nail polish for microscopy.
Viral vector design and production
Lenti-hSERT was developed based on the lentiviral construct expressing the β2 subunit of the nicotinic acetylcholine receptor under the mouse phosphoglycerol kinase (PGK) promoter, published by the Changeux group (Maskos et al., 2005). The β2 subunit was replaced with the human SERT sequence using XhoI and XbaI restriction sites. Human and mouse SERT share a 92.5% sequence identity. We used the hSERT sequence in the present study as the first step toward identifying possible sequence-specific mechanisms responsible for p38-mediated SERT translocation of the human transporter. The lenti-hSERT vector is a bicistronic construct expressing human SERT and GFP; eGFP is preceded by an internal ribosomal entry sequence (IRES2) allowing for separate translation of eGFP from the same transcript. Gene expression is under the control of the PGK promoter. The integrated virus was rendered replication incompetent by deletion of the U3 region of the 3′ long terminal repeat (Zufferey et al., 1998; Sirven et al., 2001). Sequences have been incorporated to enhance RNA stability, transgene expression, and infection of nondividing cells (Maskos et al., 2005). The viral expression plasmid was inserted into the pUC18 plasmid. As described previously (Land et al., 2009), a fee-for-service facility at the Fred Hutchinson Cancer Research Center produced the viral particles. In brief, viral particles were produced by cotransfection of the vector plasmid with a packaging plasmid and the vesicular stomatitis Indiana virus glycoprotein G protein plasmid, media was collected, and viral particles were isolated by filtration and ultracentrifugation 24–72 h following transfection. Viral titer obtained was 6.2 × 107 TU/ml. Before mouse injection, viral preparations were confirmed to be free of replication competent lentivirus by ELISA against the p24 capsid protein over a course of 4 weeks.
Stereotaxic microinjections (norBNI and lentiviral constructs)
Isoflurane-anesthetized mice were mounted on a stereotaxic alignment system (David Kopf Instruments). Mice were injected bilaterally in the ventral striatum (1.00 mm lateral, 0.98 mm anterior, 5 mm depth from bregma) or unilaterally in the dorsal raphe (0.00 mm lateral, 4.65 mm posterior, 3.85 mm depth) with 2.5 μg per side of norBNI or the lentiviral construct (dorsal raphe only), as described previously (Land et al., 2009; Smith et al., 2012). Animals were allowed to recover for at least 5 d after norBNI injection before sacrifice, or 3 weeks after lentiviral injection before CPA testing. NorBNI is a selective KOR antagonist whose effects last >21 d following a single dose (Horan et al., 1992; Bruchas et al., 2007b).
Data analysis
Data are expressed as means ± SEM. Student's unpaired, two-sample t tests were used to determine statistical differences between pairwise comparisons. Differences between groups were determined by one- and two-way ANOVA with overall α = 0.05 (p < 0.05). All post hoc pairwise comparisons were Bonferroni corrected. Concentration–response curves were fit using nonlinear regression analysis (Michaelis–Menten equation) to obtain best-fit values for Km and Vmax, and to determine whether best-fit values for each parameter were significantly different after R-FSS exposure. Statistical analyses were conducted using Graph Pad Prism 4.0.
Results
Stress-induced potentiation of cocaine CPP is mediated by GRK3 and p38α MAPK in serotonergic neurons
Previous studies have shown that KOR activation of p38 MAPK requires receptor phosphorylation by GRK3 followed by β-arrestin recruitment (Bruchas et al., 2006, 2007a), and both GRK3 and p38α MAPK expression in serotonergic neurons are required for KOR-mediated CPA, but the signal transduction pathway required for stress-induced potentiation of cocaine CPP remains unknown. To assess the role of GRK3 in KOR-mediated potentiation of cocaine CPP, GRK3 (−/−) and (+/+) littermates were exposed to one 15 min FSS on Day 3, 2 h after completion of cocaine training, and four 6 min FSS on Day 4, finishing 10 min before the final CPP posttest. GRK3 (+/+) and (−/−) control mice did not show different place preference scores [n = 13–21; unpaired, two-tailed t test between control GRK3 (+/+) and (−/−) mice; t(31) = 0.344; p > 0.05], demonstrating that lack of GRK3 did not affect basal preference for cocaine. GRK3 (+/+) mice exposed to repeated FSS showed a significant potentiation of cocaine CPP [n = 13–21; two-way ANOVA, interaction of genotype by R-FSS, F(1,57) = 9.638, p < 0.003; followed by Bonferroni's post hoc, GRK3 (+/+) littermate controls vs GRK3 (+/+) R-FSS, p < 0.05] (Fig. 1a). In contrast, GRK3 (−/−) mice repeatedly exposed to FSS did not show a change in cocaine CPP [Bonferroni's post hoc, GRK3 (−/−) control vs GRK3 (−/−) R-FSS, p > 0.05] (Fig. 1a). These data suggest that GRK3 was required for stress-induced potentiation of cocaine CPP and raise the possibility that this effect may be p38 mediated.
Figure 1.
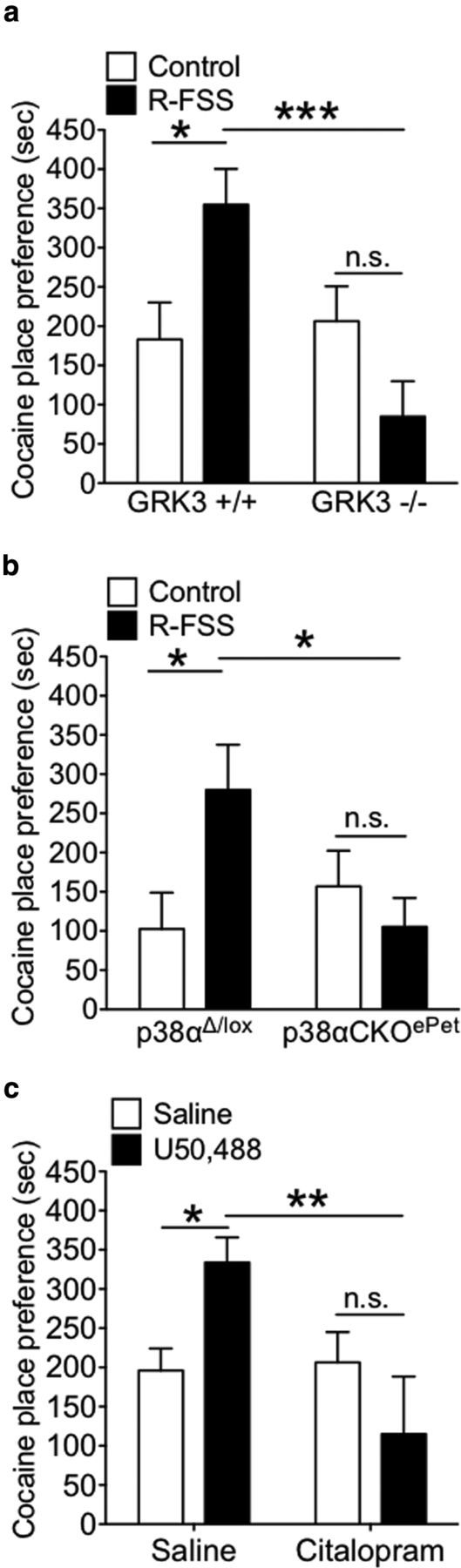
GRK3 and p38α MAPK in serotonergic neurons mediate stress-induced potentiation of cocaine CPP. a, Place preference test data demonstrating that GRK3 was required for stress-induced potentiation of cocaine CPP (two-way ANOVA followed by Bonferroni's post hoc test; n = 13–21). b, Preference test data demonstrating that p38α MAPK in serotonergic neurons was required for stress-induced potentiation of cocaine CPP (two-way ANOVA followed by Bonferroni's post hoc test; n = 14–20). c, Preference test data demonstrating that κ-mediated potentiation of cocaine CPP was blocked by pretreatment with the SSRI citalopram (two-way ANOVA followed by Bonferroni's post hoc test; n = 10–20). Data are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001. n.s., Not significant (p > 0.05).
To directly assess the role for p38 MAPK in R-FSS-induced potentiation of cocaine CPP, we generated CKO mice selectively lacking p38α in serotonergic cells (for a detailed description of CKO generation, see Materials and Methods). p38αΔ/lox and p38αCKOePet control mice did not show different place preference scores (n = 14–20; unpaired, two-tailed t test between control p38αΔ/lox and p38αCKOePet; t(32) = 0.823; p > 0.05), demonstrating that lack of p38α in serotonergic neurons did not affect basal place preference for cocaine. p38αΔ/lox and p38αCKOePet mice were exposed to one 15 min FSS on Day 3, 2 h after completion of cocaine training, and four 6 min FSS on Day 4, finishing 10 min before the final CPP posttest. p38αΔ/lox control mice exposed to repeated FSS showed a significant potentiation of cocaine CPP (n = 14–20; two-way ANOVA, interaction of genotype by R-FSS, F(1,58) = 5.729, p < 0.02; followed by Bonferroni's post hoc, p38aΔ/lox control vs p38aΔ/lox R-FSS, p < 0.05) (Fig. 1b). Alternatively, p38αCKOePet mice exposed to repeated FSS did not show a change in cocaine CPP (Bonferroni's post hoc, p38αCKOePet control vs p38αCKOePet R-FSS, p > 0.05), demonstrating that p38α MAPK in serotonergic neurons was required for stress-induced potentiation of cocaine CPP. Expression of Cre recombinase driven by the ePet promoter was previously shown to selectively excise p38α MAPK in serotonergic neurons expressing tryptophan hydroxylase (TPH2-ir), but not in non-TPH2-ir neurons in the dorsal raphe nucleus (Bruchas et al., 2011). The p38αΔ/lox mice are the appropriate controls because they have the floxed allele on the same genetic background (littermates), but express equivalent levels of p38α immunoreactivity in cells that do not express Cre (Bruchas et al., 2011).
To further investigate the role of the serotonergic system in stress-induced potentiation of cocaine CPP, mice were pretreated with either saline (10 ml/kg of body weight, i.p.) or the selective serotonin reuptake inhibitor (SSRI) citalopram (15 mg/kg, i.p.). Then, 30 min later, mice were administered either saline (10 ml/kg of body weight, i.p.) or U50,488 (5 mg/kg, i.p.) 60 min before postconditioning preference test. WT saline-pretreated and WT citalopram-pretreated control mice did not show different place preference scores (n = 10–20; unpaired, two-tailed t test between saline pretreated and citalopram pretreated; t(29) = 0.22; p > 0.05), demonstrating that pretreatment with citalopram did not affect basal preference for cocaine. Mice pretreated with saline and then injected with U50,488 showed a significant potentiation of cocaine CPP (n = 10–20; two-way ANOVA, interaction of genotype by R-FSS, F(1,57) = 7.70, p < 0.008; followed by Bonferroni's post hoc, saline plus saline vs saline plus U50,488, p < 0.05) (Fig. 1c). Conversely, mice pretreated with citalopram and then administered U50,488 did not show a change in cocaine CPP (Bonferroni's post hoc test, saline plus U50,488 vs citalopram plus U50,488, p > 0.05), demonstrating that SSRI pretreatment blocked κ agonist-induced potentiation of cocaine CPP.
Repeated swim stress, nicotine withdrawal-induced stress, or pharmacological stress increases 5-HT uptake by SERT in a norBNI-dependent manner
To determine whether stress exposure modulates 5-HT uptake by SERT, RDEV was used to measure SERT function following the addition of 5-HT to the electrochemical well containing a suspension of mouse brain synaptosomes (Hagan et al., 2010). The rate of 100 nm 5-HT uptake by synaptosomes prepared from C57BL/6 mice in the presence or absence of paroxetine was used to assess uptake mediated by SERT (Fig. 2). SERT (+/+) littermate mice showed paroxetine-sensitive 5-HT uptake, whereas SERT (−/−) mice showed no significant paroxetine-sensitive uptake (Fig. 2a, inset), confirming that SERT-specific uptake had been isolated. Whole-brain synaptosomes from wild-type mice exposed to R-FSS showed a significantly greater paroxetine-sensitive 5-HT clearance rate than synaptosomes prepared from unstressed, control C57BL/6 mice, as represented in Figure 2, a and b, and quantified in Figure 2c (n = 6–9; two-way ANOVA, effect of pretreatment, F(1,28) = 9.077, p < 0.005; effect of treatment, F(1,28) = 7.19, p < 0.012; followed by Bonferroni's post hoc, control vs R-FSS, p < 0.05) (Fig. 2c). The increase in SERT uptake rate caused by R-FSS was blocked by in vivo pretreatment with the KOR antagonist norBNI (10 mg/kg, i.p.) 24 h before the initial swim (Bonferroni's post hoc, R-FSS vs norBNI plus R-FSS, p < 0.05) and was not evident when synaptosomes were prepared 24 h following R-FSS (n = 6–8; unpaired, two-tailed t test between control and R-Recovery; t(12) = 0.48; p > 0.05) (Fig. 2c).
We showed previously that KOR activation is evident following exposure to A-FSS, but A-FSS is not sufficient to cause KOR-induced potentiation of cocaine CPP (Schindler et al., 2010). Similarly, in the current study, A-FSS exposure to one 15 min swim before synaptosomal preparation did not increase 100 nm 5-HT uptake rate by SERT compared to control mice (n = 6–8; one-way ANOVA; F(2,19) = 0.063; p > 0.05) (Fig. 2d). In this paradigm, R-FSS exposure occurred over 2 d. Thus, it is possible that A-FSS exposure might cause an increase in 5-HT uptake by SERT if a 24 h incubation period passed between A-FSS exposure and synaptosomal isolation. To investigate this possibility, mice were exposed to A-FSS, and then synaptosomes were prepared 24 h later (A-Recovery) (Fig. 2d). Synaptosomes from mice exposed to A-Recovery did not show a significantly different 100 nm 5-HT uptake rate from control mice (n = 6–8; one-way ANOVA; F(2,19) = 0.063; p > 0.05). Together, these results suggest that repeated KOR activation by R-FSS caused an increase in 100 nm 5-HT uptake by SERT that recovered 24 h after the final swim.
To assess the generality of the stress-induced changes in SERT function, we next examined changes in SERT function during withdrawal from repeated nicotine exposure, which can be considered aversive (Kenny and Markou, 2001; Koob and Le Moal, 2005), and κ antagonists attenuated the expression of both the physical (somatic signs and hyperalgesia) and affective (anxiety-related behavior and conditioned place aversion) nicotine withdrawal signs (Jackson et al., 2010). Synaptosomes prepared from mice in withdrawal 48 h following repeated exposure to nicotine (20 mg/kg/d, s.c, for 6 d), showed a significantly increased rate of 100 nm 5-HT uptake by SERT compared to synaptosomes prepared from mice administered saline (10 ml/kg of body weight, i.p.) on the same schedule (n = 7–8; two-way ANOVA, interaction of norBNI and nicotine, F(1,27) = 8.712, p < 0.007; followed by Bonferroni's post hoc, saline vs nicotine, p < 0.05) (Fig. 2e). The increase in SERT uptake rate evident during nicotine withdrawal was blocked by pretreatment with the KOR antagonist norBNI (10 mg/kg, i.p.) 1 h before the start of the nicotine treatment paradigm (Bonferroni's post hoc, nicotine vs norBNI plus nicotine, p < 0.001), and norBNI pretreatment alone had no effect on 100 nm 5-HT uptake by SERT (Bonferroni's post hoc, saline vs norBNI plus saline, p > 0.05).
Administration of the KOR agonist U50,488 was used as a pharmacological stressor and provides a more selective KOR activation than FSS exposure or withdrawal from nicotine. Synapsotomes prepared from mice administered U50,488 (5 mg/kg, i.p.) 60 min before synaptosome generation showed a significantly increased rate of 100 nm 5-HT uptake by SERT compared to synaptosomes prepared from mice administered saline (10 ml/kg of body weight, i.p.) 60 min before synaptosome isolation (n = 9–11; two-way ANOVA; effect of pretreatment, F(1,35) = 4.234, p < 0.047; effect of treatment, F(1,35) = 4.825, p < 0.035; followed by Bonferroni's post hoc, saline vs U50,488, p < 0.05) (Fig. 2f). This dose and timing were used based on behavioral studies in which U50,488 administration significantly potentiated cocaine or nicotine CPP when given 60 min before final preference testing (Schindler et al., 2010; Smith et al., 2012). The increase in SERT uptake rate by U50,488 was blocked by pretreatment with the KOR antagonist norBNI (10 mg/kg, i.p.) 24 h before U50,488 administration (Bonferroni's post hoc, U50,488 vs norBNI plus U50,488, p < 0.05), and norBNI pretreatment alone had no effect on 100 nm 5-HT uptake by SERT (Bonferroni's post hoc, saline vs norBNI plus saline, p > 0.05).
Repeated swim stress increases SERT Vmax without affecting SERT Km
An increase in 5-HT uptake by SERT following R-FSS exposure might result from a stress-induced increase in SERT Vmax (increased SERT synthesis or increased SERT expression at the plasma membrane) or a decrease in SERT Km (increased catalytic activity of SERT). To obtain values for Vmax and Km, saturation kinetic analyses of 5-HT uptake by SERT were conducted with synaptosomes prepared from control or R-FSS-exposed C57BL/6 mice. R-FSS significantly increased the rate of 5-HT uptake by SERT at 100 nm (n = 6–10; two-way ANOVA; interaction of concentration and R-FSS, F(5,74) = 2.35, p < 0.049; followed by Bonferroni's post hoc, control vs R-FSS, p < 0.05), 300 nm (Bonferroni's post hoc, control vs R-FSS, p < 0.05), and 1 μm (Bonferroni's post hoc, control vs R-FSS, p < 0.001) 5-HT, but not at 10 or 30 nm (Bonferroni's post hoc, control vs R-FSS, p > 0.05) 5-HT, compared to the rate of 5-HT uptake by SERT from control synaptosomes at the corresponding 5-HT concentration (Fig. 3a). R-FSS significantly increased SERT Vmax for 5-HT (n = 6–10; nonlinear regression followed by comparison of fits; F(1,82) = 4.91, p < 0.029) without affecting SERT Km for 5-HT (n = 6–10; nonlinear regression followed by comparison of fits; F(1,82) = 0.86, p > 0.05) (Fig. 3a, inset).
Figure 3.

R-FSS increases SERT Vmax without affecting SERT Km and does not modulate DAT or low-affinity, high-capacity transporters. a, Kinetic RDEV data demonstrating that R-FSS exposure increased SERT Vmax but did not change SERT Km (two-way ANOVA followed by Bonferroni's post hoc; n = 6–10). b, Kinetic RDEV data demonstrating that R-FSS exposure did not change the rate of DA uptake by DAT at any concentration tested (two-way ANOVA; n = 6–11). c, Kinetic RDEV data demonstrating that R-FSS did not modulate 5-HT uptake by low-affinity, high-capacity transporters at any of the concentrations tested (two-way ANOVA; n = 6–9). d, Kinetic RDEV data demonstrating that R-FSS did not modulate DA uptake by low-affinity, high-capacity transporters at any of the concentrations tested (two-way ANOVA; n = 6–11). Data are shown as mean ± SEM. *p < 0.05; ***p < 0.001.
Repeated swim stress does not increase DA uptake by DAT or 5-HT or DA uptake by low-affinity, high-capacity transporters
To obtain Vmax and Km values for DAT (not established previously for this animal species, cellular preparation, or technique), saturation kinetic analyses of DA uptake by DAT were conducted. Synaptosomes prepared from mice exposed to R-FSS did not show a significantly different rate of DA uptake by DAT at any DA concentration tested (n = 6–11; two-way ANOVA; interaction of concentration and R-FSS; F(5,75) = 1.240, p > 0.05), compared to synaptosomes prepared from control mice (Fig. 3b). Similarly, R-FSS did not significantly change Vmax (n = 6–11; nonlinear regression followed by comparison of fits; F(1,83) = 0.359, p > 0.05) or Km for DAT (n = 6–11; nonlinear regression followed by comparison of fits; F(1,83) = 0.009, p > 0.05) (Fig. 3b, inset), suggesting that R-FSS did not modulate DA uptake by DAT under these assay conditions.
In addition to uptake by their respective transporters, both 5-HT and DA can be cleared by low-affinity, high-capacity transporters (Daws, 2009; Hagan et al., 2011). The contribution to uptake of these low-affinity, high-capacity transporters increases as 5-HT concentration increases (Baganz et al., 2010; Hagan et al., 2011). To investigate whether stress-induced KOR activation regulates these alternative uptake mechanisms, we examined the uptake rate of 5-HT or DA in the presence of DAT, NET, and SERT inhibitors following repeated stress (Fig. 3c,d). Synaptosomes prepared from mice exposed to R-FSS did not show a significantly different rate of 5-HT uptake by low-affinity, high-capacity transporters at any concentration of 5-HT tested (n = 6–9; two-way ANOVA; interaction of concentration and R-FSS; F(5,74) = 0.717, p > 0.05), compared to synaptosomes prepared from control mice (Fig. 3c). Similarly, no effect of R-FSS was seen at any concentration of DA tested compared to synaptosomes prepared from control mice (n = 6–11; two-way ANOVA; interaction of concentration and R-FSS; F(5,73) = 0.259, p > 0.05) (Fig. 3d). 5-HT at 1 μm and DA at 3 μm are still below saturating concentrations for these low-affinity, high-capacity transporters (Fig. 3c,d), and it is therefore possible that R-FSS has effects at concentrations higher then those used in the current study; however, these results suggest that KOR activation by repeated stress selectively increased SERT function without affecting DAT or the low-affinity, high-capacity transporters.
Repeated swim stress increases SERT surface expression
An increase in SERT Vmax could arise from an increase in SERT surface expression or an increase in SERT synthesis. To determine whether R-FSS increased SERT surface expression, purified synaptosomes from control, R-FSS, norBNI, norBNI plus R-FSS, and R-Recovery-exposed mice were biotinylated to selectively label cell surface proteins. As a control for the specificity of cell-surface labeling by the cell impermeant form of biotin, surface, intracellular, and total samples were extracted with Laemmli's buffer and processed for Western blotting with the calnexin antibody. Calnexin is an integral protein of the endoplasmic reticulum and thus should not be available for labeling by biotin (Samuvel et al., 2005). As shown in Figure 4a, calnexin immunoreactivity is seen in the nonbiotinylated fraction and in the total protein samples, but was absent in the surface samples, confirming that the majority of synaptosomes in each preparation were intact and biotin labeling of intracellular proteins did not occur under these assay conditions.
Figure 4.

R-FSS increases SERT Vmax by increasing SERT surface expression. a, Representative Western blot images demonstrating biotinylation of only surface proteins. Calnexin is an intracellular protein and thus should not appear in the surface samples. b, Representative Western blot images and corresponding analysis showing that R-FSS increased SERT surface expression in a norBNI-dependent manner that recovered 24 h after swim (two-way ANOVA followed by Bonferroni's post hoc; n = 6–8). c, Representative Western blot images and corresponding analysis showing that the R-FSS did not change total SERT expression. Data are shown as mean ± SEM. *p < 0.05; **p < 0.01. Surf, Surface; FT, flow through (intracellular sample); total, total protein (both surface and intracellular).
To determine whether R-FSS increased SERT surface expression, surface proteins were extracted from intact synaptosomes with Laemmli's buffer and processed for Western blotting with the SERT antibody. We found that R-FSS increased surface SERT immunoreactivity compared to unstressed controls (n = 6–8; two-way ANOVA; effect of pretreatment, F(1,24) = 7.284, p < 0.013; effect of treatment, F(1,24) = 10.02, p < 0.004; followed by Bonferroni's post hoc, control vs R-FSS, p < 0.01) (Fig. 4b). The increase in SERT surface expression by R-FSS was blocked by pretreatment with the KOR antagonist norBNI (10 mg/kg, i.p.) 24 h before the initial swim (Bonferroni's post hoc, R-FSS vs norBNI plus R-FSS, p < 0.05) and was not evident when synaptosomes were prepared 24 h after R-FSS (n = 6–8; unpaired, two-tailed t test between control R-Recovery; t(12) = 0.339; p > 0.05) (Fig. 4b).
To determine whether R-FSS increased total SERT expression, proteins were extracted from intact synaptosomes with Laemmli's buffer and processed for Western blotting with the SERT antibody. We found no change in total SERT immunoreactivity following treatment (n = 6; two-way ANOVA; interaction of norBNI and R-FSS; F(1,24) = 0.528, p > 0.05) (Fig. 4c), suggesting that the increase in SERT Vmax following R-FSS was not a result of increased SERT synthesis. Together, these data suggest that R-FSS increases SERT Vmax for 5-HT by increasing SERT plasma membrane surface expression.
GRK3 and p38α MAPK are required for R-FSS-induced increases in SERT function and surface expression
Activation of p38 MAPK has been shown previously to regulate SERT function and surface membrane expression (Zhu et al., 2004, 2005; Samuvel et al., 2005). In addition, KOR activation of p38 MAPK was shown previously to occur by a GRK3-arrestin-dependent mechanism in vivo and in vitro (Bruchas et al., 2006). To assess the role of GRK3 in KOR-mediated SERT regulation, GRK3 (−/−) and (+/+) littermates were exposed to R-FSS or remained in the home cage before synaptosomal generation. R-FSS significantly increased the rate of 100 nm 5-HT uptake by SERT in GRK3 (+/+) littermate controls [n = 6–8; two-way ANOVA; interaction of genotype and R-FSS, F(1,24) = 16.82, p < 0.0004; followed by Bonferroni's post hoc, GRK3 (+/+) control vs GRK3 (+/+) R-FSS, p < 0.001] (Fig. 5a). Alternatively, GRK3 (−/−) mice exposed to R-FSS did not show an increase in SERT uptake rates as compared to GRK3 (−/−) control mice [Bonferroni's post hoc, GRK3 (−/−) control vs GRK3 (−/−) R-FSS, p > 0.05] (Fig. 5a).
Figure 5.
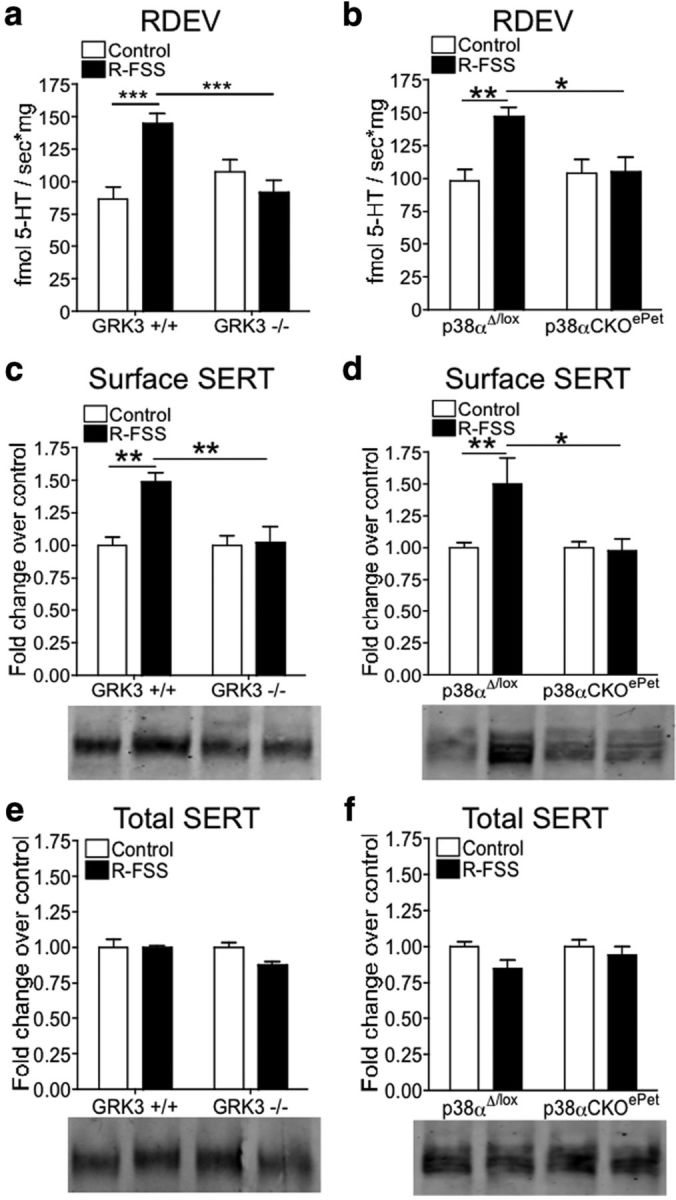
R-FSS-induced increases in 5-HT uptake by SERT and SERT surface expression require GRK3 and p38a MAPK in serotonergic neurons. a, RDEV data demonstrating that the R-FSS-induced increase in the rate of 5-HT uptake by SERT required GRK3 (two-way ANOVA followed by Bonferroni's post hoc test; n = 6–8). b, RDEV data demonstrating that R-FSS increased the rate of 5-HT uptake by SERT in p38αΔ/lox but not in p38αCKOePet mice (two-way ANOVA followed by Bonferroni's post hoc test; n = 6–8). c, Representative Western blot images and corresponding analysis showing that R-FSS increased SERT surface expression in a GRK3-dependent manner (two-way ANOVA followed by Bonferroni's post hoc test; n = 4). d, Representative Western blot images and corresponding analysis showing that R-FSS increased SERT surface expression in a p38α MAPK-dependent manner (two-way ANOVA followed by Bonferroni's post hoc test; n = 5–8). e, Representative Western blot images and corresponding analysis showing that the GRK3 genotype or R-FSS did not change total SERT expression (two-way ANOVA; n = 4). f, Representative Western blot images and corresponding analysis showing that the p38α MAPK genotype or R-FSS did not change total SERT expression (two-way ANOVA; n = 4–8). Data are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
These data suggest that GRK3 was required for KOR-induced increases in 5-HT uptake by SERT and raise the possibility that this effect may be mediated by KOR activation of p38 MAPK. To test this hypothesis, p38αΔ/lox and p38αCKOePet mice were exposed to R-FSS or remained in their home cage before synaptosomal generation. R-FSS increased the rate of 100 nm 5-HT uptake by SERT in p38αΔ/lox mice (n = 6–8; two-way ANOVA; interaction of genotype and R-FSS, F(1,24) = 6.073, p < 0.021; followed by Bonferroni's post hoc, p38αΔ/lox control vs p38αΔ/lox R-FSS, p < 0.01) compared to control mice of the same genotype (Fig. 5b). R-FSS had no effect on the rate of 100 nm 5-HT uptake by SERT in p38αCKOePet mice (Bonferroni's post hoc, p38αCKOePet control vs p38αCKOePet R-FSS, p > 0.05) (Fig. 5b), demonstrating that p38α was required for R-FSS effects on SERT function.
We next investigated whether GRK3 was required for R-FSS-induced increase in SERT surface expression. We found that R-FSS increased surface SERT immunoreactivity compared to controls in the GRK3 (+/+) genotype [n = 4; two-way ANOVA; interaction of genotype and R-FSS, F(1,12) = 7.267, p < 0.02; followed by Bonferroni's post hoc, GRK3 (+/+) control vs GRK3 (+/+) R-FSS, p < 0.01] (Fig. 5c). The increase in SERT surface expression by R-FSS was not seen in the GRK3 (−/−) genotype [Bonferroni's post hoc, GRK3 (−/−) control vs GRK3 (−/−) R-FSS, p > 0.05] (Fig. 5c), suggesting that GRK3 was required for stress-induced increase in surface SERT expression.
We used a similar strategy to determine whether p38 MAPK in serotonergic neurons was required for R-FSS-induced increase in SERT surface expression by testing p38αΔ/lox and p38αCKOePet mice. We found that R-FSS increased surface SERT immunoreactivity compared to controls in the p38αΔ/lox genotype (n = 5–8; two-way ANOVA; interaction of genotype and R-FSS, F(1,22) = 4.604, p < 0.043; followed by Bonferroni's post hoc, p38αΔ/lox control vs p38αΔ/lox R-FSS, p < 0.01) (Fig. 5d). The increase in SERT surface expression by R-FSS was not seen in the p38αCKOePet genotype (Bonferroni's post hoc, p38αCKOePet control vs p38αCKOePet R-FSS, p > 0.05) (Fig. 5d), demonstrating that p38α MAPK in serotonergic neurons was required for R-FSS effects on SERT surface expression.
To assess whether GRK3 or p38α MAPK expression changes total SERT levels, total protein was extracted with Laemmli's buffer and processed for Western blotting with the SERT antibody. As for WT mice, we found no change in total SERT immunoreactivity across GRK3 genotypes or following R-FSS (n = 4; two-way ANOVA; interaction of genotype and R-FSS, F(1,12) = 2.676, p > 0.05) (Fig. 5e). Similarly, we found no change in total SERT immunoreactivity across p38α genotypes or following R-FSS (n = 6–8; two-way ANOVA; interaction of genotype and R-FSS, F(1,18) = 0.853, p > 0.05) (Fig. 5f). Together, these data suggest that the increase in SERT function and surface expression following R-FSS was mediated by GRK3 and p38α MAPK in serotonergic neurons.
Repeated swim stress selectively increases SERT surface expression in the ventral striatum
SERT expression is evident in multiple brain regions (Torres et al., 2003), and the previous experiments in the present study were conducted using whole-brain synaptosomes. To determine whether R-FSS affected SERT in all of the serotonergic projections equivalently, we compared synaptosomes prepared from grossly dissected brain regions. Surprisingly, we found that R-FSS significantly increased surface SERT immunoreactivity compared to controls only in the ventral striatum (VS; n = 6; unpaired, two-tailed t test; t(10) = 2.86, p < 0.017) (Fig. 6a, red box). R-FSS did not change surface SERT immunoreactivity compared to controls in the medial prefrontal cortex (mPFC; n = 4; unpaired, two-tailed t test; t(6) = 0.171, p > 0.05), hippocampus (n = 6; unpaired, two-tailed t test; t(10) = 0.312, p > 0.05), dorsal striatum (DS; n = 6; unpaired, two-tailed t test; t(10) = 0.773, p > 0.05), amygdala (n = 4; unpaired, two-tailed t test; t(6) = 0.372, p > 0.05), or dorsal raphe (n = 4; unpaired, two-tailed t test; t(6) = 0.776, p > 0.05) (Fig. 6a). Similar to whole-brain synaptosomes, we found no change in total SERT immunoreactivity across the brain regions examined: mPFC (n = 6; unpaired, two-tailed t test; t(10) = 0.294, p > 0.05), hippocampus (n = 6; unpaired, two-tailed t test; t(10) = 1.487, p > 0.05), DS (n = 6; unpaired, two-tailed t test; t(10) = 0.073, p > 0.05), VS (n = 6; unpaired, two-tailed t test; t(10) = 1.573, p > 0.05), amygdala (n = 3–4; unpaired, two-tailed, t test; t(5) = 0.886, p > 0.05), and DR (n = 4; unpaired, two-tailed t test; t(6) = 1.053, p > 0.05) (Fig. 6b). Together, these data demonstrate that R-FSS significantly increased surface SERT expression specifically in the ventral striatum without changing total SERT levels or affecting cell surface SERT in the other brain regions tested.
Figure 6.
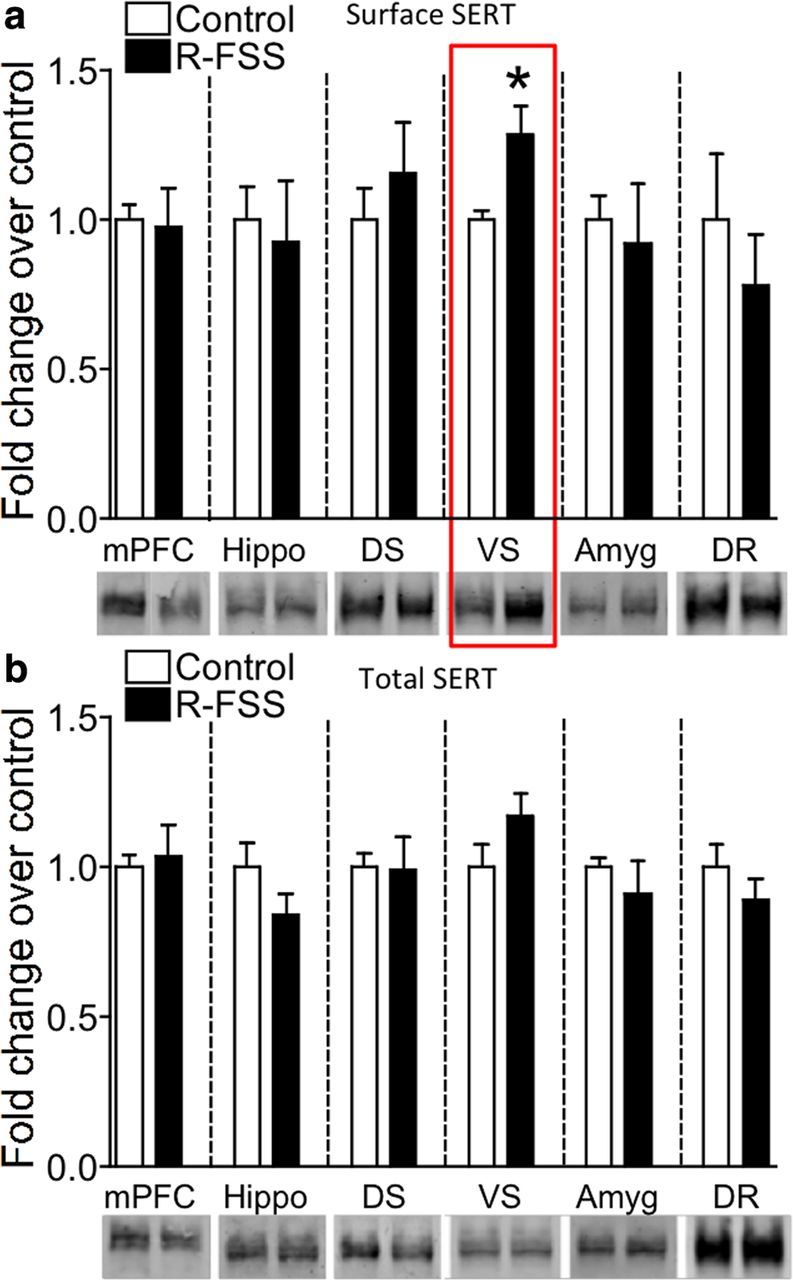
SERT surface expression in selectively increased in the ventral striatum following R-FSS. a, Representative Western blot images and corresponding analysis showing that R-FSS increased SERT surface expression only in the ventral striatum (unpaired, two-tailed t test; n = 4–6). b, Representative Western blot images and corresponding analysis showing that R-FSS did not change total SERT expression in any of the brain regions investigated (two-way ANOVA; n = 3–6). Data are shown as mean ± SEM. *p < 0.05. Hippo, Hippocampus, Amyg, amygdala.
Repeated swim stress increases surface SERT expression in the ventral striatum through activation of local κ-opioid receptors
KORs are expressed on the cell bodies of serotonergic DR neurons and on the afferent nerve terminals projecting to the VS (Tao and Auerbach, 2002; Land et al., 2009). Thus it is possible that KOR activation in the DR or VS could mediate the increase in surface SERT seen following R-FSS. To determine which pool of KORs mediate this effect, we stereotaxically injected the long-lasting KOR antagonist norBNI into the DR or VS. NorBNI injected into the DR did not significantly affect the increased surface SERT immunoreactivity in the VS caused by R-FSS (n = 3–5; two-way ANOVA; interaction of brain region and R-FSS, F(1,12) = 13.50, p < 0.003; followed by Bonferroni's post hoc, norBNI DR control vs norBNI DR R-FSS, p < 0.01) (Fig. 7a). In contrast, norBNI injection in the nucleus accumbens completely blocked the stress-induced increase in SERT surface expression in the VS (Bonferroni's post hoc, norBNI VS control vs norBNI VS R-FSS, p > 0.05) (Fig. 7a). No change in total SERT immunoreactivity was evident (n = 3–5; two-way ANOVA; interaction of brain region and R-FSS, F(1,12) = 0.477, p > 0.05) (Fig. 7b). These results suggest that local KOR in the VS mediate the stress-induced increase in surface SERT expression.
Figure 7.
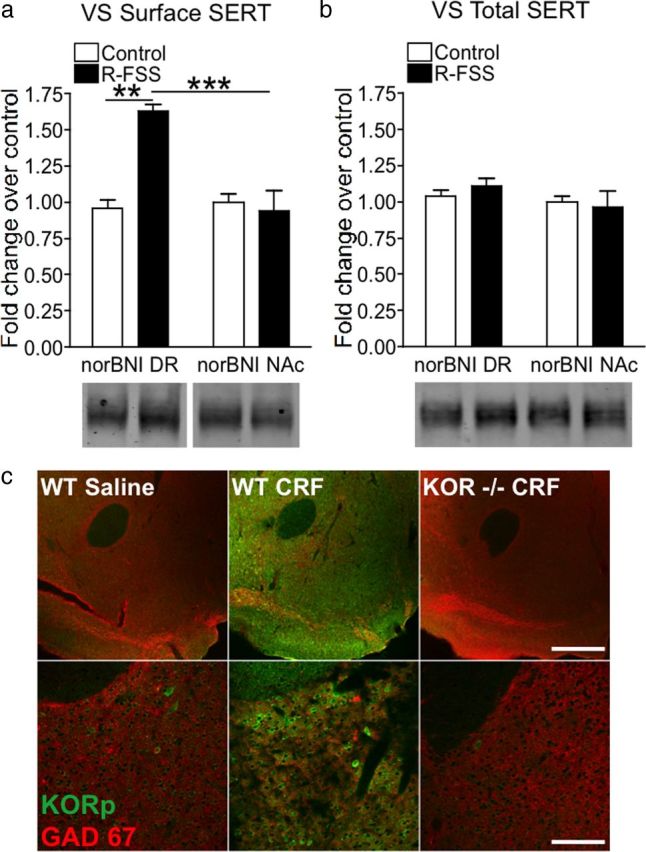
Local KORs mediate increases SERT surface expression in the ventral striatum following R-FSS. a, Representative Western blot images and corresponding analysis showing that R-FSS increased SERT surface expression in the ventral striatum when norBNI was microinjected into the dorsal raphe but not into the nucleus accumbens (two-way ANOVA followed by Bonferroni's post hoc test; n = 3–5). b, Representative Western blot images and corresponding analysis showing that norBNI microinjection into the dorsal raphe or nucleus accumbens or R-FSS did not change total SERT expression in the ventral striatum (two-way ANOVA; n = 3–5). c, Low power (10×) and high power (40×) representative fluorescent images displaying KORp immunoreactivity (green) and GAD67 (red) in the nucleus accumbens of animals treated with 0.9% saline or CRF (1 μg/5 μl). CRF-induced dynorphin release increased KORp immunoreactivity in WTs compared to saline controls. CRF-induced KORp immunoreactivity was specific, as it is absent in animals lacking KOR. Data are shown as mean ± SEM. **p < 0.01 comparing norBNI DR control with norBNI DR R-FSS; ***p < 0.003 comparing norBNI DR R-FSS with norBNI NAc R-FSS. Scale bars: c, top, 400 μm; bottom, 100 μm.
Stress exposure results in the release of CRF, leading to the release of endogenous dynorphin and subsequent activation of KOR (Land et al., 2008). KOR activation leads to the phosphorylation of serine-369 on its C-terminal tail by GRK3 (McLaughlin et al., 2003b). To visualize activated KOR using immunohistochemistry, an antibody that recognizes phosphorylated serine-369 was developed (McLaughlin et al., 2003b). To investigate whether CRF causes dynorphin release in the NAc, wild-type and KOR (−/−) were injected intracerebroventricularly with either saline or CRF (1 μg/5 μl). Immunohistochemical analysis showed that CRF caused an increase in KORp immunoreactivity within the NAc that was absent in KOR (−/−), suggesting that stress leads to the release of dynorphin and activation of KOR within the NAc (Fig. 7c).
KOR-mediated CPA requires SERT
These results suggested that stress-induced release of dynorphin within the nucleus accumbens activates p38α MAPK in the serotonergic projections from the dorsal raphe nucleus and that the local translocation of SERT from an endosomal vesicle to the plasma membrane mediates the stress response. Consistent with this concept, we found that SERT (+/−) and (−/−) mice did not show U50,488 CPA, whereas wild-type SERT (+/+) littermates showed robust CPA [n = 7–12; one-way ANOVA; F(2,27) = 4.023, p < 0.031; followed by Bonferroni's post hoc, SERT (+/+) vs (+/−), p < 0.05; SERT (+/+) vs (−/−), p < 0.05], demonstrating that SERT was required for κ-mediated aversion (Fig. 8b). We next generated a bicistronic lentiviral vector (Maskos et al., 2005; Land et al., 2009) to express hSERT-GFP under the PGK promoter (lenti-SERT) in SERT (−/−) mice (Fig. 8a). To assess whether KOR regulation of SERT was sufficient for U50,488 CPA, we stereotaxically injected lenti-SERT into the DR of SERT (−/−) mice. We had demonstrated previously that lentiviral-mediated gene transfer did not nonspecifically affect this behavior (Land et al., 2009). Lenti-SERT-injected mice showed a significant recovery of κ-mediated CPA (n = 4–7; unpaired, two-tailed t test; t(9) = 2.857, p < 0.019). To confirm that SERT expression driven by the lenti-SERT vector restored the normal translocation response, we generated whole-brain synaptosomes and found that SERT surface expression was increased following R-FSS (Fig. 8c). These data further support the suggested involvement of serotonergic projections from the DR to the VS in κ-mediated CPA.
Figure 8.
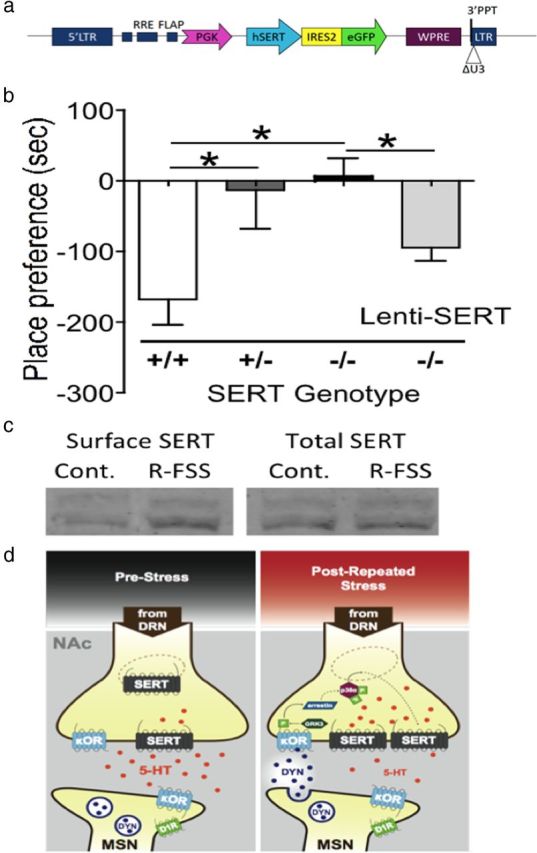
SERT is required for κ-mediated conditioned place aversion. a, Schematic of lentiviral construct for expression of SERT-GFP. LTR, Long terminal repeat; RRE, Rev response element; FLAP, 99 bp DNA “flap” (enhances infection of nondividing cells); PGK, human PGK promoter; WPRE, woodchuck hepatitis B virus posttranscriptional regulatory element (enhances RNA stability and transgene expression); ΔU3, deletion of U3 region of 3′ long terminal repeat (renders integrated virus replication incompetent). b, Preference test data demonstrating that SERT is required for conditioned place aversion to the KOR agonist U50,488 (one-way ANOVA followed by Bonferroni's post hoc test; n = 7–12). c, Representative Western blot images showing that R-FSS increased SERT surface expression when lenti-SERT was expressed in SERT (−/−) mice. d, Diagram depiction modeling the proposed mechanism by which stress-induced KOR and p38α MAPK activation cause increase in surface SERT expression within the ventral striatum and a subsequent decrease in extracellular serotonin levels. *p < 0.05 for the comparisons marked.
Discussion
The present study elucidates a novel signal transduction cascade within the afferent nerve terminals projecting from the dorsal raphe to the ventral striatum that is activated by the endogenous dynorphin opioids following repeated stress exposure and underlies prodepressive and proaddictive behaviors in mice. Although both SERT and KOR regulation have been strongly implicated in the aversive effects of stress exposure, the present study describes a molecular link for these two mediators and identifies the brain region involved. Together these data suggest that a component of KOR-mediated stress behaviors results from modulation of serotonergic tone within the ventral striatum and requires both GRK3 and p38α MAPK activation.
Although the FSS assay is a well-established behavioral stress model, and withdrawal from drugs of abuse including cocaine or nicotine is stressful (Parsons et al., 1995; Turchan et al., 1998; Kenny and Markou, 2001; Awtry and Werling, 2003; Koob and Le Moal, 2005; Jackson et al., 2010), stress-induced regulation of SERT and a possible role for KOR have not been established. Numerous studies have examined the effects of various stressors on 5-HT turnover (an indirect measure of serotonergic activity), 5-HT release using microdialysis, [3H]-5-HT uptake in brain tissue, and [3H]-paroxetine binding to SERT in multiple rodent species and strains with conflicting results (Watanabe et al., 1993; Kirby et al., 1995; Adell et al., 1997; Berton et al., 1999; Connor et al., 1999a,b; Martin et al., 2000; El Yacoubi et al., 2003; Racca et al., 2005; Lee et al., 2007). Importantly, no previous studies have identified a role for κ-mediated regulation of SERT in swim stress or withdrawal from repeated nicotine, or used the FSS paradigm that we have shown previously can cause an norBNI-sensitive increase in immobility, odorant-swim stress aversion, and potentiation of cocaine CPP and of nicotine CPP.
Using the sensitive RDEV method for quantifying neurotransmitter uptake kinetics (Hagan et al., 2010), we found that various potent stressors increased the rate of 5-HT uptake by SERT in an norBNI-dependent manner. SERT Km and Vmax can also be modulated by other GPCRs such as the adenosine receptor (AR), the serotonin 1B receptor (5-HT1B), and the α2 adrenergic receptor, in addition to PKC, PKG, and p38 MAPK (Steiner et al., 2008; Ramamoorthy et al., 2011). Similar to the reported effects of AR agonist treatment and PKG activation, we found that R-FSS increased SERT Vmax without affecting Km, suggesting that repeated stress increases surface SERT expression. Interestingly, pharmacological stimulation of AR3 increased SERT surface expression by an increase in SERT exocytosis and not a decrease in endocytosis of existing surface SERT (Zhu et al., 2004). Parallel experiments will be required to determine whether stress-induced increase in surface SERT expression mediated by KOR activation was due to increased exocytosis, however, the current results suggest that p38 MAPK may phosphorylate an undefined substrate in the vesicle fusion machinery to stimulate SERT translocation. Previously published results have shown that DAT can be modulated by stress exposure and by numerous GPCRs and kinases (Ramamoorthy et al., 2011); however, in the present study, we found that R-FSS did not significantly affect DAT. Nevertheless, a role for KOR regulation of dopamine in aversion is not excluded by the current study.
Although an acute 15 min FSS exposure causes dynorphin release and subsequent KOR activation (as demonstrated by norBNI-sensitive stress-induced analgesia), acute KOR activation was not sufficient to cause an norBNI-sensitive increase in immobility, potentiation of cocaine CPP, or p38 MAPK activation (McLaughlin et al., 2003a; Bruchas et al., 2007a; Schindler et al., 2010). Sustained KOR activation was required for GRK3 and p38 activation, and both were required for KOR-mediated immobility and CPA (Bruchas et al., 2007a). Additionally, p38α MAPK in serotonergic neurons has been shown to mediate aversive stress responses (Bruchas et al., 2011), and in the current study we integrate these previous results to demonstrate that stress-induced potentiation of cocaine CPP and increases in SERT surface expression in the nerve terminals of the serotonergic neurons are also GRK3 and p38α MAPK dependent.
Stress-induced potentiation of the rewarding effects of other drugs of abuse including nicotine (Smith et al., 2012) and ethanol (Sperling et al., 2010) are possibly mediated by similar mechanisms, but further studies will be required to determine whether the actions of other psychostimulants, including amphetamine, also require KOR-mediated effects on SERT function. Together, these data suggest that the prodepressive and proaddictive effects of stress share common signaling mechanisms. The idea that stress exposure results in a dysphoric state that leads to potentiation of drug seeking and drug reinstatement was suggested previously (Koob, 2008; Bruchas et al., 2010; Schindler et al., 2010), and the present study supports the concept that these behavioral responses may be a consequence of transient hyposerotonergic tone in the nucleus accumbens. While dysphoria (like pain) is an internal emotional state that cannot be directly or objectively quantified, humans taking κ-selective agonists report dysphoria (Pfeiffer et al., 1986), and dysphoria can result in behavioral aversion, which can be objectively measured.
R-FSS increases dynorphin release (Shirayama et al., 2004) and KOR activation within the NAc (Land et al., 2008). cAMP response element-binding protein (CREB) within the NAc is increased in response to stress and positively regulates dynorphin gene expression in that brain region (Carlezon et al., 1998; Pliakas et al., 2001). Overexpression of CREB within the NAc produces prodepressive effects, and this is blocked by the KOR antagonist norBNI, suggesting that CREB-induced expression of dynorphin mediates the prodepressive effects observed in the present study (Carlezon et al., 1998; Pliakas et al., 2001). Ultrastructural localization studies demonstrated that dynorphin is in GABAergic D1 type medium spiny neurons, and dynorphin-expressing terminals appose KOR-containing terminals within the NAc (Svingos et al., 1999; Ma et al., 2003; Hara et al., 2006). Thus, local release of dynorphin from MSNs may activate KORs expressed on DR afferents to the NAc, leading to increased serotonin uptake and a hyopserotonergic tone.
Both increases and decreases in serotonergic tone within the NAc have been previously linked to stress responses and may underlie several neuropsychiatric diseases. Olfactory bulbectomized rats, a model of depression, are hyposerotonergic in the NAc (Connor et al., 1999a), and Wistar–Kyoto rats, hyperresponsive to stress, demonstrate decreased serotonergic responsivity in the NAc (De La Garza and Mahoney, 2004). Additionally, infusion of the SSRI fluoxetine into the NAc decreased immobility and increased active coping mechanisms in the forced swim test (Chau et al., 2011), and infusion of the SSRI sertraline into the NAc abolished the cocaine-withdrawal-induced increase in cocaine CPP (Harris et al., 2001). Depletion of NAc serotonin by 5,7-dihydroxytryptamine increased the incentive value of cocaine as assessed by breakpoint (Loh and Roberts, 1990), increased morphine self-administration (Smith et al., 1987), and increased responding for a conditioned reward (Fletcher et al., 1999). Conversely, systemic blockade of 5-HT(2C) receptors inhibit nicotine self-administration (Fletcher et al., 2012). Air puff, a mild stressor, increased the 5HT metabolite 5-hydroxindole-3-acetic acid within the NAc (Merali et al., 1997). Thirty minutes of forced swim stress caused a prolonged elevation in NAc serotonin levels as measured by microdialysis (Kirby et al., 1995), but one 15 min swim followed 24 h later by one 5 min swim resulted in no change to NAc serotonin levels (Kirby and Lucki, 1997). Systemic salvinornin A, a KOR agonist, decreased NAc extracellular levels of DA but not 5-HT in rats (Carlezon et al., 2006), but not in mice (Zhang et al., 2005). The mixed results are potentially due to differences in measurement technique used, species/strain differences, and, most importantly, the type and timing of the stress exposure.
Extracellular serotonin levels are thought to regulate mood (Coppen and Doogan, 1988; Haenisch and Bonisch, 2011), and increased SERT function has been shown to increase behavioral despair in mice (Zhu et al., 2010). Here we demonstrated that KOR-mediated aversion is absent in SERT knock-out mice, demonstrating an integral role for SERT in this behavior. Additionally, 5-HT depletion has been shown to increase cocaine seeking (Walsh and Cunningham, 1997). Kappa-mediated aversion was blocked by pretreatment with the SERT inhibitor citalopram (Bruchas et al., 2011), footshock-induced reinstatement of alcohol self-administration was blocked by pretreatment with the SERT inhibitor fluoxetine (Lê et al., 1999), and in the present study we demonstrate that κ-mediated potentiation of cocaine CPP was also blocked by pretreatment with citalopram, supporting the hypothesis that stress- and κ-mediated effects occur at least in part through regulation of the serotonergic system. Additional studies are required to elucidate the downstream effects of a stress-induced hyposerotonergic state in the ventral striatum. Additional work is also required to identify which of the serotonin receptor(s) are involved, whether they are located presynaptically or postsynaptically, and how they locally affect NAc signal processing.
In conclusion, the present study demonstrates that repeated but not acute swim stress increases SERT function and ventral striatum surface expression through a KOR- and p38α MAPK-dependent manner, and implicates this molecular process in stress-induced behaviors such as immobility, aversion, and addiction risk. Additionally, this study raises the possibility that KOR antagonists may promote stress resilience, and further implicates the κ receptor in the negative aspects of the stress response.
Footnotes
This work was supported by United States Public Health Service Grants DA07278, DA20570, MH63303, and DA030074. We thank Drs. Caron and Lefkowitz (Duke University) for the GRK3 (−/−) mice, and Dr. Pintar (University of Medicine and Dentistry of New Jersey) for the KOR(−/−) mice. Dr. Evan Deneris (Case Western Reserve University) provided the ePET1-Cre mice, and the floxed p38α (p38αlox) transgenic mice were provided by Dr. K. Otsu (Osaka University) through the RIKEN Bioresearch Center. We thank Hannah DeMeritt for genotyping the SERT mice and maintaining the breeding colony. We also thank Michele Kelly, Emma Martinelli, and Sujin Lee for technical assistance, and Dr. Peter Groblewski for providing comments on this manuscript.
The authors declare no competing financial interests.
References
- Adell A, Casanovas JM, Artigas F. Comparative study in the rat of the actions of different types of stress on the release of 5-HT in raphe nuclei and forebrain areas. Neuropharmacology. 1997;36:735–741. doi: 10.1016/s0028-3908(97)00048-8. [DOI] [PubMed] [Google Scholar]
- Awtry TL, Werling LL. Acute and chronic effects of nicotine on serotonin uptake in prefrontal cortex and hippocampus of rats. Synapse. 2003;50:206–211. doi: 10.1002/syn.10259. [DOI] [PubMed] [Google Scholar]
- Baganz N, Horton R, Martin K, Holmes A, Daws LC. Repeated swim impairs serotonin clearance via a corticosterone-sensitive mechanism: organic cation transporter 3, the smoking gun. J Neurosci. 2010;30:15185–15195. doi: 10.1523/JNEUROSCI.2740-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, Durand M, Aguerre S, Mormède P, Chaouloff F. Behavioral, neuroendocrine and serotonergic consequences of single social defeat and repeated fluoxetine pretreatment in the Lewis rat strain. Neuroscience. 1999;92:327–341. doi: 10.1016/s0306-4522(98)00742-8. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007a;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007b;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette WB, Bailey MD, Kukoyi S, Blakely RD, Trowbridge CG, Justice JB., Jr Human norepinephrine transporter kinetics using rotating disk electrode voltammetry. Anal Chem. 1996;68:2932–2938. doi: 10.1021/ac960022x. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Béguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Chau DT, Rada PV, Kim K, Kosloff RA, Hoebel BG. Fluoxetine alleviates behavioral depression while decreasing acetylcholine release in the nucleus accumbens shell. Neuropsychopharmacology. 2011;36:1729–1737. doi: 10.1038/npp.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor TJ, Kelliher P, Harkin A, Kelly JP, Leonard BE. Reboxetine attenuates forced swim test-induced behavioural and neurochemical alterations in the rat. Eur J Pharmacol. 1999a;379:125–133. doi: 10.1016/s0014-2999(99)00492-6. [DOI] [PubMed] [Google Scholar]
- Connor TJ, Song C, Leonard BE, Anisman H, Merali Z. Stressor-induced alterations in serotonergic activity in an animal model of depression. Neuroreport. 1999b;10:523–528. doi: 10.1097/00001756-199902250-00015. [DOI] [PubMed] [Google Scholar]
- Coppen AJ, Doogan DP. Serotonin and its place in the pathogenesis of depression. J Clin Psychiatry. 1988;49(Suppl):4–11. [PubMed] [Google Scholar]
- Daws LC. Unfaithful neurotransmitter transporters: focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol Ther. 2009;121:89–99. doi: 10.1016/j.pharmthera.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Garza R, 2nd, Mahoney JJ., 3rd A distinct neurochemical profile in WKY rats at baseline and in response to acute stress: implications for animal models of anxiety and depression. Brain Res. 2004;1021:209–218. doi: 10.1016/j.brainres.2004.06.052. [DOI] [PubMed] [Google Scholar]
- Earles C, Schenk JO. Rotating disk electrode voltammetric measurements of dopamine transporter activity: an analytical evaluation. Anal Biochem. 1998;264:191–198. doi: 10.1006/abio.1998.2850. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Bouali S, Popa D, Naudon L, Leroux-Nicollet I, Hamon M, Costentin J, Adrien J, Vaugeois JM. Behavioral, neurochemical, and electrophysiological characterization of a genetic mouse model of depression. Proc Natl Acad Sci U S A. 2003;100:6227–6232. doi: 10.1073/pnas.1034823100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher PJ, Korth KM, Chambers JW. Selective destruction of brain serotonin neurons by 5,7-dihydroxytryptamine increases responding for a conditioned reward. Psychopharmacology (Berl) 1999;147:291–299. doi: 10.1007/s002130051170. [DOI] [PubMed] [Google Scholar]
- Fletcher PJ, Rizos Z, Noble K, Soko AD, Silenieks LB, Lê AD, Higgins GA. Effects of the 5-HT2C receptor agonist Ro60-0175 and the 5-HT2A receptor antagonist M100907 on nicotine self-administration and reinstatement. Neuropharmacology. 2012;62:2288–2298. doi: 10.1016/j.neuropharm.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Haenisch B, Bönisch H. Depression and antidepressants: insights from knockout of dopamine, serotonin or noradrenaline re-uptake transporters. Pharmacol Ther. 2011;129:352–368. doi: 10.1016/j.pharmthera.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Hagan CE, Neumaier JF, Schenk JO. Rotating disk electrode voltammetric measurements of serotonin transporter kinetics in synaptosomes. J Neurosci Methods. 2010;193:29–38. doi: 10.1016/j.jneumeth.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan CE, Schenk JO, Neumaier JF. The contribution of low-affinity transport mechanisms to serotonin clearance in synaptosomes. Synapse. 2011;65:1015–1023. doi: 10.1002/syn.20929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Yakovleva T, Bakalkin G, Pickel VM. Dopamine D1 receptors have subcellular distributions conducive to interactions with prodynorphin in the rat nucleus accumbens shell. Synapse. 2006;60:1–19. doi: 10.1002/syn.20273. [DOI] [PubMed] [Google Scholar]
- Harris GC, Altomare K, Aston-Jones G. Preference for a cocaine-associated environment is attenuated by augmented accumbal serotonin in cocaine withdrawn rats. Psychopharmacology (Berl) 2001;156:14–22. doi: 10.1007/s002130100693. [DOI] [PubMed] [Google Scholar]
- Heinz A, Ragan P, Jones DW, Hommer D, Williams W, Knable MB, Gorey JG, Doty L, Geyer C, Lee KS, Coppola R, Weinberger DR, Linnoila M. Reduced central serotonin transporters in alcoholism. Am J Psychiatry. 1998;155:1544–1549. doi: 10.1176/ajp.155.11.1544. [DOI] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WM, Leurs R, Timmerman H, Pintar JE. Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res. 2000;880:102–108. doi: 10.1016/s0006-8993(00)02776-1. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Carroll FI, Negus SS, Damaj MI. Effect of the selective kappa-opioid receptor antagonist JDTic on nicotine antinociception, reward, and withdrawal in the mouse. Psychopharmacology (Berl) 2010;210:285–294. doi: 10.1007/s00213-010-1803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Neurobiology of the nicotine withdrawal syndrome. Pharmacol Biochem Behav. 2001;70:531–549. doi: 10.1016/s0091-3057(01)00651-7. [DOI] [PubMed] [Google Scholar]
- Kirby LG, Lucki I. Interaction between the forced swimming test and fluoxetine treatment on extracellular 5-hydroxytryptamine and 5-hydroxyindoleacetic acid in the rat. J Pharmacol Exp Ther. 1997;282:967–976. [PubMed] [Google Scholar]
- Kirby LG, Allen AR, Lucki I. Regional differences in the effects of forced swimming on extracellular levels of 5-hydroxytryptamine and 5-hydroxyindoleacetic acid. Brain Res. 1995;682:189–196. doi: 10.1016/0006-8993(95)00349-u. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhar MJ. Molecular pharmacology of cocaine: a dopamine hypothesis and its implications. Ciba Found Symp. 1992;166:81–89. doi: 10.1002/9780470514245.ch6. discussion 89–95. [DOI] [PubMed] [Google Scholar]
- Laasonen-Balk T, Kuikka J, Viinamäki H, Husso-Saastamoinen M, Lehtonen J, Tiihonen J. Striatal dopamine transporter density in major depression. Psychopharmacology (Berl) 1999;144:282–285. doi: 10.1007/s002130051005. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Poulos CX, Harding S, Watchus J, Juzytsch W, Shaham Y. Effects of naltrexone and fluoxetine on alcohol self-administration and reinstatement of alcohol seeking induced by priming injections of alcohol and exposure to stress. Neuropsychopharmacology. 1999;21:435–444. doi: 10.1016/S0893-133X(99)00024-X. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim HJ, Kim JG, Ryu V, Kim BT, Kang DW, Jahng JW. Depressive behaviors and decreased expression of serotonin reuptake transporter in rats that experienced neonatal maternal separation. Neurosci Res. 2007;58:32–39. doi: 10.1016/j.neures.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Müller CR, Hamer DH, Murphy DL. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- Lira A, Zhou M, Castanon N, Ansorge MS, Gordon JA, Francis JH, Bradley-Moore M, Lira J, Underwood MD, Arango V, Kung HF, Hofer MA, Hen R, Gingrich JA. Altered depression-related behaviors and functional changes in the dorsal raphe nucleus of serotonin transporter-deficient mice. Biol Psychiatry. 2003;54:960–971. doi: 10.1016/s0006-3223(03)00696-6. [DOI] [PubMed] [Google Scholar]
- Loh EA, Roberts DC. Break-points on a progressive ratio schedule reinforced by intravenous cocaine increase following depletion of forebrain serotonin. Psychopharmacology (Berl) 1990;101:262–266. doi: 10.1007/BF02244137. [DOI] [PubMed] [Google Scholar]
- Ma J, Ye N, Lange N, Cohen BM. Dynorphinergic GABA neurons are a target of both typical and atypical antipsychotic drugs in the nucleus accumbens shell, central amygdaloid nucleus and thalamic central medial nucleus. Neuroscience. 2003;121:991–998. doi: 10.1016/s0306-4522(03)00397-x. [DOI] [PubMed] [Google Scholar]
- Malison RT, Price LH, Berman R, van Dyck CH, Pelton GH, Carpenter L, Sanacora G, Owens MJ, Nemeroff CB, Rajeevan N, Baldwin RM, Seibyl JP, Innis RB, Charney DS. Reduced brain serotonin transporter availability in major depression as measured by [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane and single photon emission computed tomography. Biol Psychiatry. 1998;44:1090–1098. doi: 10.1016/s0006-3223(98)00272-8. [DOI] [PubMed] [Google Scholar]
- Martin C, Duclos M, Mormède P, Manier G, Chaouloff F. Hippocampal and striatal [(3)H]5-HT reuptake under acute stressors in two rat strains differing for their emotivity. Neurosci Lett. 2000;288:246–248. doi: 10.1016/s0304-3940(00)01246-5. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloëz-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003a;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Xu M, Mackie K, Chavkin C. Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J Biol Chem. 2003b;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Chavkin C. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J Biol Chem. 2004;279:1810–1818. doi: 10.1074/jbc.M305796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z, Lacosta S, Anisman H. Effects of interleukin-1beta and mild stress on alterations of norepinephrine, dopamine and serotonin neurotransmission: a regional microdialysis study. Brain Res. 1997;761:225–235. doi: 10.1016/s0006-8993(97)00312-0. [DOI] [PubMed] [Google Scholar]
- Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LH, Koob GF, Weiss F. Serotonin dysfunction in the nucleus accumbens of rats during withdrawal after unlimited access to intravenous cocaine. J Pharmacol Exp Ther. 1995;274:1182–1191. [PubMed] [Google Scholar]
- Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ. G protein-coupled receptor kinase 3 (GRK3) gene disruption leads to loss of odorant receptor desensitization. J Biol Chem. 1997;272:25425–25428. doi: 10.1074/jbc.272.41.25425. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimeis mediated by kappa opioid receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Racca S, Spaccamiglio A, Esculapio P, Abbadessa G, Cangemi L, DiCarlo F, Portaleone P. Effects of swim stress and alpha-MSH acute pre-treatment on brain 5-HT transporter and corticosterone receptor. Pharmacol Biochem Behav. 2005;81:894–900. doi: 10.1016/j.pbb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Shippenberg TS, Jayanthi LD. Regulation of monoamine transporters: role of transporter phosphorylation. Pharmacol Ther. 2011;129:220–238. doi: 10.1016/j.pharmthera.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuvel DJ, Jayanthi LD, Bhat NR, Ramamoorthy S. A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci. 2005;25:29–41. doi: 10.1523/JNEUROSCI.3754-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk JO, Wright C, Bjorklund N. Unraveling neuronal dopamine transporter mechanisms with rotating disk electrode voltammetry. J Neurosci Methods. 2005;143:41–47. doi: 10.1016/j.jneumeth.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Schindler AG, Li S, Chavkin C. Behavioral stress may increase the rewarding valence of cocaine-associated cues through a dynorphin/kappa-opioid receptor-mediated mechanism without affecting associative learning or memory retrieval mechanisms. Neuropsychopharmacology. 2010;35:1932–1942. doi: 10.1038/npp.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MM, Krueger KC, Deneris ES. A differentially autoregulated Pet-1 enhancer region is a critical target of the transcriptional cascade that governs serotonin neuron development. J Neurosci. 2005;25:2628–2636. doi: 10.1523/JNEUROSCI.4979-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- Sirven A, Ravet E, Charneau P, Zennou V, Coulombel L, Guétard D, Pflumio F, Dubart-Kupperschmitt A. Enhanced transgene expression in cord blood CD34(+)-derived hematopoietic cells, including developing T cells and NOD/SCID mouse repopulating cells, following transduction with modified trip lentiviral vectors. Mol Ther. 2001;3:438–448. doi: 10.1006/mthe.2001.0282. [DOI] [PubMed] [Google Scholar]
- Smith JE, Shultz K, Co C, Goeders NE, Dworkin SI. Effects of 5,7-dihydroxytryptamine lesions of the nucleus accumbens on rat intravenous morphine self-administration. Pharmacol Biochem Behav. 1987;26:607–612. doi: 10.1016/0091-3057(87)90173-0. [DOI] [PubMed] [Google Scholar]
- Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C. Stress-induced activation of the dynorphin/kappa-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci. 2012;32:1488–1495. doi: 10.1523/JNEUROSCI.2980-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sora I, Hall FS, Andrews AM, Itokawa M, Li XF, Wei HB, Wichems C, Lesch KP, Murphy DL, Uhl GR. Molecular mechanisms of cocaine reward: combined dopamine and serotonin transporter knockouts eliminate cocaine place preference. Proc Natl Acad Sci U S A. 2001;98:5300–5305. doi: 10.1073/pnas.091039298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP. Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacology (Berl) 2010;210:199–209. doi: 10.1007/s00213-010-1844-5. [DOI] [PubMed] [Google Scholar]
- Steiner JA, Carneiro AM, Blakely RD. Going with the flow: trafficking-dependent and -independent regulation of serotonin transport. Traffic. 2008;9:1393–1402. doi: 10.1111/j.1600-0854.2008.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Colago EE, Pickel VM. Cellular sites for dynorphin activation of kappa-opioid receptors in the rat nucleus accumbens shell. J Neurosci. 1999;19:1804–1813. doi: 10.1523/JNEUROSCI.19-05-01804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113–115. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Tao R, Auerbach SB. Opioid receptor subtypes differentially modulate serotonin efflux in the rat central nervous system. J Pharmacol Exp Ther. 2002;303:549–556. doi: 10.1124/jpet.102.037861. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Turchan J, Przewlocka B, Lason W, Przewlocki R. Effects of repeated psychostimulant administration on the prodynorphin system activity and kappa opioid receptor density in the rat brain. Neuroscience. 1998;85:1051–1059. doi: 10.1016/s0306-4522(97)00639-8. [DOI] [PubMed] [Google Scholar]
- Walsh SL, Cunningham KA. Serotonergic mechanisms involved in the discriminative stimulus, reinforcing and subjective effects of cocaine. Psychopharmacology (Berl) 1997;130:41–58. doi: 10.1007/s002130050210. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Sakai RR, McEwen BS, Mendelson S. Stress and antidepressant effects on hippocampal and cortical 5-HT1A and 5-HT2 receptors and transport sites for serotonin. Brain Res. 1993;615:87–94. doi: 10.1016/0006-8993(93)91117-b. [DOI] [PubMed] [Google Scholar]
- Wellman CL, Izquierdo A, Garrett JE, Martin KP, Carroll J, Millstein R, Lesch KP, Murphy DL, Holmes A. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci. 2007;27:684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ. Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: agonist actions at kappa opioid receptors. Psychopharmacology. 2005;179:551–558. doi: 10.1007/s00213-004-2087-0. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Hewlett WA, Feoktistov I, Biaggioni I, Blakely RD. Adenosine receptor, protein kinase G, and p38 mitogen-activated protein kinase-dependent up-regulation of serotonin transporters involves both transporter trafficking and activation. Mol Pharmacol. 2004;65:1462–1474. doi: 10.1124/mol.65.6.1462. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD. p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem. 2005;280:15649–15658. doi: 10.1074/jbc.M410858200. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology. 2010;35:2510–2520. doi: 10.1038/npp.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]