

Abstract
Transcription factor expression levels, which sensitively reflect cellular development and disease state, are typically monitored via cumbersome, reagent-intensive assays that require relatively large quantities of cells. Here we demonstrate a simple, quantitative approach to their detection based on a simple, electrochemical sensing platform. This sensor sensitively and quantitatively detects its target transcription factor in complex media (e.g., 250 μg/ml crude nuclear extracts) in a convenient, low-reagent process requiring only 10 μl of sample. Our approach thus appears a promising means of monitoring transcription factor levels.
Monitoring fluctuating transcription factor (TF) expression levels provides an important assessment of the state of cell populations (e.g., human embryonic stem cell differentiation into neural precursors is often assessed by monitoring the TFs Oct4 and Sox11). Unfortunately, however, traditional methods of measuring TF concentration (e.g., ELISA, Western blots), binding activity (e.g., gel-shift assays), or transcript levels (e.g., quantitative PCR), are cumbersome and time-consuming2. Alternative methods have been developed for in vivo (e.g., genetically encoded reporter constructs) and in vitro (e.g., microcantilever-based assays3, surface plasmon resonance imaging4, and electrochemical impedence spectroscopy (EIS)5) detection. These newer techniques, however, are associated with their own challenges: the former require extensive modification of the cells and the latter require extensive sample processing due to their difficulty functioning in complex media. The routine measurement of TF binding thus remains challenging.
Recent years have seen the development of a broad class of electrochemical biosensors, which we have termed “E-DNA sensors,” that employ redox-tagged, electrode bound probe oligonucleotides. Signal generation in this class of sensor is linked to binding-induced changes in the efficiency with which an attached redox-tag approaches the electrode perform. Because such changes are generally not mimicked by the non-specific adsorption of interferants to the electrode surface and because electroactive contaminants are rare in biological samples6, such sensors perform well in complex media, such as crude cellular extracts, undiluted blood serum7, and unmodified foodstuffs8, suggesting they may be among the more promising of the reagentless biosensor architectures9.
Among the many classes of targets that have proven amenable to detection using E-DNA type sensors6 are DNA-binding proteins. Previous sensors for these targets, however, relied on the steric bulk of the bound target to lower the efficiency of electron transfer10,11. This leads to “signal-off” behavior (i.e., binding reduces current), which suffers from limited gain, greater probability of false positive results (due, for example, to probe degradation), and relatively poor detection limits. In response, we present here a new concept for the electrochemical detection of transcription factors and other DNA binding proteins that is reagentless, “signal-on,” and selective enough and miniaturizable enough to deploy directly in 10 μl samples of crude nuclear extracts.
The new sensor relies on a structure-switching mechanism. Specifically, the sensor probe, an electrode-bound, methylene-blue-modified DNA, forms two stable, but rapidly interconverting conformations (Figure 1)12,13. One of these conformations, which contains the double-stranded TF binding site, positions a redox-active reporter, methylene blue, adjacent to the electrode, leading to efficient electron transfer. The second conformation, which lacks the TF binding site, positions the methylene blue away from the electrode, reducing electron transfer efficiency. In absence of the TF target, the non-signaling, non-binding state is thermodynamically favored. Upon TF binding the conformational equilibrium is pushed towards the binding-competent state14, leading to a large increase in Faradaic current signal (Figure 1).
Figure 1.

The structure-switching electrochemical TF sensor is based on the use of a redox modified DNA probe, which is in equilibrium between two conformations (non-binding, left, and binding, right). Binding of the TF to its consensus sequence, shown in red, shifts the population towards binding conformation, placing the methylene blue redox tag close to the electrode surface and thus increasing its electron transfer rate (eT). Thus, in the presence of the target TF, TATA-binding protein (TBP), a robust current signal increase is observed at the redox potential characteristic of methylene blue.
Although inspired by an earlier, structure-switching optical beacon15, our new electrochemical sensor avoids the difficulties associated with optical measurements in complex samples. It does, however, require careful consideration of design elements specific to electrochemical sensors in this broad class6. For example, if the redox reporter is separated from the electrode in the target-free state via a single-stranded region, then we observe an unacceptably high background current (>100 nA). Because of this phenomenon, we cannot directly adopt the solution-phase architecture to an electrochemical read-out (Supporting Information). Likewise, because the gain of electrochemical sensors is typically less than that of fluorescent sensors, the equilibrium constant for the conformational switch must be more carefully optimized: if the equilibrium constant is too small, binding will be inhibited as it must overcome a very unfavorable conformational free energy; if it is too large, most of the switch will be in the signaling conformation even in the absence of target, leading to excessive background14. For the architecture presented here we have used UNAfold 16 to design a probe with a predicted switching free energy of ΔΔG of −1.11 kJ/mol, which is near optimal for such sensors14.
To demonstrate this new sensing architecture, we have designed a sensor against the common eukaryotic transcription factor TATA binding protein (TBP). To fabricate the device, we followed previously established protocols for sensors in this class17. Briefly, we reduced a disulfide-terminated oligonucleotide with an internal methylene blue (sequence: 5′- Thiol – GAA TAG GTT CCT ATA AAA GGT TGG TTT TAT AAA CCT A T-mb C CTA TTC-3′) to the free thiol by reaction with tris(2-carboxyethyl)phosphine. We then incubated an electrochemically polished gold rod electrode in a solution of the reduced probe at 25 nM. We then backfilled the surface by incubating it in 2 mM 6-mercapto-1-hexanol for 60 min. to generate a stable self-assembled monolayer. This procedure results in a surface coverage of ~1011 molecules/cm2, resulting in mean probe-to-probe separation great enough (~30 nm) such that interactions among neighboring probes are effectively negligible12,18. In addition to ensuring optimal probe spacing and the formation of a stable probe-gold bond, the backfill layer likely also serves to mildly passivate the bare gold, reducing non-specific interactions between the probe and the surface19. In use, the sensor is interrogated using square wave voltammetry (60 Hz frequency, 50 mV amplitude, see Supporting information) and monitoring the resultant methylene blue peak at −0.22 V (vs. Ag/AgCl). We find that the sensor responds rapidly and robustly to its target: titrating the sensor with increasing amounts of TBP (in 50 mM sodium phosphate, 150 mM NaCl, pH 7.4) produces a ~200% increase in signaling current (Figure 2b, n > 12). Although Figure 1, modeled on two-dimensional folding of the sensor, suggests a specific geometry for the surface-bound probe DNA, the actual structure of the probe may be more complex20. The conformation-linked signaling mechanism proposed, a binding induced change in probe geometry that alters the efficiency with which the attached methylene blue approaches the surface, is consistent both with our data, with the free energy calculations we performed in the design of the sensor, and with prior sensors based upon the folding behavior of DNA attached to similar surfaces under similar conditions7,8,11. The dissociation constant obtained from this titration, 121 ± 20 nM, is in close agreement with the 110 ± 24 nM dissociation constant calculated from measurements of the kinetic association and dissociation rates of the sensor (Supporting Information). It is worth noting that this value, however, differs from the affinity of TBP for simple duplex DNA, reflecting the energetic cost associated with the conformational change that drives signaling.
Figure 2.
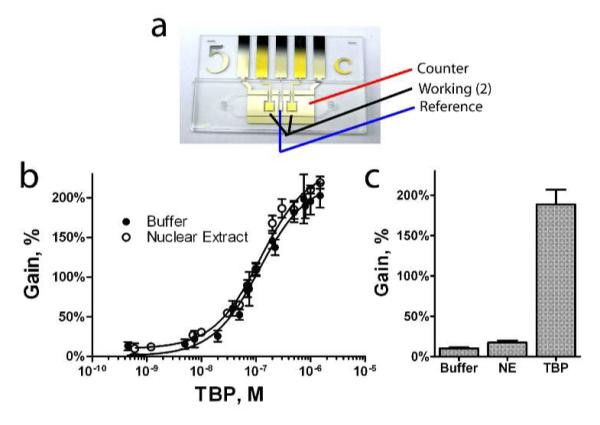
A microfluidic sample chamber containing two sensors supports the detailed measurement of the TF TBP binding in ~10 ul samples. a, the microfluidic chips used in this study. b, the dose-response behavior of the electrochemical TF sensor to the protein TBP is robust and sensitive in buffer or in 250 μg/ml HeLa nuclear extract (NE), displaying an affinity of 121 ± 20 nM. c, Endogenous TBP levels in nuclear extract can be quantitatively obtained using Eq. 1, comparing sensor response in extract to response in pure buffer (Smin, baseline signal) and in the presence of saturating levels of TBP (Smax, maximum response). NE analyzed by this method was shown to contain 4 ± 2 nM of TBP.
Regular monitoring of TF expression in cell cultures is only feasible if the sample requirements for each assay are low and the sensor performs well in crude nuclear extracts. To this end, we have investigated the use of microfluidic systems with a sample volume of 10 μl and two working electrode surfaces (Figure 2a) as an alternative to the rod electrodes we employed in the above proof-of-principle studies. Each chip was assembled from three modular, separately fabricated layers – the electrode substrate, the chamber layer, and the fluidic via substrate21 (Supporting Information). Using these microfluidic devices, we find that the electrochemical TF sensor functions effectively even when employed in media as complex as 250 μg/ml nuclear cell extract. TBP titrated against a sensor equilibrated in HeLa nuclear extract displayed binding response very similar to that observed in buffer (Figure 2b). The binding curve is, however, slightly offset from the curve obtained in buffer; previous experiments have shown that such offset is due to the presence of endogenous TBP present in this extract, as it is a ubiquitous core TF necessary for transcription15,22.
The sensor supports the convenient quantification of transcription factors in cell extract. To do so, we measure the sensor′s response in buffer, cell extract and, finally, extract to which a large excess of exogenous TBP has been added. These measurements can be used to calculate the concentration of TF in the sample, C, via the relationship:
| (1) |
where Smin is the minimum signal response, Smax is the maximum signal response, and Ssamp is the signal response seen in the sample of interest. Performing a sequential addition and electrochemical measurement series in one microfluidic chip, measuring buffer, extract, and after further addition of 1 μM TBP (Figure 2c), we determined the endogenous TBP concentration to be 4 ± 2 nM. While this is clearly near the limit of detection of this first-generation device, it is in close agreement with the results of prior studies15. We believe that the detection limit could be improved by further narrowing the free-energy difference between the probe′s two lowest energy conformations, by further optimizing probe surface coverage, and by the use of a cell extract depleted of the protein of interest (e.g., via the addition of an excess of unmodified binding-site DNA) to provide improved background measurements.
The electrochemical sensing approach presented here, which is likely generalizable to the detection of other TFs15, may provide a convenient platform for the routine assessment of transcription factor activities in cultured cell populations. The approach requires only the ability to design structure-switching sensors containing the relevant binding sequence (which are well documented for thousands of TFs23) which, as demonstrated here, is quite straightforward.
Supplementary Material
Acknowledgements
We wish to thank the members of the Plaxco lab for helpful discussion. This work was supported by ARO Institute for Collaborative Biotechnologies and NIH grants R01EB007689 and R01EB009764. A.J.B. is a Tri-County Blood Bank Santa Barbara Foundation Fellow. A.V.B. is a Fond Québécois de la Recherche sur la Nature et les Technologies Fellow.
Footnotes
Supporting Information Available: Electrochemistry conditions and parameters, details of unsuitable sensor designs, kinetic results, and microfluidic chip preparation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Cai J, Chen J, Liu Y, Miura T, Luo Y, Loring JF, Freed WJ, Rao MS, Zeng X. Stem Cells. 2006;24:516. doi: 10.1634/stemcells.2005-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lymperopoulos K, Crawford R, Torella JP, Heilemann M, Hwang LC, Holden SJ, Kapanidis AN. Angew. Chem. Int. Ed. 2010;49:1316. doi: 10.1002/anie.200904597. [DOI] [PubMed] [Google Scholar]
- (3).Huber F, Hegner M, Gerber C, Güntherodt HJ, Lang HP. Biosens. Bioelectron. 2006;21:1599. doi: 10.1016/j.bios.2005.07.018. [DOI] [PubMed] [Google Scholar]
- (4).Smith EA, Erickson MG, Ulijasz AT, Weisblum B, Corn RM. Langmuir. 2003;19:1486. [Google Scholar]
- (5).Bogomolova A, Komarova E, Reber K, Gerasimov T, Yavuz O, Bhatt S, Aldissi M. Anal. Chem. 2009;81:3944. doi: 10.1021/ac9002358. [DOI] [PubMed] [Google Scholar]
- (6).Lubin AA, Plaxco KW. Acc. Chem. Res. 2010;43:496. doi: 10.1021/ar900165x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW. J. Am. Chem. Soc. 2006;128:3138. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- (8).Cash KJ, Ricci F, Plaxco KW. J. Am. Chem. Soc. 2009;131:6955. doi: 10.1021/ja9011595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu J, Cao Z, Lu Y. Chem. Rev. 2009;109:1948. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ricci F, Bonham AJ, Mason AC, Reich NO, Plaxco KW. Anal. Chem. 2009;81:1608. doi: 10.1021/ac802365x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gorodetsky A, Ebrahim A, Barton J. J. Am. Chem. Soc. 2008;130:2924. doi: 10.1021/ja7106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).White RJ, Phares N, Lubin AA, Xiao Y, Plaxco KW. Langmuir. 2008;24:10513. doi: 10.1021/la800801v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vallee-Belisle A, Plaxco K. Curr. Opin. Struct. Biol. 2010;20:518. doi: 10.1016/j.sbi.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vallee-Belisle A, Ricci F, Plaxco KW. Proc. Natl. Acad. Sci. U.S.A. 2009;106:13802. doi: 10.1073/pnas.0904005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vallée-Bélisle A, Bonham AJ, Reich NO, Ricci F, Plaxco KW. J. Am. Chem. Soc. 2011;133:13836. doi: 10.1021/ja204775k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Markham NR, Zuker M. Methods Mol. Biol. 2008;453:3. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- (17).Lubin AA, Vander Stoep Hunt B, White RJ, Plaxco KW. Anal. Chem. 2009;81:2150. doi: 10.1021/ac802317k. [DOI] [PubMed] [Google Scholar]
- (18).Ricci F, Lai RY, Heeger AJ, Plaxco KW, Sumner JJ. Langmuir. 2007;23:6827. doi: 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Boon EM, Salas JE, Barton JK. Nat. Biotechnol. 2002;20:282. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- (20).Aldaye FA, Palmer AL, Sleiman HF. Science. 2008;321:1795. doi: 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]
- (21).Ferguson B, Buchsbaum S, Swensen J, Hsieh K, Lou X, Soh H. Anal. Chem. 2009;81:6503. doi: 10.1021/ac900923e. [DOI] [PubMed] [Google Scholar]
- (22).Borggrefe T, Davis R, Bareket-Samish A, Kornberg RD. J. Biol. Chem. 2001;276:47150. doi: 10.1074/jbc.M109581200. [DOI] [PubMed] [Google Scholar]
- (23).Bryne JC, Valen E, Tang MHE, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. Nucleic Acids Res. 2008;36:D102. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.