

Abstract
Lipoprotein receptor signaling regulates the positioning and differentiation of postmitotic neurons during development and modulates neuronal plasticity in the mature brain. Depending on the contextual situation, the lipoprotein receptor ligand Reelin can have opposing effects on cortical neurons. We show that Reelin increases growth cone motility and filopodia formation, and identify the underlying signaling cascade. Reelin activates the Rho GTPase Cdc42, known for its role in neuronal morphogenesis and directed migration, in an apolipoprotein E receptor 2-, Disabled-1-, and phosphatidylinositol 3-kinase-dependent manner. We demonstrate that neuronal vesicle trafficking, a Cdc42-controlled process, is increased after Reelin treatment and further provide evidence that the peptidergic VIP/PACAP38 system and Reelin can functionally interact to promote axonal branching. In conclusion, Reelin-induced activation of Cdc42 contributes to the regulation of the cytoskeleton of individual responsive neurons and converges with other signaling cascades to orchestrate Rho GTPase activity and promote neuronal development. Our data link the observation that defects in Rho GTPases and Reelin signaling are responsible for developmental defects leading to neurological and psychiatric disorders.
Introduction
During development of the neocortex, migrating neurons are navigated to their final destinations where they differentiate by the coordinated activity of multiple extracellular cues that act on specific membrane receptors (Hatten, 1999; Rakic, 2006; Ayala et al., 2007; Kawauchi and Hoshino, 2008). Reelin, one of these signaling molecules, controls cortical layering by signaling through the very-low-density lipoprotein receptor (Vldlr) and the apolipoprotein-E-receptor-2 (Apoer2) (Tissir and Goffinet, 2003; Herz and Chen, 2006). Reelin induces tyrosine phosphorylation of the adaptor protein Disabled-1 (Dab1). Dab1 activates, besides other signaling pathways, class I phosphatidylinositol-3-kinase (PI3K) (Stolt and Bock, 2006). Mouse mutants lacking Dab1, both Src family kinases Src and Fyn, or the Reelin receptors Vldlr and Apoer2 phenocopy the Reelin-deficient reeler mutant (Howell et al., 1997; Trommsdorff et al., 1999; Kuo et al., 2005). Various lines of evidence indicate that Reelin targets cytoskeletal elements (Beffert et al., 2002; Assadi et al., 2003; Chai et al., 2009) and herewith also can participate in neuronal morphogenesis. Reelin induces spine formation (Niu et al., 2008; Pujadas et al., 2010) and is involved in axonal development (Borrell et al., 1999).
The Rho family members RhoA, Rac, and Cdc42 are key regulators of many cellular processes during neural development, including neurite outgrowth, neurite branching, polarity, and vesicle trafficking (Horton and Ehlers, 2003; Hall, 2005; Jaffe and Hall, 2005; Konno et al., 2005; Ridley, 2006). In principle, extracellular guidance molecules that promote neurite outgrowth or motility activate Rac and Cdc42 to stop neurite growth or to induce growth cone collapse they activate RhoA.
Rac stimulates actin dynamics through the WASP family verprolin-homologous protein (or WAVE) by direct interaction with the actin-related protein 2/3 (Arp2/3) complex and profilin II. Cdc42 interacts with the Arp2/3 complex via N-WASP, which induces process formation and neurite outgrowth (Govek et al., 2005; Kholmanskikh et al., 2006; Heasman and Ridley, 2008). Cdc42 and mPar6αPar3 (Par for partitioning defective) orientate the nucleus, the centrosome, and the Golgi apparatus during neuronal growth cone motility (Etienne-Manneville and Hall, 2001; Shi et al., 2003).
Cdc42 and Rac also contribute to activation of the p21-activated kinase (PAK)/LIM-kinase1 (LIMK) pathway, which inhibit actin filament disassembly by phosphorylating actin-depolymerizing factor (ADF)/cofilin leading to growth inhibition. The fine tuning of these convergent and divergent pathways downstream of Rho GTPases is fundamental for correct neuronal development (Ng and Luo, 2004).
Several roles have been proposed for Reelin during neocortical development including functioning as a chemoattractant, a stop, or detachment signal, or as a radial glial elongation factor (Hack et al., 2002; Zhao et al., 2004; Soriano and Del Río, 2005; Förster et al., 2006; Cooper, 2008). Despite detailed knowledge about the morphological effects of Reelin on neuronal migration, cortical layering, and differentiation, the molecular mechanisms by which Reelin regulates these processes on a cellular level remain elusive. Reelin may stimulate growth cone motility and extension, while in a different context it may induce growth cone arrest, thereby controlling cytoskeletal plasticity. Here, we propose a role for Cdc42 in Reelin-mediated neuronal development and thus link lipoprotein receptor signaling with Rho GTPases.
Materials and Methods
Materials.
PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) was purchased from Biomol Research Laboratories, PP3 (4-amino-7-phenylpyrazol[3,4-d]pyrimidine), NMDA, dizocilpine (MK801), Wiskostatin, and the Akt inhibitor VIII were obtained from Calbiochem; ARA014418, SB-216763, LY294002, and wortmannin were obtained from Sigma. Secramine was a gift of T. Kirchhausen (Harvard University, Cambridge, MA).
Plasmids.
The expression plasmids encoding the GST-PAK–Cdc42/Rac interactive binding (CRIB) and GST-mDia1 were obtained from Dr. J. Collard (Netherlands Cancer Institute, Amsterdam, The Netherlands). The expression plasmid Apoer2-DsRed was constructed by cloning the full-length sequence of murine Apoer2 into the pDsRed-Monomer-N2 vector. The Raichu 1054 and 1011 plasmids were obtained from Dr. M. Matsuda (Kyoto University, Kyoto, Japan).
Animals.
Dab1, Apoer2, Vldlr, and Apoer2/Vldlr-double knock-out mice were maintained in accordance with the institutional guidelines of the University of Freiburg. Genotypes were confirmed by PCR analysis of genomic DNA. Wild-type (WT) NMRI mice were obtained from the Animal Facility of the University of Freiburg. For timed pregnancies, mating pairs were set up in the evening and checked for vaginal plugs the following morning; day of plug was deemed embryonic day 0.5 (E0.5).
Production of recombinant protein.
Preparation of Reelin-containing supernatants and control supernatants. HEK-293 cells stably transfected with full-length Reelin cDNA and control cells transfected with a control vector (MOCK) were grown in DMEM with 10% FBS and 0.9 g/L G418 (Geneticin, Applichem) for 2 d to reach full confluence. The medium was then replaced by serum-free DMEM, and the cells were incubated for 2 d at 37°C in 5% CO2. Conditioned medium was collected and centrifuged. The supernatant was concentrated 10-fold using 100 kDa cutoff centrifugal filters (Millipore), sterile filtered, and stored at −80°C until used. Concentrated mock samples were subjected to the same purification procedure, and the according fractions were collected. Reelin- and MOCK-conditioned supernatants were tested for their ability to stimulate Reelin-dependent phosphorylation of Dab1 and AKT. Approximately 5 nm Reelin was applied for measurement of growth cone motility, and ∼0.5 nm Reelin was applied for measurement of filopodia formation and axonal branching. Mock fractions were applied to neurons in exactly the same relative concentrations as Reelin samples. Glutathione S-transferase (GST)-receptor-associated protein (RAP) and GST control proteins were produced as described previously (Herz et al., 1991).
Preparation of toxins.
Full-length CNF1 was expressed in BL21 cells growing in minimal medium (40 mmol/L Na2HPO4, 20 mmol/L KH2PO4, 18 mmol/L NH4Cl, 8 mmol/L NaCl, 1 mmol/L MgSO4, 100 μmol/L CaCl2, 50 nmol/L H3BO3, 7 nmol/L FeSO4, 7 nmol/L CdCl2, 3 nmol/L ZnSO4, 2 nmol/L MnCl2, 1 nmol/L NiCl2, 1 nmol/L NaMoO4, 0.5 nmol/L CuCl2, 13 nmol/L EDTA, 50 mmol/L glucose, and 3 μmol/L thiamine). At OD600 0.6, protein production was induced with 0.2 mmol/L isopropyl β-d-thiogalactoside. Cells were harvested after 3–6 h, and the GST-fusion proteins were purified by means of glutathione Sepharose (Pharmacia) at 4°C. Since the toxin was unstable during thrombin cleavage, it was used as a GST-fusion protein.
Neuronal cell culture.
Primary cultures of mouse cortical neurons were prepared from brains of mice at E15.5. Dissociated cells were seeded in DMEM plus 10% FBS with a low endotoxin level (<0.5 EU/ml) on 10 mm glass coverslips or for biochemical experiments on 10 or 3.5 cm cell culture dishes coated with poly-l-lysine. After 1 d in culture, the incubation medium was replaced by serum-free Neurobasal medium (Invitrogen), supplemented with B27 (Invitrogen) and 1 mm GlutaMax (Lonza), pH 7.3. Cultures were incubated at 37°C in a humidified atmosphere with low oxygen conditions (6.5% CO2 and 9% O2). Neurons showed a density of ∼4 × 104 cells/cm2 when experiments were performed. Cortical cultures contained <1% astrocytes, as determined by GFAP immunohistochemistry for astrocytes and β-tubulin III immunohistochemistry for neurons. Neurons were transfected using a neuronal Nucleofector Kit (Amaxa) according to the manufacturer's instructions.
Organotypic hippocampal slice cultures.
Hippocampal slice cultures from P1 wild-type rats were prepared as described previously (Zhao et al., 2004). Briefly, hippocampi were sliced (300 μm) perpendicular to their longitudinal axis, placed onto Millipore membranes, and transferred to a six-well plate with 1 ml/well nutrition medium (25% heat-inactivated horse serum, 25% HBSS, 50% minimal essential medium, 2 mm glutamine, pH 7.2, 5% CO2 at 37°C). The medium was changed every 2 d and replaced by serum-free Neurobasal/B27 medium 12 h before treatment with Reelin or Mock-conditioned medium after 14 d. Five slices per treatment condition were pooled, lysed and subjected to Cdc42 effector pull-down assays, as described below.
Immunocytochemical staining.
Cultured cortical neurons were fixed for 12 min with 4% paraformaldehyde in a cytoskeletal stabilization buffer (CSB) containing 80 mm Pipes, 5 mm EGTA, 1 mm MgCl2 and 4% polyethylene glycol (molecular weight, 35,000 kDa). After washing with CSB, neurons were permeabilized for 2 min with 0.1% (v/v) Triton X-100 in CSB. Normal goat or normal donkey serum was used to block nonspecific binding. The following primary antibodies were used: monoclonal mouse anti-β-tubulin III (1:4000; Sigma); rabbit polyclonal anti-Apoer2 against the entire ligand binding domains of Apoer2 (Ab 186, 1:500); mouse monoclonal anti-Cdc42 (B-8, 1:50 Santa Cruz Biotechnology); and anti-Reelin antibody G10 (1:1000, Millipore). The resulting immune complexes were visualized with an Alexa Fluor 488 F(ab′)2 fragment of donkey or goat anti-mouse or anti-rabbit IgG, respectively (Invitrogen). For F-actin staining, cells were stained with Alexa 594-conjugated phalloidin.
Reelin stripe assay.
Microcontact printing was performed using polydimethylsiloxane stamps in an adaptation of published procedures (Delamarche et al., 1997). Stamps were coated for 1 h with ∼0.5 nm Reelin, then washed with doubly distilled H2O, dried with a nitrogen stream, and pressed onto poly-l-lysine-coated slides.
GTPase effector pull-down assays.
The GST-fusion protein was expressed in Escherichia coli BL21 cells grown at 37°C. Expression was induced by adding 0.1 mm isopropyl β-d-thiogalactoside at OD600 1.0. Two hours after induction, the cells were collected and lysed by sonication in lysis buffer (50 mm Tris-HCl, pH 8.0, 2 mm MgCl2, 2 mm DTT, 10% glycerol, and 1 mm PMSF). The lysate was centrifuged at 14,000 × g, and the supernatant was used for purification of the GST-PAK-CRIB-/GST-C21-domain and GST-mDia1-domain by affinity purification using glutathione-Sepharose beads (GE Healthcare). Beads loaded with the GST-fusion proteins were washed twice with GST-fishing-buffer [50 mm Tris, pH 7.4, 100 mm NaCl, 2 mm MgCl2, 10% glycerol, 1% (v/v) Nonidet P-40, and 25 μg/ml aprotinin] at 4°C.
GST-PAK-CRIB/GST-mDia domain pull-down experiments.
Approximately 1 × 106 neurons were used for GTP-Rac and GTP-Cdc42 and ∼4 × 106 neurons were used for GTP-RhoA experiments. Cells were treated with drugs as indicated in the Results section and harvested after addition of 250 μl of GST-fishing-buffer. The detergent-soluble supernatant was recovered after centrifugation for 15 min at 14,000 × g and 4°C. GTP-Rac, GTP-Cdc42, or GTP-RhoA proteins were precipitated for 1 h with 20 μl of GST-PAK/GST- C21 mDia /GST fusion protein at 4°C. The complexes were washed three times with ice-cold PBS, resuspended, and boiled with Laemmli buffer. Bound Rac and Cdc42 proteins were detected by Western blotting using anti-Rac1 (1:1000, BD Bioscience), anti-Cdc42 (1:1000, Millipore), and anti-RhoA (1:200, Santa Cruz Biotechnology).
Immunoblotting.
After drug treatment, neurons were washed twice with PBS and harvested in ice-cold lysis buffer [50 mm Tris, pH 7.4, 100 mm NaCl, 2 mm MgCl2, 10% glycerol, 1% (v/v) Nonidet P-40, and a phosphatase and protease inhibitor mixture (Sigma)]. Lysates were cleared by centrifugation at 4°C and 20,000 × g for 20 min, adjusted for protein content, mixed with 4× concentrated gel-loading buffer (125 mm Tris, pH 6.8, 50% glycerol, 4% SDS, 5% β-mercaptoethanol), boiled, separated by SDS-gel electrophoresis (50 μg of protein/lane), and transferred to Hybond-C extra nitrocellulose membrane (GE Healthcare). Membranes were blocked in PBS, pH 7.4, with 0.05% Tween 20 and 5% nonfat dry milk or 5% BSA and incubated overnight at 4°C with antibodies diluted in blocking buffer. Secondary anti-mouse or anti-rabbit antibodies coupled to horseradish peroxidase (1:10,000, GE Healthcare) were applied for 1 h at room temperature. Bound IgG was detected by enhanced chemoluminescence. The following antibodies were used: rabbit anti-phospho-Ser473Akt (1:1000, Cell Signaling Technology); mouse monoclonal anti-phospho-tyrosine 4G10 (1:3000, Millipore); a rabbit anti-Dab1 antibody raised against the 13 C-terminal amino acids of murine Dab1 (1:1000, Trommsdorff et al., 1999); rabbit anti phospho-Ser3cofilin (1:1000, Cell Signaling Technology); and rabbit anti-actin (1:20,000, Sigma) as a loading control.
Time-lapse light microscopy.
Cortical neurons were plated on poly-l-lysine-coated glass-bottom microwell dishes (MatTek). For time-lapse light microscopy, neurons were incubated in a custom-made chamber at 37°C in a humidified atmosphere (6.5% CO2, 9% O2), staged on a Zeiss Axiovert 200 microscope system equipped with a digital camera (Coolsnap HQ, RoperScientific). Differential interference contrast (DIC)-images were acquired every 30 s with a Zeiss DIC × 40, A:1.4 oil-immersion objective for up to 8 h. To minimize phototoxicity, an automatic shutter was used (50 ms/frame). The MetaMorph 7.1 software (Universal Imaging) was used to acquire and process the resulting stacks of images. Only stage II and stage II+ neurons at day in vitro (DIV) 1 were selected for analysis. To quantify motility, stacks (time-lapse movies) were automatically thresholded to binarize the information of the images. The distal 15 μm neurites were defined and marked. The numbers of pixel changes compared with the respective previous image of a stack were determined by using the “region measurements” function of the MetaMorph software. To compare motility of the same growth cone of the “control period” with the “treatment period,” pixel changes of one or two 15 min intervals of the control period and within the first 45 min of the treatment period were determined. Afterward, the SDs of pixel changes of the 15 min intervals were divided by the average of the total pixel area to normalize the motility of growing structures. When GFP-labeled neurons were used, the grayscale image of the fluorescence signal was also binarized by thresholding. To analyze the motility compared with fluorescence resonance energy transfer (FRET) activity, the yellow fluorescent protein (YFP) image was thresholded to generate a binary mask with a value of zero outside the cell and a value of one inside the cell. The aspects of a resulting binarized image of a DIC, FRET, or GFP image were different as the degree of coverage of the thresholded to original image was different, but this fact did not influence quantification, as relative pixel differences were analyzed. All of these three thresholding methods obtained similar results. To express motility of the treatment period in a relative context to the control period, the average of the control period was set as 1. For each treatment group, the neurites from at least n ≥ 6 different neurons were analyzed. With this method, only motility changes without any information about the origin of motility within one neuron can be described.
To determine the number of light-microscopic dense membrane carriers, phase contrast images were captured at 1 s intervals with a Zeiss X 63 Phase III, A:1.4 oil-immersion objective for 3 min. The same neurons were investigated during the control and the treatment period. Membrane traffic was analyzed by measuring the number of membrane carriers flowing through a neurite segment of 10 μm. This segment was located immediately proximal to the neurite shaft. Values of the mean were calculated and sorted in descending order. Fifteen minutes after Reelin or vector-control treatment, the membrane traffic was analyzed again in the same neuron. For normalization the average of the control period was set as 100%, and the average of the treatment period is expressed in a relative context to the control period. The velocity of membrane carriers was measured using the MetaMorph 7.1 software.
For determination of the number of mitochondria, neurons were loaded with 50 nm MitoTracker Orange (Invitrogen) for 20 min. Fluorescence from MitoTracker was imaged and measured with the above-described microscope using an oil DIC immersion objective (Plan-Apochromat 100×/1.4) and a Cairn-OptoScan monochromator system with a Ludl filter wheel (Ludl Electronic Products). Images were taken every second over 3 min. To reduce phototoxicity, a 5 nm bandwidth of the fluorescent light and a binning of 2 were used to reduce exposure time. Nevertheless, phototoxicity made it impossible to measure the same neurons after the addition of Reelin; therefore, >40 independent control and Reelin neurons were analyzed. The numbers of mitochondria flowing through a neurite segment of 10 μm were counted. This segment was located immediately proximal to the neurite shaft.
Morphometry of fixed cells.
A Zeiss LSM 510 VIS confocal system was used to acquire z-stacks of representative neurons; afterward, vertical projections were performed. For quantification, neurons were imaged with the Zeiss Axiovert 200 microscope system. Images were processed with the MetaMorph software. After thresholding, growth cones were defined and marked, and then the areas of the distal 15 μm of the neurites (in μm2) were determined by using the “region measurements” function of the MetaMorph software.
Silencing of Cdc42.
shRNAs directed against murine Cdc42 were cloned into a pEGFP-shRNA plasmid (Dai et al., 2005). The hairpin sequence is 5′-GATCC-X-TTCAAGAGC-Y-TTTTTTGGAAA-3′, with X and Y sense and antisense sequence, respectively. The following sequences were used: X = GTGTGTTGTTGTTGGTGAT, GACTACGACCGCTAAGTTA, CTAGTATGTTTCTCAGTGG, and GTCTGTGATGGAACTCATG.
FRET.
Raichu-Cdc42 or Rac1 respectively contains Cdc42 or Rac1, and the CRIB domain of Pak1 flanked with YFP on the N terminus and cyan fluorescent protein (CFP) on the C terminus (Itoh et al., 2002). In the absence of nucleotide exchange factor (GEF) activity, GDP is bound to Cdc42 so it does not bind the CRIB domain, and there is no FRET between the YFP and CFP. Upon activation of a GEF to load GTP on Cdc42, Cdc42-GTP binds the CRIB domain, bringing YFP and CFP into close proximity to allow FRET. YFP/CFP ratio images were calculated to represent FRET efficiency, which correlated with the activities of Cdc42. E15.5 embryonic cortical neurons were transfected directly after preparation using the Amaxa Nucleofector System (Lonza) with a vector encoding Raichu-Cdc42 or Cdc42 and incubated for 1 d at 37°C. Neurons were washed and preincubated in Neurobasal medium containing B27 supplement. Fluorescence from Raichu-Cdc42 was imaged and measured using the above-described microscope through an oil-immersion objective (Plan-Apochromat 40×/1.4) For emission ratio imaging, the following filter sets were used (Chroma): CFP: D436/20, D480/40; FRET: D436/20, D535/25; YFP: D500/20, D535/25. Images were taken every 30 s. At each time point, three images were recorded with the following exposure times: CFP (100 ms), FRET (100 ms), and YFP (100 ms) at binning of 2 × 2 or when necessary 3 × 3. MetaMorph software was used to perform image analysis. In brief, all images were first shading corrected and background subtracted. The YFP image had the largest signal-to-noise ratio and therefore provided the best distinction between the cell and the background. Hence, the YFP image was thresholded to generate a binary mask with a value of zero outside the cell and a value of one inside the cell. After multiplication by this mask, the FRET image was divided by the CFP image to yield a ratio image reflecting Cdc42 activation throughout the cell. A linear pseudocolor lookup table was applied, and the ratio values were normalized to the lower scale value to visualize Cdc42 activity. To quantify activities, intensities of FRET signals were analyzed with help of the MetaMorph software. Three different series of experiments were performed: a “control series,” where after 30 min 37°C warm Neurobasal medium was added to the neurons for an additional 30 min; a “Reelin” series, where Reelin-containing Neurobasal medium was added for 30 min; and a “MOCK” series, where Neurobasal medium containing supernatant of MOCK-transfected HEK293 cells was added. Calculated FRET intensities of control neurons were averaged and used as the reference value for normalization, and therefore were set for each time point as 100%.
Statistical analysis.
All data are expressed as the mean ± SEM. The Mann–Whitney U test and the Kruskal–Wallis test (for multiple comparisons) were used. If samples showed normal distribution, ANOVA and Scheffé's test or Student's t test were used.
Results
Reelin induces growth cone motility
To investigate cellular effects of Reelin, we determined the growth cone motility of dissociated cultured cortical neurons in vitro as a functional read out for signaling cascades on a single cell level (Kawauchi and Hoshino, 2008). In time-lapse microscopic studies, the neurites and growth cones of individual cortical stage II neurons regularly showed spontaneous motility (Fig. 1A,B; supplemental Movie 1, available at www.jneurosci.org as supplemental material). Using morphological criteria, it was impossible to discriminate between axons and minor processes, although one neurite often exhibited higher motility in the control phase and was therefore regarded as the putative axon (Bradke and Dotti, 1997). Analysis of stage III cortical neurons showed that Reelin affected axons (arrow) and minor processes (supplemental Movie 2, available at www.jneurosci.org as supplemental material) in a similar manner. As the effect of Reelin on motility was stronger in stage II neurons, we used these neurons for further experiments. Bath application of Reelin increased motility within 5–10 min, and neurites developed large, very motile growth cones (Fig. 1A,B,D). Motility differences within one neuron were quantified using the MetaMorph software, which allowed for the transformation of visible motility changes of a stack (movie) into a relative numeric value (Fig. 1B,C).
Figure 1.

Reelin induces neurite motility. A, Time-lapse DIC microscopy of a stage-II mouse cortical neuron derived from a wild-type brain E15.5, DIV1, 16 h after plating, before (Cont.) and after bath application of ∼5 nm Reelin. Images were taken at 30 s intervals. Exemplarily, four subsequent micrograph images of a growth cone within the middle of the control period and four subsequent micrographs within the middle of the Reelin treatment period are shown. B, For quantification, all images of a stack (supplemental Movie 1, available at www.jneurosci.org as supplemental material) were binarized by thresholding and pixel differences were analyzed automatically using MetaMorph software. C, The calculated relative motility of untreated neurons was set as “1.” Supernatants of vector (MOCK-control) transfected HEK293 cells induced no additional growth cone motility. Means ± SEM; n ≥ 6; ***p < 0.001 vs untreated or MOCK-controls. D–G, Effects of Reelin on growth cone morphology. Neurons were treated with Reelin for 30 min and stained for F-actin and β-tubulin III (scale bar, 20 μm) (D) or (E) for the indicated times. The areas of the growth cones, representing the distal 15 μm of the neurites, were determined using the MetaMorph software. Pooled data from three individual experiments were plotted; bars represent mean (n = 50; **p < 0.01, ***p < 0.001 vs controls). F, Neurons were treated with Reelin, the GSK3b inhibitors ARA014418 and SB-216763 for 30 min, LY294002, or wortmannin were added 30 min before the treatment with Reelin. Bars represent mean (n = 50; ***p < 0.001 vs controls). G, Percentage of neurons with one (black columns) or more (white columns) axon-like growth cones with a fanned morphology (means ± SEM; n = 12 quantifications of different coverslips, *p < 0.05 vs controls).
To further analyze this effect of Reelin on the neuronal cellular architecture, we added Reelin to the incubation medium for up to 120 min (Fig. 1E). These Reelin-treated neurons showed enlarged growth cones with abundant filopodia as visualized by staining for F-actin (Fig. 1D). To quantify these Reelin effects, the size (area) of F-actin stained growth cones was measured. Growth cones more than doubled from 35 ± 2.9 μm2 of control neurons to 71 ± 8.4 μm2 after 30 min of Reelin treatment (Fig. 1E). The maximum effect occurred after 30 min.
Many effects on the reorganization of the actin cytoskeleton of growth cones are mediated by PI3K. Since Reelin activates PI3K via Dab1, we tested whether we could block the effects of Reelin on the actin cytoskeleton by inhibiting PI3K activity. After preincubation with wortmannin (100 nm) or LY294002 (20 μm), two chemically unrelated inhibitors, for 30 min, no Reelin effects on growth cone morphology were seen (Fig. 1F).
Reelin stimulates serine-9 phosphorylation of glycogen synthase kinase-3β (GSK3β), which decreases GSK3β activity (Beffert et al., 2002). GSK3β phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones, and chronic inhibition of GSK3β is associated with morphological changes in growth cones (Goold et al., 1999; Kim et al., 2006). However, pharmacological inhibition of GSK3β did not induce changes in growth cone morphology, indicating that GSK3β inhibition was not involved here (Fig. 1F).
One of the first steps of axon determination in a neuronal culture can be observed after staining the actin and tubulin cytoskeleton. Axonal growth cones have a pronounced fanned morphology. The growth cones of the minor neurites, which later become the dendrites, are smaller, and filopodia protrude at acute angles from the tip of the growth cones. Next, we studied how Reelin treatment affected the number of axon-like growth cones with a fanned morphology per cell. As shown in Figure 1G, Reelin treatment resulted in an increase of neurons with more than one large growth cone per cell.
Reelin stimulates motility via Apoer2
Reelin signaling is transmitted through the Apoer2, Vldlr, and the intracellular adaptor protein Dab1, resulting in activation of Src family tyrosine kinases (D'Arcangelo et al., 1999; Hiesberger et al., 1999; Arnaud et al., 2003a; Bock and Herz, 2003). To further verify a causative connection between Reelin application and enhanced neurite motility, and to examine whether the canonical Reelin signaling cascade is involved, we measured Reelin-induced motility in the presence of bath-applied GST-RAP (10 μg/ml), PP2 (1 μm) (Fig. 2A), or the Reelin antibody CR50 (Fig. 2B). RAP is a molecular chaperone for LDLR (low-density lipoprotein receptor) family members that blocks Reelin binding (Herz et al., 1991) PP2 is a Src family protein tyrosine kinase inhibitor (Hanke et al., 1996), and the Reelin antibody CR50 is a Reelin-neutralizing antibody, which blocks Reelin signaling (Fig. 2B) (Ogawa et al., 1995; D'Arcangelo et al., 1999). All treatments abolished Reelin-induced motility. In addition, Reelin did not increase motility in neuronal cultures from mice lacking the Dab1 adapter protein (Fig. 2C).
Figure 2.
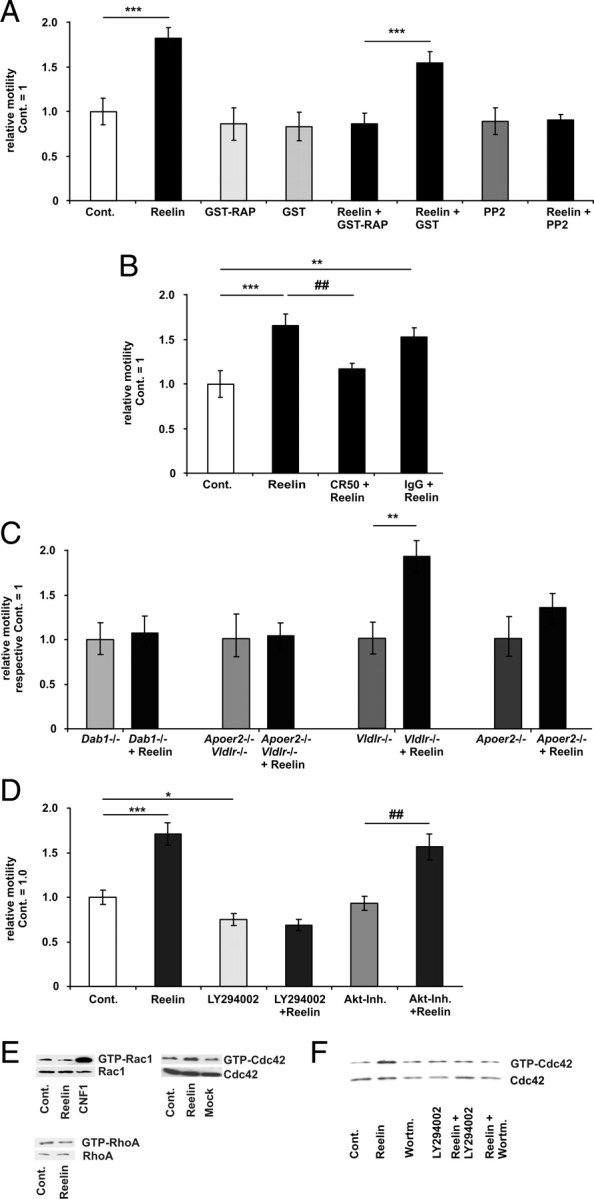
Reelin stimulates motility via Apoer2 and Cdc42. A, The motility of WT neurons was determined before and after bath application of ∼5 nm Reelin, GST-RAP, GST, Reelin, PP2, or in combination as indicated. The relative motilities of the control (Cont.) periods of each treatment group were set as 1 and were compared with the respective treatment periods. Means ± SEM; n ≥ 6; ***p < 0.001 vs control; ###p < 0.001 vs GST-RAP. B, Before the addition of Reelin, Reelin was preincubated with the Reelin-neutralizing CR-50 antibody or control mouse IgG antibody at a concentration of 200 μg/ml. Means ± SEM; n ≥ 6; ***p < 0.001, **p < 0.01 vs control; ##p < 0.01 vs Reelin. C, The motility of Dab1−/−, Apoer2−/−, Vldlr−/−, and Vldlr−/− and Apoer2−/− (double knock-out) neurons was determined before and after bath application of Reelin. Means ± SEM; n ≥ 6; **p < 0.01 vs Vldlr−/−. D, The motility of WT neurons was determined before and after bath application of Reelin, LY294002, Akt inhibitor (Akt-Inh.) VIII, or in combination, as indicated (means ± SEM; n ≥ 6, *p < 0.05, ***p < 0.001 vs controls; ##p < 0.01 vs Akt-Inh.). E, WT neurons were treated with Reelin (15 min). Afterward GTP-Rac1 or GTP-Cdc42 levels were determined by a PAK-CRIB and GTP-RhoA levels by a mDia1 pull-down assay. As a positive control for Rac activation, we applied the Rac1 activating E. coli cytotoxic necrotizing factor 1 (CNF1 toxin). F, WT neurons were treated with Reelin (15 min), wortmannin (45 min), or Reelin was present during the last 15 min of wortmannin treatment (45 min). GTP-Cdc42 was determined with a PAK-CRIB pull-down assay.
To investigate the relative contributions of both Reelin receptors, cultures from lipoprotein receptor-deficient mice (Trommsdorff et al., 1999) were used. Reelin did not increase motility in cultures from Vldlr/Apoer2 double knock-out mice or from Apoer2 single knock-out mice. However, Reelin still increased motility in cultures of Vldlr single knock-out mice (Fig. 2C), indicating that the effect of Reelin on growth cone motility is mediated mainly via Apoer2, which was predominantly expressed at the distal ends of the neurites in cultured cortical neurons (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and (Beffert et al., 2002). Because knock-out mice might compensate for their defects by altering other signaling pathways, we performed a rescue experiment for the Apoer2 knock-out mice. We transfected cultures from Apoer2 knock-out mice with a plasmid encoding for Apoer2. Reelin again induced motility (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Reelin activates Cdc42 via PI3K
In hippocampal neurons, inhibition of PI3K activity reduces neurite motility (Leemhuis et al., 2004). To examine whether PI3K was also involved, we used the PI3K inhibitors LY294002 (20 μm) or wortmannin (100 nm). PI3K inhibition with LY294002 slightly reduced baseline growth cone motility compared with controls and abolished the effect of Reelin on motility (Fig. 2D). To further explore the role of PI3K-dependent signaling mechanisms, we used the Akt1/Akt2 inhibitor VIII (100 nm). As shown in Figure 2D, inhibition of Akt activity had no effect on Reelin-dependent growth cone motility.
The large number of direct PI3K effectors or of PtdIns(3,4,5)P3 and PtdIns (3,4)P2, the lipid products of PI3K, creates an extremely complex signaling web downstream of PI3K activation (Cantrell, 2001). However, some key players are beginning to emerge, linking PI3K activity to specific cellular responses (Hawkins et al., 2006). These include small GTPases of the Rho family, which regulate the cytoskeletal and membrane rearrangements required for cell movement (Wymann and Marone, 2005). Here, the Rho GTPase Rac1, beside other small GTPases, has a well known role in mediating growth cone motility (Obermeier et al., 1998; Meyer and Feldman, 2002). Reelin did not enhance the amount of GTP-bound Rac1 in a CRIB pull-down assay in cortical cultures, confirming previous results (Ballif et al., 2004). When we looked at Rac activity by FRET analysis of neurons transfected with Raichu-Rac1, Reelin increased the Rac1-FRET signal in a subset of neurons. We detected a strong basal FRET signal at the perikaryon of stage II neurons and a slight increase within neurites after addition of Reelin (supplemental Movie 3A, available at www.jneurosci.org as supplemental material). Moreover, a few remaining stage I neurons in culture showed an increased FRET signal over the whole cell after addition of Reelin (supplemental Movie 3B, available at www.jneurosci.org as supplemental material).
Incubation of cortical neurons with Reelin consistently increased the amount of GTP-bound Cdc42, indicating that Reelin activated Cdc42 (see Figs. 2E,F, 3B, 5C, 6A,D, 9F). Reelin had no effect on RhoA activity (Fig. 2E). Next, we studied whether the activation of Cdc42 by Reelin was dependent on PI3K activation. Pretreatment with the PI3K inhibitors wortmannin (100 nm) or LY294002 (20 μm) abolished Reelin-induced Cdc42 activation (Fig. 2F).
Figure 3.
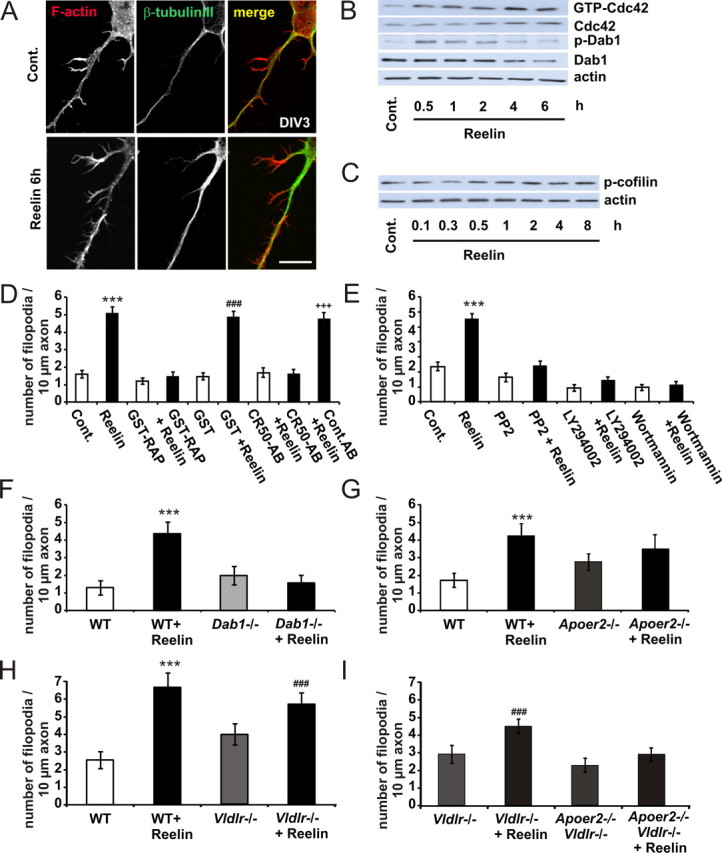
Reelin induces axonal filopodia. A, Neurons were treated with ∼0.5 nm Reelin for 6 h and stained for F-actin and β-tubulin III. Scale bar, 10 μm. B, In cortical neurons, Reelin enhanced the GTP binding of Cdc42 in a time-dependent manner, as determined by PAK-CRIB pull-down experiments. Protein levels of tyrosine-phosphorylated Dab1 detected with the anti-p-tyrosine-antibody 4G10, total Dab1, and actin in cellular lysates from control (Cont.)- or Reelin-treated neurons were measured by immunoblotting. C, Neurons were treated with Reelin for up to 8 h. Protein levels of Ser3-phosphorylated n-cofilin and actin in cellular lysates from control- or Reelin-treated neurons were measured by immunoblotting. D, E, Axonal filopodia formation of WT neurons was determined with or without bath application of Reelin, GST-RAP, GST, CR50, IgG (D), and PP2, LY294002, and wortmannin (E), or in combination, as indicated. Means ± SEM; n ≥ 30; ***p < 0.001 vs controls; ###p < 0.001 vs GST; +++p < 0.001 vs CR50-AB. F–I, Filopodia formation of Dab1−/− (F), Apoer2−/− (G), Vldlr−/− (H), and Vldlr−/−, Apoer2−/− (double knock-out; here, Vldlr−/− act as “controls” because double knock-out mice are bred on a Vldlr−/− background) (I) was determined with or without bath application of Reelin (means ± SEM; n ≥ 30; ***p < 0.001 vs WT; ###p < 0.001 vs Vldlr−/−).
Figure 5.

The peptidergic VIP/PACAP38 system and Reelin functionally interact to promote axonal branching. A, B, Effects of ∼0.5 nm Reelin, the Rho kinase inhibitor Y27632 and VIP on the F-actin and microtubular cytoskeleton in cortical neurons. A, Neurons were treated at DIV3 for 6 h with Reelin, Y27632, and VIP, or the respective combinations. Neurons were stained for F-actin and β-tubulin III. Scale bar, 10 μm. B, The numbers of β-tubulin-positive axonal braches were quantified. Means ± SEM; n ≥ 50; *p < 0.05 vs controls; ###p < 0.001 vs Reelin; +++p < 0.001 vs Y27632; §§§p < 0.001 vs VIP. C, Neurons were treated with Reelin (30 min), VIP (30 min), or VIP and Reelin. Afterward, GTP-Cdc42 levels were determined by a PAK-CRIB pull-down assay. Protein levels of tyrosine-phosphorylated Dab1 were measured with the anti-p-tyrosine antibody 4G10, phosphorylated Akt were measured with a serine 473-phosphorylated Akt antibody and actin in cellular lysates from control, and treated neurons were measured as an input control. D, The motility of neurons was determined before and after bath application of VIP and after the additional application of Reelin, as indicated (means ± SEM; n ≥ 5; *p < 0.05 vs controls or VIP). Cont., Control.
Figure 6.

Reelin activates Cdc42 via Apoer2. A–D, GTP-bound Cdc42 was determined after 15 min of treatment with Reelin in neuronal cultures (DIV6-7) from Apoer2−/− (A), Vldlr−/− (B), Vldlr−/−, and Apoer2−/− and Vldlr−/− (double knock-out) (C), and from Dab1−/− mice (D).
Figure 9.

Dab1 degradation limits Reelin-dependent neurite motility. A, Cultured cortical neurons were treated with Reelin for up to 120 min. Protein levels of tyrosine-phosphorylated Dab1 detected with the anti-p-tyrosine-antibody 4G10, total Dab1, serine 473-phosphorylated Akt, Ser3-phosphorylated cofilin, and actin in cellular lysates from control- or Reelin-treated neurons were measured by immunoblotting. B, In cortical neurons, Reelin enhances the GTP binding of Cdc42, as determined by PAK-CRIB pull-down experiments in a time-dependent manner (upper lanes). Respective inputs are shown in the lower lanes. C, Reelin regulates neurite motility in a time-dependent manner. Neurite motility of cortical neurons was determined after bath application of Reelin over 480 min. The relative motility before Reelin application was set as 1. Afterward, motility was measured in 30 min intervals. Means ± SEM; n = 15 different neurons from n = 5 different neuronal cultures; *p < 0.05 vs controls. D, Reelin did not desensitize neurites to other motility stimuli. Cortical neurons were pretreated for 120 min with Reelin or MOCK. Afterward, neurite motility was determined over a 30 min interval, and relative motility was set as 1. After bath application of Reelin or NMDA (10 μm), the relative motility of the same neurons was measured again. Means ± SEM; n ≥ 6; ***p < 0.001 vs after: Mock 2 h; ##p < 0.01 vs after: Reelin 2 h). E, Reelin induces growth cone motility independently of NMDAR. The motility of WT cortical neurons (DIV1-2) was determined before and after bath application of Reelin, NMDA, MK801, or in combination, as indicated (means ± SEM; n ≥ 6, ***p < 0.001 vs controls; ###p < 0.001 vs MK-801). F, WT cortical neurons were treated (15 min) with Reelin, NMDA, MK801, or in combination, as indicated; afterward, GTP-Cdc42 levels were determined by a PAK-CRIB pull-down assay. GTP-bound Cdc42 is shown in the upper lanes, and total Cdc42 in the lower lanes. Cont., Control.
To determine whether Cdc42 plays a role in Reelin-induced growth cone motility, small interfering RNA was used to knock down Cdc42 in cortical neurons. These Cdc42-deficient neurons did not respond with increased motility after Reelin application (supplemental Fig. 3, supplemental Movie 4, available at www.jneurosci.org as supplemental material).
Cdc42 activation in a neuronal culture can be associated with an increased number of filopodia and neurite branching (Govek et al., 2005; Ridley, 2006). As we only observed filopodia motility at axons (look at the arrow in supplemental Movie 2, available at www.jneurosci.org as supplemental material), we treated cortical neurons (DIV3) with Reelin for 6 h and determined the number of axonal filopodia with or without Reelin treatment (Fig. 3A). The number of axonal filopodia after Reelin treatment at least doubled compared with controls in neurons from WT animals (Fig. 3D–G). As prolonged exposure of neurons to Reelin is known to desensitize the Reelin signaling pathway (Arnaud et al., 2003b; Bock et al., 2004; Feng et al., 2007), we used one tenth of the Reelin used for motility assays. To test whether Cdc42 was still activated and Dab1 was still phosphorylated, we performed a CRIB pull-down assay and a Western blot, which showed that Reelin stimulated Cdc42 activity up to 6 h (Fig. 3B) and phosphorylated Dab1 under these conditions.
ADF/cofilins, which sever and depolymerize aged actin filaments, are central regulators of cytoskeletal dynamics in filopodia formation. The function of ADF/cofilins, in addition to replenishing the cytoplasmic actin monomer pool in neurons, is to control the proper length of actin filaments and thus to prevent formation of abnormal protrusions (Hotulainen et al., 2009). Therefore, we measured whether Reelin affected ADF/cofilin phosphorylation. As shown in Figure 3C, Reelin increased ADF/cofilin phosphorylation, indicating that Reelin might be involved in filopodia stabilization.
To examine whether the canonical Reelin signaling cascade is involved in filopodia formation, we measured Reelin-induced filopodia formation in the presence of bath-applied GST-RAP (10 μg/ml), the Reelin-neutralizing antibody CR50 (Fig. 3D), the Src family protein tyrosine kinase inhibitor PP2 and the PI3Ks inhibitors LY294002 and wortmannin (Fig. 3E). All treatments abolished Reelin-induced filopodia formation. In addition, Reelin did not increase the number of filopodia in neuronal cultures from mice lacking the Dab1 adapter protein (Fig. 3F), the Apoer2 receptor (Fig. 3G), or both Reelin-receptors Apoer2 and Vldlr (Fig. 3I). Reelin was still able to increase filopodia formation in neuronal cultures from mice lacking the Vldlr (Fig. 3H).
To study neuronal morphogenesis of cortical neurons when Reelin is given as a substrate that mimics the Reelin-rich marginal zone, dissociated cortical neurons were incubated on coverslips coated with stripes (50 μm width) of recombinant Reelin, alternating with control stripes. Neuromorphogenesis on the striped substrate was studied 36 h after plating. Outgrowing neuronal processes did not show a preference for a specific substrate. Thus, many neurons were found to give rise to processes on either the control substrate or the Reelin substrate (Fig. 4A,B). Neurons with contact to the Reelin stripe had longer axons and more axonal filopodia. The number of minor processes or minor process filopodia and the total length of the minor processes were not affected by the Reelin substrate (Fig. 4C).
Figure 4.

A, Reelin as matrix induces morphological changes. Neurons were cultured on striped matrix of Reelin and control stripes for 36 h. Neurons were partially located on the Reelin stripe as visualized with the Reelin antibody G10 (green) and on the control stripes (scale bar, 10 μm). Axons growing on the Reelin stripe have longer axons and show more filopodia. B, Higher magnification of the F-actin staining of the Reelin stripe; between the two green lines, for better visualization of the axonal filopodia without the Reelin-staining (scale bar, 10 μm). C, Quantification of axonal length, total length of minor processes, axonal filopodia, minor processes, and minor process filopodia. Means ± SEM; n ≥ 27; **p < 0.01 vs controls. D, Reelin activates Cdc42 in organotypic rat hippocampal slices cultures. Slice cultures were treated with Reelin (30 min); afterward GTP-Cdc42 levels were determined by a PAK-CRIB pull-down assay. As input control, actin and Cdc42 were determined by Western blot. Contr., Control; proc., process.
Next, we investigated whether Cdc42 activates Reelin in organotypic hippocampal slice cultures. Therefore, we treated P1 (DIV14) rat hippocampal slice cultures with Reelin for 30 min. Reelin enhanced the amount of GTP-bound Cdc42 in a CRIB pull-down assay, indicating that Reelin is also capable of inducing Cdc42 activity in a different context (Fig. 4D).
The number of axonal branches did not increase after 6 h of Reelin treatment alone (Fig. 5B). However, when Rho kinase was inhibited with Y27632 (10 μm), Reelin was also capable to increase the number of axonal branches (Fig. 5B); Y27632 alone did not influence the number of axonal branches compared with controls. The neuropeptides vasoactive intestinal peptide (VIP) and pituitary-adenylyl-cyclase-activating polypeptide-38 (PACAP38), which have an established role in neuronal development (Waschek, 2002; Dickson and Finlayson, 2009; Vaudry et al., 2009), can activate Cdc42 and Rac1, but also decrease RhoA and Rho kinase activity in neural cells (Meyer et al., 2005; Henle et al., 2006). As VIP-mediated Rho kinase inhibition induces the elongation of dendrites and axons by stabilizing microtubules (Leemhuis et al., 2007), we used VIP alone and in combination with Reelin. In line with previous results, VIP (10 nm) alone did not influence neurite branching (Fig. 5A,B). The combined treatment of VIP and Reelin increased the number of axonal branches, however, indicating that functional interaction of Reelin and PACAP38/VIP signaling is capable of inducing neurite branching within 6 h of treatment (Fig. 5A,B). To test whether the neuronal signaling molecules VIP and Reelin use the same signaling pathway, we treated neurons with both substances alone or in combination and determined Dab1 and Akt phosphorylation. In addition, we performed a CRIB pulldown-assay to measure Cdc42 activty. As shown in Figure 5C, Reelin and VIP activated Cdc42, but only Reelin activated the canonical Reelin signaling cascade by phosphorylation of Dab1 and Akt. To test whether VIP affected neurite motility, we treated neurons first with VIP and in addition with Reelin. As shown in Figure 5D, VIP did not affect neurite motility, indicating that both molecules cooperate by acting on different signaling pathways.
The ability of Reelin to induce motility and filopodia formation via Apoer2 raised the question of whether both Vldlr and Apoer2 equally contribute to Reelin-induced Cdc42 activation. Therefore, we used neuronal cultures from mice lacking Vldlr, Apoer2, or both receptors. The ability of Reelin to increase Cdc42 activity was markedly reduced in Apoer2-deficient neurons (Fig. 6A), still present in Vldlr-deficient neurons (Fig. 6B) and completely abolished in Vldlr/Apoer2 double knock-out (Fig. 6C) as well as Dab1 knock-out cultures (Fig. 6D). In contrast to the contribution of Apoer2 to motility, Apoer2-deficient neurons still showed a modest increase of Cdc42 activity after Reelin treatment. These results pointed to other Reelin-dependent cellular effects that are mediated via Cdc42.
To further investigate the functional roles of Cdc42-dependent regulation in growth cone motility, Cdc42 activity and motility were assessed in parallel by FRET analysis of neurons transfected with Raichu-Cdc42 (supplemental Movie S3, available at www.jneurosci.org as supplemental material). As the subcellular localization of these Raichu probes may not be the same as those of the authentic Cdc42 (Nakamura et al., 2006), we analyzed the growth cone and the distal 15 μm of a neurite and the whole neuron. In Raichu-Cdc42-transfected cortical neurons, motility and Cdc42 activity were both increased after addition of Reelin (Fig. 7A–C; supplemental Movie 5, available at www.jneurosci.org as supplemental material). We never observed differences in Cdc42 activation between the neurites of one neuron.
Figure 7.

Reelin activates Cdc42-dependent cellular motility. A, Activity of Cdc42 in a motile neuron was visualized by using FRET (Raichu-Cdc42). YFP/CFP ratio images were calculated to represent FRET efficiency (normalized EA), which is not identical to but is correlated with the activities of Cdc42 (top). The YFP stacks were used to determine motility in parallel over time (bottom). B, To demonstrate the increase of Cdc42 activity, which is associated with neurite motility, four different time points are presented in A (blue lines, I-IV) and in B. The corresponding movie of the stack is presented in supplemental Movie S5, available at www.jneurosci.org as supplemental material. Experiments were performed at least five times and similar results were obtained. C, EAs in distal neurites were averaged from two time intervals of 5 min, 15 min after the addition of Mock or Reelin (means ± SEM; n ≥ 6; **p < 0.01 vs Mock). D, Effects of Wiskostatin and Secramine on Reelin-induced motility. The motility of WT neurons was determined before and after bath application of Reelin, Wiskostatin, Secramine, or in combination, as indicated (means ± SEM; n ≥ 6; **p < 0.01 vs controls; #p < 0.05 vs Reelin). Cont., Control.
Reelin regulates Cdc42-dependent cellular processes
To corroborate our finding that Cdc42 is involved in Reelin-induced motility, we either directly inhibited the activation of Cdc42 or blocked effects of Cdc42 on the actin cytoskeleton. Secramine acutely inhibits activation of Cdc42 by a mechanism dependent upon the guanine dissociation inhibitor RhoGDI (Pelish et al., 2006). Wiskostatin is a chemical inhibitor of the Cdc42 effector neural Wiskott-Aldrich syndrome protein (N-WASP), which stabilizes N-WASP in its autoinhibited state (Peterson et al., 2004). Preincubation of cortical cultures with Wiskostatin (5 μm) or Secramine (100 nm) abolished the effect of Reelin on neurite motility (Fig. 7D).
Cdc42 but not RhoA or Rac1 localizes in part to the Golgi apparatus, together with its targets N-WASP, IQGAP, and the Golgi vesicle coat protein coatomer (Erickson et al., 1996; Ridley, 2006). Cdc42 and N-WASP-controlled actin polymerization regulates Golgi-to-endoplasmic reticulum transport (Egea et al., 2006) and thus is a central event of the multiple steps of vesicle trafficking (Luna et al., 2002). Localized accumulation of signaling endosomes or vesicles amplified by a BDNF-chemotactic gradient, which depends on PI3K and Rho GTPase activation, is important for the correct polarization of the neuron for directed migration (Zhou et al., 2007). A selective bulk-flow of organelles and trans-Golgi network (TGN)-derived vesicles can be observed morphologically. Therefore, we analyzed the level of membrane trafficking along a selected neurite segment of the same neuron before and after bath application of Reelin by using phase contrast microscopy (Fig. 8A; supplemental Movie 6, available at www.jneurosci.org as supplemental material) (Bradke and Dotti, 1997). After Reelin treatment, total membrane traffic, which is normally directed preferentially to axons (Deitch and Banker, 1993), increased by 24% ± 8.9 (Fig. 8B). Moreover, these new membrane carriers were more homogeneously distributed in all processes (Fig. 8C). However, the overall velocity of anterograde and retrograde membrane transport was not changed by Reelin (Fig. 8D). As these dense membrane carriers partly represent mitochondria, we investigated whether the increase in membrane traffic corresponded to an increase in the number of mitochondria. Therefore, we visualized the mitochondria with MitoTracker and followed them with fluorescence time-lapse microscopy (supplemental Fig. 4, supplemental Movie 7, available at www.jneurosci.org as supplemental material). The number of mitochondria that were concentrated in the axons of control cells was not changed after Reelin treatment. In addition, they were not homogeneously distributed in all processes. Therefore, we assume that the increase in dense membrane carriers after Reelin treatment represented an increase of organelles or vesicles derived from the TGN.
Figure 8.

Reelin increases vesicle traffic. A, Traffic of vesicles and membrane compartments in all neurites was imaged by phase-contrast microscopy. Images were collected every second over a 3 min interval (see also supplemental Movie S6, available at www.jneurosci.org as supplemental material). Scale bars: left, 10 μm; right, 2 μm. B, The numbers of membrane carriers traveling within 3 min through a defined neurite segment were counted for all neurites. Numbers were sorted according to the number of membrane carriers counted during the control period. In control neurons, the first column represents the traffic in the axon, which is defined as the neurite with the most intense traffic. The total number per neuron of the control period was set as 100% and was compared with a 3 min interval of the same neuron 15 min after addition of Reelin (means ± SEM; n = 20; **p < 0.01; ***p < 0.001 vs controls). C, To compare the relative distribution of membrane carriers between axons (1.00 neurite) and dendrites (2.00 and 3.00 neurites), both the total number of vesicles of the Reelin period and the control period were set as 100% (means ± SEM; n = 20; *p < 0.05 vs controls). D, Frequency distribution of the velocities of the membrane carriers were measured. One hundred fifty or more membrane carriers were tracked for each condition, and the speed of anterograde or retrograde transport was measured. The overlapping peaks in the frequency distribution diagrams show that there is no significant change in the distribution of velocities measured in control neurons and Reelin-treated neurons. Cont., Control.
Dab1 degradation in response to Reelin blocks Reelin-dependent growth cone motility
Prolonged exposure of neurons to Reelin reduces Dab1 protein levels by means of polyubiquitination and proteasomal targeting of phosphorylated Dab1 (Arnaud et al., 2003b; Bock et al., 2004; Feng et al., 2007).
To investigate whether Reelin-dependent neurite motility is influenced by degradation of Dab1, we first treated cultured cortical neurons for up to 120 min. We confirmed an increase in tyrosine phosphorylation of Dab1 within 5 min with its maximum after 15–30 min, and afterward a decrease in Dab1 phosphorylation (Fig. 9A) and total Dab1 protein levels. Reelin also increased the level of GTP-bound Cdc42 within 5 min (Fig. 9B). The strongest Cdc42 activation was seen after 15 min, which was back to control level after 120 min. This time course was similar to the effects of Reelin on Dab1 and Akt phosphorylation and can be explained by Dab1 degradation (Fig. 9A). The level of Ser3-phosphorylated cofilin increased within the 120 min of Reelin treatment, indicating that the actin turnover remained in equilibrium under the present experimental conditions (Endo et al., 2003). Phosphorylation of cofilin did not follow the time course of Dab1 phosphorylation and Cdc42 activation.
To study the time course of Reelin-induced motility, we continuously measured motility after addition of Reelin in intervals of 30 min up to 480 min (Fig. 9C). Motility increased within the first 30 min and afterward decreased again. The lowest motility, which was significantly below control level, was observed after 150–180 min, but we never observed a complete arrest (Fig. 9C).
To examine whether Dab1 degradation in response to Reelin renders neurites insensitive to Reelin and other stimuli, we pretreated the neuronal cultures with Reelin for 120 min and then again added either fresh Reelin or NMDA (10 μm). NMDA receptor (NMDAR) stimulation induces neurite motility by activating the Rho GTPases Rac1 and Cdc42 via the Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Konur and Ghosh, 2005; Henle et al., 2006). After preincubation with Reelin (120 min), addition of fresh Reelin did not alter motility, whereas stimulation of NMDAR increased motility. These results indicate that Reelin-dependent motility is self-limiting, but that the cellular motility machinery is still intact and susceptive to other stimuli (Fig. 9D).
Reelin can also regulate NMDAR activity through a mechanism that requires tyrosine phosphorylation of Dab1 and NR1 and thereby potentiates calcium influx through NMDARs in cortical neurons (Chen et al., 2005). To exclude the possibility that the Reelin effect on motility was actually mediated via NMDAR, we aimed to block Reelin effects on motility and Cdc42 activity by the NMDAR open channel blocker MK801. However, preincubation of cortical neurons with MK801 (100 nm) did not abolish the effects of Reelin but prevented the effects of NMDA on motility and Cdc42 activity (Fig. 9E,F).
Discussion
Mutations of genes regulating neuronal development are causative for a large number of neurological and psychiatric disorders (Lewis and Levitt, 2002). In the developing brain, neuronal motility, which includes process formation, growth cone motility, vesicle transport, and migration, is one of the basal cellular developmental mechanisms. Motility is controlled by a variety of extrinsic cues that have to be transformed by transmembrane receptors into changes of the actin and tubulin cytoskeleton (Hatten, 1999; Schaar and McConnell, 2005; Rakic, 2006). One of these factors is the lipoprotein receptor ligand Reelin. Reelin can increase, silence, and direct neuronal motility, depending on the contextual situation (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (Förster et al., 2002; Zhao et al., 2004; Hack et al., 2007; Chai et al., 2009).
We have identified that the Reelin and Cdc42 signaling cascades interact to regulate neuronal development, pointing to Cdc42 as an important cellular effector of Reelin.
It is not surprising that defects in Rho GTPases and Reelin signaling were independently described as being responsible for neurodevelopmental defects leading to neurological and psychiatric disorders (Bothwell and Giniger, 2000; Fatemi, 2005; Govek et al., 2005).
Reelin activates Cdc42
Reelin induces cofilin phosphorylation, which is mediated by LIMK (Chai et al., 2009). LIMK kinase is activated through phosphorylation by PAK, which in turn is activated by Rac/Cdc42 or Rho kinase for RhoA. Based on our experiments, there is no evidence that Reelin activates RhoA, as RhoA activity did not change and Reelin-induced growth cone motility or enlargement of growth cones would rather point to RhoA inhibition instead of activation. In a CRIB pull-down assay, Reelin did not elevate Rac1 activity (Fig. 2E), but Reelin induced motility, which would point to Rac activation.
The most coherent results were obtained for Cdc42. Reelin consistently activated Cdc42 (Figs. 2E,F, 3B, 5C, 6A,D, 9F), and induced filopodia formation (Figs. 3, 4), neurite branching (Fig. 5), and neuronal vesicle trafficking (Fig. 8), all of which are typical cellular Cdc42-dependent effects. Cdc42 participated in the effect of Reelin on motility (Fig. 7A), even though we cannot rule out the involvement of Rac1 here.
Differential subcellular localization and activity of Cdc42 and the Par complex are critical for axon specification (Shi et al., 2003). By activating Cdc42 via Apoer2 at the tips of all neurites (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), Reelin seems to interfere with axon–dendrite differentiation of cultured cortical neurons (Fig. 1G).
Cdc42-dependent cellular effects of Reelin
N-WASP links Cdc42 activity to changes of the actin cytoskeleton and is normally activated by binding of the Rho GTPase to its CRIB motif. N-WASP is an activator of the Arp2/3 complex that initiates new actin polymerization at the leading edge of cells, which is essential for cell motility (Meyer and Feldman, 2002; Jaffe and Hall, 2005). Dab1 can also directly bind to and activate N-WASP, which presumably results in the activation of a conformational change similar to that induced by Cdc42 (Suetsugu et al., 2004). Dab1 therefore appears to act both dependent and independently of Cdc42. Our results showing increased basal Cdc42 activity in Dab1−/− neurons support this view (Fig. 6D). Higher Cdc42 activity probably compensates for the missing Dab1 impact on N-WASP.
Regulation of vesicle trafficking, which depends on PI3K and Cdc42 activation, is essential for the correct polarization of the neuron during glial-guided directed migration (Ridley, 2006; Sakakibara and Horwitz, 2006; Solecki et al., 2006; Zhou et al., 2007). Reelin increased the number of vesicles or membrane carriers arising from the trans-Golgi network (Fig. 8), further emphasizing its importance in the positioning of neurons and suggesting a role of Reelin-mediated Cdc42 activation during migration.
Reelin has different effects depending on the contextual situation
Cofilin, an essential regulator of actin filament dynamics, is highly expressed in growth cones (Endo et al., 2003). Increased phosphorylation of n-cofilin is observed in cortical lysates from reeler mice when they are stimulated with Reelin (Chai et al., 2009). This overphosphorylation of cofilin is most likely the result of a lack of Dab1 downregulation and related to the recent finding that functional Reelin receptors are much more abundant in reeler mice (Uchida et al., 2009). In our wild-type neurons, we see an increase of n-cofilin phosphorylation after Reelin treatment (Figs. 3C, 9A), which might be important for the stabilization of filopodia (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (Hotulainen et al., 2009), but motility induced by other stimuli than Reelin is not silenced (Fig. 9C). Elevation of GTPase activity by NMDAR stimulation, which is mediated by Rac1 and Cdc42 (Fig. 5F) via the Ca2+/CaMKII (Konur and Ghosh, 2005; Henle et al., 2006), led to an increase in motility in Reelin-desensitized neurons (Fig. 9E). This indicates that the cellular motility machinery of the neurons remains susceptive to other stimuli and emphasizes that the Reelin-signaling cascade is part of a signaling network regulating Cdc42 activity.
By embedding Rho GTPases into different effector domain complexes, they can mediate opposing cellular effects. The selection of these pathways could be achieved by specifically engaging different GEFs. For example, different GEFs activating the same GTPase can have opposite effects in modifying LIMK activity (Ng and Luo, 2004). How Reelin-stimulated GEFs selectively couple Cdc42 to different downstream effector pathways remains to be determined.
The VIP/PACAP38 signaling cascade also contributes to control Rho GTPase activity (Waschek, 2002; Henle et al., 2006). We provide evidence that Reelin and a peptidergic system can work together to promote neuronal development by inducing axonal branching (Fig. 3). A physiological role for the observed synergistic effect of Rho GTPase-modulating signaling cascades with Reelin on axonal branching is supported by observations in reeler mice, where decreased axonal branching of entorhinal axons was noted (Borrell et al., 1999).
Of note, Reelin-induced neurite motility and filopodia formation were virtually unaffected in neurons from Vldlr knock-out mice, whereas lack of Apoer2 blocked the effect of Reelin (Figs. 2C, 3G). Vldlr seems to function as a stop signal for migrating neurons, whereas Apoer2 has been shown to be important for the positioning of late-generated neurons in the developing neocortex (Hack et al., 2007). Our results mirror the observation that the two Reelin receptors have divergent roles during migration on a cellular level. We suggest that differences in the localization of Vldlr and Apoer2, which is concentrated in neurite growth cones (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), play an important role in the differential activation of Cdc42 via both receptors, and that this difference contributes to the divergent physiological functions of both Reelin receptors. An issue of concern is the exact location of Vldlr to different cellular compartments. Attempts to localize Vldlr have been hampered by the low specificity of antibodies.
Reelin as an attractive factor or stop signal—or both
Reelin has a topical distribution; it is concentrated in the marginal zone and is likely to build up a gradient by its fragments that diffuse for larger distances than the unprocessed full-length 400 kDa molecule (Förster et al., 2006; Jossin et al., 2007). It seems that in the marginal zone the motility of radially migrating neurons silences as they grow toward this Reelin-enriched region (Chai et al., 2009). At the same time, the tips of apical dendrites branch to form the apical tuft in layer I. This increased branching in layer I points to an increased process motility in the Reelin-enriched marginal zone (Förster et al., 2002). Different models of Reelin action on individual migrating neurons have been proposed to account for its well characterized effects on cortical structure. These include the interpretation of Reelin as a stop and as a detachment, permissive, attractive, or branch-inducing signal (for a review, see Cooper, 2008). In contrast to Reelin itself, functional Reelin receptors are found predominantly in the intermediate zone or the subventricular zone of the developing neocortex. The action of Reelin, which is also present in the subventricular/intermediate zone, could start already just as neurons begin to migrate (Yoshida et al., 2006; Uchida et al., 2009). Activation of Cdc42, a central regulator for directed migration on the cellular level, points to the putative role of Reelin as an attractive factor when neurons start to migrate.
Desensitization is a key feature of most signaling pathways as it allows a system to reset and thus regain sensitivity to repeated stimulation. Many studies demonstrated that the Reelin pathway displays such a negative feedback mechanism, which also includes regulation of Cdc42 activity (Fig. 9B). This can explain the conflicting roles of Reelin in the regulation of neuronal motility and migration. Parallel to the degradation of Dab1, Reelin-dependent motility is silenced (Fig. 9A,C). It is conceivable that this reduced motility contributes to the detachment of migrating neurons from their radial guidance scaffold when they reach the Reelin-enriched marginal zone.
In summary, we show that Reelin-mediated lipoprotein signaling influences the cellular machinery that is necessary for coordinated actin and microtubule dynamics, leading to changes in growth cone motility, filopodia formation, neurite branching, and vesicle trafficking. This suggests that Cdc42 is a central cellular regulator of Reelin signaling information during neuronal development and differentiation. The Reelin signaling cascade participates in a more complex signaling network involving other Rho GTPase-activating signaling cascades with implications for its role in the mature CNS.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft, the Forschungskommission of the Medical Faculty of Freiburg University, and the Landesgraduiertenförderung of Baden-Württemberg. We thank J.G. Collard (The Netherlands Cancer Institute, Amsterdam, The Netherlands), M. Matsuda (Kyoto University, Kyoto, Japan) and F. Dai (Department of Anatomy, Freiburg University, Freiburg, Germany) for plasmids, J. Nimpf and N. Andrade (Max F. Perutz Laboratories, Vienna, Austria) for the a186 antibody, and T. Kirchhausen (Harvard University, Cambridge, MA) for secramine, S. Zhao (Department of Anatomy, Freiburg University, Freiburg, Germany) for hippocampal slice cultures, Gudula Schmidt (Institute of Pharmacology, Freiburg University, Freiburg, Germany) for CNF1, J. Göldner (Zentrum für Neurowissenschaften, Freiburg, Germany) for excellent technical assistance, H.E. Blum (Department of Internal Medicine II, Freiburg University, Freiburg, Germany) for support, and P. May (Zentrum für Neurowissenschaften, Freiburg University, Freiburg, Germany) for critical reading of the manuscript.
References
- Arnaud L, Ballif BA, Förster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr Biol. 2003a;13:9–17. doi: 10.1016/s0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Cooper JA. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol Cell Biol. 2003b;23:9293–9302. doi: 10.1128/MCB.23.24.9293-9302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, Wynshaw-Boris A, Herz J, D'Arcangelo G, Clark GD. Interaction of reelin signaling and Lis1 in brain development. Nat Genet. 2003;35:270–276. doi: 10.1038/ng1257. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Ballif BA, Arnaud L, Arthur WT, Guris D, Imamoto A, Cooper JA. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol. 2004;14:606–610. doi: 10.1016/j.cub.2004.03.038. [DOI] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- Bock HH, Herz J. Reelin activates SRC family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/s0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, May P, Bergner O, Herz J. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein disabled-1. J Biol Chem. 2004;279:33471–33479. doi: 10.1074/jbc.M401770200. [DOI] [PubMed] [Google Scholar]
- Borrell V, Del Río JA, Alcántara S, Derer M, Martínez A, D'Arcangelo G, Nakajima K, Mikoshiba K, Derer P, Curran T, Soriano E. Reelin regulates the development and synaptogenesis of the layer-specific entorhino-hippocampal connections. J Neurosci. 1999;19:1345–1358. doi: 10.1523/JNEUROSCI.19-04-01345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothwell M, Giniger E. Alzheimer's disease: neurodevelopment converges with neurodegeneration. Cell. 2000;102:271–273. doi: 10.1016/s0092-8674(00)00032-5. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron. 1997;19:1175–1186. doi: 10.1016/s0896-6273(00)80410-9. [DOI] [PubMed] [Google Scholar]
- Cantrell DA. Phosphoinositide 3-kinase signalling pathways. J Cell Sci. 2001;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- Chai X, Förster E, Zhao S, Bock HH, Frotscher M. Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine 3. J Neurosci. 2009;29:288–299. doi: 10.1523/JNEUROSCI.2934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Beffert U, Ertunc M, Tang TS, Kavalali ET, Bezprozvanny I, Herz J. Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci. 2005;25:8209–8216. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008;31:113–119. doi: 10.1016/j.tins.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Dai F, Yusuf F, Farjah GH, Brand-Saberi B. RNAi-induced targeted silencing of developmental control genes during chicken embryogenesis. Dev Biol. 2005;285:80–90. doi: 10.1016/j.ydbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- Deitch JS, Banker GA. An electron microscopic analysis of hippocampal neurons developing in culture: early stages in the emergence of polarity. J Neurosci. 1993;13:4301–4315. doi: 10.1523/JNEUROSCI.13-10-04301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarche E, Bernard A, Schmid H, Michel B, Biebuyck H. Patterned delivery of immunoglobulins to surfaces using microfluidic networks. Science. 1997;276:779–781. doi: 10.1126/science.276.5313.779. [DOI] [PubMed] [Google Scholar]
- Dickson L, Finlayson K. VPAC and PAC receptors: from ligands to function. Pharmacol Ther. 2009;121:294–316. doi: 10.1016/j.pharmthera.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Egea G, Lázaro-Diéguez F, Vilella M. Actin dynamics at the Golgi complex in mammalian cells. Curr Opin Cell Biol. 2006;18:168–178. doi: 10.1016/j.ceb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K. Control of growth cone motility and morphology by LIM kinase and slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci. 2003;23:2527–2537. doi: 10.1523/JNEUROSCI.23-07-02527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JW, Zhang C, Kahn RA, Evans T, Cerione RA. Mammalian Cdc42 is a brefeldin A-sensitive component of the Golgi apparatus. J Biol Chem. 1996;271:26850–26854. doi: 10.1074/jbc.271.43.26850. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Fatemi SH. Reelin glycoprotein: structure, biology and roles in health and disease. Mol Psychiatry. 2005;10:251–257. doi: 10.1038/sj.mp.4001613. [DOI] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Müller U, Frotscher M. Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:13178–13183. doi: 10.1073/pnas.202035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster E, Zhao S, Frotscher M. Laminating the hippocampus. Nat Rev Neurosci. 2006;7:259–267. doi: 10.1038/nrn1882. [DOI] [PubMed] [Google Scholar]
- Goold RG, Owen R, Gordon-Weeks PR. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J Cell Sci. 1999;112:3373–3384. doi: 10.1242/jcs.112.19.3373. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Hack I, Bancila M, Loulier K, Carroll P, Cremer H. Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat Neurosci. 2002;5:939–945. doi: 10.1038/nn923. [DOI] [PubMed] [Google Scholar]
- Hack I, Hellwig S, Junghans D, Brunne B, Bock HH, Zhao S, Frotscher M. Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development. 2007;134:3883–3891. doi: 10.1242/dev.005447. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Henle F, Fischer C, Meyer DK, Leemhuis J. Vasoactive intestinal peptide and PACAP38 control N-methyl-D-aspartic acid-induced dendrite motility by modifying the activities of Rho GTPases and phosphatidylinositol 3-kinases. J Biol Chem. 2006;281:24955–24969. doi: 10.1074/jbc.M604114200. [DOI] [PubMed] [Google Scholar]
- Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS. 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. J Biol Chem. 1991;266:21232–21238. [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD. Neuronal polarity and trafficking. Neuron. 2003;40:277–295. doi: 10.1016/s0896-6273(03)00629-9. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Llano O, Smirnov S, Tanhuanpää K, Faix J, Rivera C, Lappalainen P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185:323–339. doi: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M. Activation of Rac and Cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol Cell Biol. 2002;22:6582–6591. doi: 10.1128/MCB.22.18.6582-6591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. RHO GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Gui L, Goffinet AM. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J Neurosci. 2007;27:4243–4252. doi: 10.1523/JNEUROSCI.0023-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Hoshino M. Molecular pathways regulating cytoskeletal organization and morphological changes in migrating neurons. Dev Neurosci. 2008;30:36–46. doi: 10.1159/000109850. [DOI] [PubMed] [Google Scholar]
- Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat Neurosci. 2006;9:50–57. doi: 10.1038/nn1619. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno D, Yoshimura S, Hori K, Maruoka H, Sobue K. Involvement of the phosphatidylinositol 3-kinase/Rac1 and Cdc42 pathways in radial migration of cortical neurons. J Biol Chem. 2005;280:5082–5088. doi: 10.1074/jbc.M408251200. [DOI] [PubMed] [Google Scholar]
- Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005;46:401–405. doi: 10.1016/j.neuron.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Kuo G, Arnaud L, Kronstad-O'Brien P, Cooper JA. Absence of Fyn and Src causes a Reeler-like phenotype. J Neurosci. 2005;25:8578–8586. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis J, Boutillier S, Barth H, Feuerstein TJ, Brock C, Nürnberg B, Aktories K, Meyer DK. Rho GTPases and phosphoinositide 3-kinase organize formation of branched dendrites. J Biol Chem. 2004;279:585–596. doi: 10.1074/jbc.M307066200. [DOI] [PubMed] [Google Scholar]
- Leemhuis J, Henle F, Meyer DK. VIP induces the elongation of dendrites and axons in cultured hippocampal neurons: role of microtubules. Peptides. 2007;28:1700–1705. doi: 10.1016/j.peptides.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Luna A, Matas OB, Martínez-Menárguez JA, Mato E, Durán JM, Ballesta J, Way M, Egea G. Regulation of protein transport from the Golgi complex to the endoplasmic reticulum by CDC42 and N-WASP. Mol Biol Cell. 2002;13:866–879. doi: 10.1091/mbc.01-12-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DK, Fischer C, Becker U, Göttsching I, Boutillier S, Baermann C, Schmidt G, Klugbauer N, Leemhuis J. Pituitary adenylyl cyclase-activating polypeptide 38 reduces astroglial proliferation by inhibiting the GTPase RhoA. J Biol Chem. 2005;280:25258–25266. doi: 10.1074/jbc.M501630200. [DOI] [PubMed] [Google Scholar]
- Meyer G, Feldman EL. Signaling mechanisms that regulate actin-based motility processes in the nervous system. J Neurochem. 2002;83:490–503. doi: 10.1046/j.1471-4159.2002.01185.x. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Kurokawa K, Kiyokawa E, Matsuda M. Analysis of the spatiotemporal activation of rho GTPases using Raichu probes. Methods Enzymol. 2006;406:315–332. doi: 10.1016/S0076-6879(06)06023-X. [DOI] [PubMed] [Google Scholar]
- Ng J, Luo L. Rho GTPases regulate axon growth through convergent and divergent signaling pathways. Neuron. 2004;44:779–793. doi: 10.1016/j.neuron.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Niu S, Yabut O, D'Arcangelo G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J Neurosci. 2008;28:10339–10348. doi: 10.1523/JNEUROSCI.1917-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier A, Ahmed S, Manser E, Yen SC, Hall C, Lim L. PAK promotes morphological changes by acting upstream of Rac. EMBO J. 1998;17:4328–4339. doi: 10.1093/emboj/17.15.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, Macia E, Feng Y, Shair MD, Kirchhausen T. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat Chem Biol. 2006;2:39–46. doi: 10.1038/nchembio751. [DOI] [PubMed] [Google Scholar]
- Peterson JR, Bickford LC, Morgan D, Kim AS, Ouerfelli O, Kirschner MW, Rosen MK. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat Struct Mol Biol. 2004;11:747–755. doi: 10.1038/nsmb796. [DOI] [PubMed] [Google Scholar]
- Pujadas L, Gruart A, Bosch C, Delgado L, Teixeira CM, Rossi D, de Lecea L, Martínez A, Delgado-García JM, Soriano E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J Neurosci. 2010;30:4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. A century of progress in corticoneurogenesis: from silver impregnation to genetic engineering. Cereb Cortex. 2006;16:i3–i17. doi: 10.1093/cercor/bhk036. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Sakakibara A, Horwitz AF. Mechanism of polarized protrusion formation on neuronal precursors migrating in the developing chicken cerebellum. J Cell Sci. 2006;119:3583–3592. doi: 10.1242/jcs.03080. [DOI] [PubMed] [Google Scholar]
- Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci U S A. 2005;102:13652–13657. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Govek EE, Hatten ME. mPar6α controls neuronal migration. J Neurosci. 2006;26:10624–10625. doi: 10.1523/JNEUROSCI.4060-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano E, Del Río JA. The cells of Cajal-Retzius: still a mystery one century after. Neuron. 2005;46:389–394. doi: 10.1016/j.neuron.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Stolt PC, Bock HH. Modulation of lipoprotein receptor functions by intracellular adaptor proteins. Cell Signal. 2006;18:1560–1571. doi: 10.1016/j.cellsig.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Suetsugu S, Tezuka T, Morimura T, Hattori M, Mikoshiba K, Yamamoto T, Takenawa T. Regulation of actin cytoskeleton by mDab1 through N-WASP and ubiquitination of mDab1. Biochem J. 2004;384:1–8. doi: 10.1042/BJ20041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Uchida T, Baba A, Pérez-Martínez FJ, Hibi T, Miyata T, Luque JM, Nakajima K, Hattori M. Downregulation of functional reelin receptors in projection neurons implies that primary Reelin action occurs at early/premigratory stages. J Neurosci. 2009;29:10653–10662. doi: 10.1523/JNEUROSCI.0345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev. 2009;61:283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- Waschek JA. Multiple actions of pituitary adenylyl cyclase activating peptide in nervous system development and regeneration. Dev Neurosci. 2002;24:14–23. doi: 10.1159/000064942. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol. 2005;17:141–149. doi: 10.1016/j.ceb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133:537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- Zhao S, Chai X, Förster E, Frotscher M. Reelin is a positional signal for the lamination of dentate granule cells. Development. 2004;131:5117–5125. doi: 10.1242/dev.01387. [DOI] [PubMed] [Google Scholar]
- Zhou P, Porcionatto M, Pilapil M, Chen Y, Choi Y, Tolias KF, Bikoff JB, Hong EJ, Greenberg ME, Segal RA. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55:53–68. doi: 10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]