

Abstract
Kahalalide F (KF) and the regioisomer isoKF are novel anticancer drugs of marine origin and currently under clinical investigation. Here we report the synthesis of two new KF analogs with significant in vitro and in vivo antifungal and antitumor activities. The primary amine hydrogen of ornithine in KF has been replaced with 4-fluoro-3-methylbenzyl and morpholin-4-yl-benzyl via reductive N-alkylation. The TGI of these analogs using the NCI-60 cell line screening revealed promising results when compared to paclitaxel. The result of in vivo hollow fiber and animal toxicity assays are presented.
Keywords: Kahalalide F, Anti-cancer, Anti-infective, Structure-activity relationship, Marine natural products
1. Introduction
Marine natural products have been a source of new lead drugs for the treatment of deadly diseases for nearly four decades. The search for superior drugs for the treatment of cancer continues to focus successfully on naturally-occurring sources for anticancer therapy. Cytotoxic peptides are synthesized by a wide variety of plants, animals and microbes. The sacoglossan mollusks of the genus Elysia have been intensively investigated for their biologically active natural products which display significant pharmacological utility. The depsipeptides called the kahalalides were first isolated from Elysia rufescens and more recently Elysia ornata and are reported to have antimalarial, anticancer, antituberculosis and antifungal activities.1 Later it was discovered that the green alga Bryopsis pennata, on which the mollusk feeds is also a reasonable source of depsipeptides but in lower concentration.2 This suggested that the kahalalides are likely secondary metabolites produced by an associated microorganism or from the diet of the green alga B. pennata and later from which a Vibrio sp. strain ER1A was identified as a source of 1.1f
A diverse array of depsipeptides including kahalalides A-F, isoKF, K, O-Q, and three linear peptides, kahalalides G, H, and J, ranging from a C31 tripeptide to a C75 tridecapeptide have been isolated from the mollusk.3 Recently, two additional new cyclic depsipeptides, 5-OHKF and norKA were isolated from the green alga Bryopsis pennata.4 Kahalalide F (1), isoKF and other 1 related analogs are the most promising compounds of the kahalalide family due to their significant anticancer activities. The NCI-COMPARE analysis and evidence of a positive therapeutic outcome in some patients with solid tumors including prostate cancer supports the need for further SAR studies for this class.5 IsoKF is currently under-going phase II development in liver, melanoma and non-small cell lung cancer (NSCLC).6 A phase II trial for the treatment of patients with severe psoriasis is also ongoing using 1 and in addition 1 is active against several pathogenic microorganisms that cause the opportunistic infections associated with HIV/AIDS.7
Due to the limited availability of Elysia for the isolation of 1 and the expenses associated with D amino acids the cost of commercial production of 1 maybe a limitation however the production of 1 may eventually be cost effective by the discovery of a Vibrio sp found to produce 1.1f As a result the modification of 1 using semisynthesis has merit.
1 alters the function of the lysosomal membrane, a characteristic that differentiates it from all other known antitumor agents. The COMPARE analysis in a panel of 60 human cancer cell lines for cell proliferation pathways reveals that KF is part of the list of new chemicals that interact with the Erb/Her-neu pathway.8 This specific interaction has been described in a translational program that has confirmed a selective downregulation of ErbB3 expression by 1 treatment. Sensitivity to 1 significantly correlates with baseline expression levels of ErbB3 (HER3), but not of other ErbB receptors, in a panel of established cell lines from different origins.9 These findings suggest that ErbB3 may be a potential marker for 1 sensitivity in patients. Studies demonstrate that 1 induces cell necrosis in vivo (oncosis) and shows selectivity for tumor cells compared with healthy cells in vitro.10
As shown in figure 1, kahalalide F is a head-to-side-chain cyclic depsipeptide that terminates in a short chain fatty acid conjugated to the N-terminus.11 Since the quantities of the natural product available were inadequate for future additional clinical studies convergent strategies for solid phase synthetic routes were developed.12 As a part of our structure-activity relationship studies of 1 and isoKF analogs, we have recently reported the first solution phase semi-synthetic modification and lead optimization of 1 that can be adapted to a drug generated through fermentation.13
Figure 1.

The structural formula for kahalalide F and isokahalalide F
Earlier we reported a series of ornithine derivatives and the SAR for these alterations the molecule.13 Two molecules selected from this earlier study in which the terminal amine of ornithine of the parent molecule 1 is substituted with 4-fluoro-3-methylbenzyl and morpholin-4-yl-benzyl were evaluated by NCI using in vitro cytotoxicity and in vivo animal models. The results of these studies are reported here.
Results and Discussion
Chemistry
Two new 1/isoKF analogs were synthesized via a solution phase promoted reductive N-alkylation of the primary amino group of ornithine.
In this study we focused our attention on the primary amine of ornithine as the key functional group for modification beginning with the natural product since they play a crucial role in the bioactivity of this class of compounds. Secondary amines in particular are extremely important pharmacophores of numerous biologically active compounds, which have been extensively investigated in the area of drug discovery. Therefore, two selected aldehydes were subjected to a standard reductive alkylation.14 The reductive alkylation of 1 with aldehydes was best performed under optimized conditions by which the parent molecule was exposed to 5 equivalents of aldehyde in methanol for 30 minutes at room temperature prior to portion wise addition of two equivalents of triacetoxyborohydride at the same conditions to give the desired products in good yields. Dialkylated derivative of analog 2 (compound 3) was isolated in low yield while no trace of bis(morpholin-4-yl-benzyl)-KF was observed (Scheme 1 and Table 1).
Scheme 1.

Synthesis of mono- and di-alkyl-N-substituted-KF based on the stepwise reductive alkylation.
Table 1.
| Compound | R1 | R2 | Yield (%) |
|---|---|---|---|
| 2 | H |
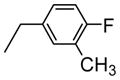
|
68.7 |
| 3 |
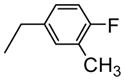
|
|
6.6 |
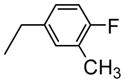
| 4 | H |
|
69.7 |
The structures of N-alkylated-KF analogs were confirmed with spectroscopic and MS techniques. The positive-ion high-resolution ESIMS of analogs 2–4 showed accurate [M+H]+ ion peaks in positive mode, in accordance with their molecular formula (Table 2). In comparison with KF, the 1H-NMR spectra of these compounds clearly exhibited a downfield shift of CH2 (δ) signals on L-ornithine [δH= 2.74 (1) to 2.94 (2) and 2.84 (4) ppm], indicating the presence of N-substituted moieties.
Table 2.
Selected 1H-NMR and DEPT 135° data for 1 and its N-alkylated-KF analogs (2,4) in DMSO-d6.
| Compound | HRESIMS [M+H]+ | 1H-NMR | 13C-NMR | ||||
|---|---|---|---|---|---|---|---|
| L-Ornithine
|
Derivative (R) residue | L-Ornithine
|
Derivative (R) Residue | ||||
| NHR | C H2(δ) | CH2(δ) | NHCH2R | ||||
| 1 | 1477.9408 | 7.62 (m, 2H, NH2) | 2.74 (m) | 7.19–7.28 (m, 5H, Ph) | 38.7 | - | 126.8 (Ph), 128.5 (2C, Ph), 129.5 (Ph), 130.1 (Ph) |
| 2 | 1599.9945 | 7.63 (m) | 2.94 (m) | 6.75–7.28 (m, 8H, Ph) | 38.7 | 47.6 | 14.6 (CH3), 114.7 (Ph), 115.0 (Ph), 126.9 (Ph), 127.5 (Ph), 128.6 (Ph), 129.7 (Ph), 130.2 (Ph), 131.6 (Ph) |
| 4 | 1653.0366 | 7.63 (m) | 2.84 (bs) | 3.12 (m, 4H), 3.73 (m, 4H), 6.98–7.28 (m, 9 H, Ph) | 38.4 | 47.6 | 48.5 (2×CH2, Mor), 66.4 (2×CH2, Mor), 115.2 (2C, Ph), 126.9 (Ph), 128.6 (2C, Ph), 129.7 (Ph), 130.3 (Ph), 131.3 (2C, Ph) |
Abbreviations: Ph=phenyl, Mor=morpholine
The 1H-NMR spectrum of 4-Fluoro-3-methylbenzylamino-KF (2) showed eight characteristic proton signals in the aromatic region assignable to an extra three protons corresponding to the monosubstituted 4-fluoro-3-methylbenzyl moiety in combination with five aromatic protons of phenylalanine [δH= 6.75–7.28 (m, 8H) ppm], which was confirmed unambiguously by three new aromatic signals in the DEPT 135° spectrum [δC= 114.8, 127.5, and 131.6 ppm].
The structure of morpholin-4-yl-benzylamino-KF (4) was confirmed by comparing its 1H-NMR data with 1 in which the aromatic protons of analog 4 resonated at 6.98–7.28 (m, 9 H) ppm, along with chemical shifts at 3.12 (4H) and 3.73 (4H) ppm, consistent with four methylene groups on morpholine moiety. The latest data are in agreement with corresponding chemical shifts from the DEPT 135° which are observed at 48.5 (2C), and 66.4 (2C) ppm for morpholine, and 115.2 (2C) and 131.3 (2C) ppm for aromatic moiety, respectively.
Biological Activity
Activity in opportunistic infections
The bioactivity of 1 and its analogs were tested in vitro for their activity against several microorganisms that cause opportunistic infections including Escherichia coli, Pseudomonas aeruginosa, Methicillin resistant Staphylococcus aureus, Candida albicans, Cryptococcus neoformans, Mycobacterium intracellulare and Aspergillus fumigatus. These depsipeptides do not demonstrate activity against E. coli, P. aeruginosa, or Methicillin resistant S. aureus (data has not shown). Neither 1 nor its analogs displayed significant activity against M. intracellulare. Interestingly, 1 and its analogs exhibit activity against the yeast type fungus (C. albicans), dimorphic fungus (C. neoformans) and filamentous fungi (A. fumigatus and Fusarium spp.). The in vitro activity against fungi is shown in table 3.
Table 3.
In Vitro Data of antimicrobial activities
| Compound |
C. albicans ATCC 90028 (μM) |
C. neoformans ATCC 90113 (μM) |
A. fumigatus ATCC 90906 (μM) |
M. intracellulare ATCC 23068 (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| IC50 | MIC | MFC | IC50 | MIC | MFC | IC50 | MIC | MFC | IC50 | MIC | MBC | |
| 1 | 3.02 ± 0.04 | 10 | 20 | 1.53 ± 0.38 | 5 | 5 | 3.21 ± 0.05 | 10 | 20 | >20 | >20 | >20 |
| 2 | 3.64± 0.31 | 10 | 20 | 0.95± 0.15 | 5 | 10 | 3.09± 0.05 | 10 | 10 | >20 | >20 | >20 |
| 4 | 5.76± 0.27 | 20 | >20 | 1.73 ± 0.15 | 5 | 10 | 5.00 ± 0.74 | 10 | 20 | >20 | >20 | >20 |
| Amp B | 0.25 ± 0.04 | 0.625 | 1.25 | 0.79 ± 0.05 | 0.016 | 0.06 | 1.32 ± 0.06 | 2.5 | 2.5 | NT | NT | NT |
| Cipro | NT | NT | NT | NT | NT | NT | NT | NT | NT | 0.42 ± 0.07 | 1.0 | >1 |
NT= Not Tested
Amphotericin B and ciprofloxacin are used for positive control antifungal and antibacterial standards, respectively.
IC50 is the concentration that affords 50% inhibition of growth.
MIC (Minimum Inhibitory Concentration) is the lowest test concentration that allows no detectable growth.
MFC/MBC (Minimum Fungicidal/Bactericidal Concentration) is the lowest test concentration that kills 100% of the organism.
Analogs 2 and 4 exhibit improved activity against C. albicans, C. neoformans, and A. fumigates relative to 1. Analog 2 showed highest activity against C. neoformans (IC50= 0.95 μM) when compared to the parent molecule (IC50= 1.53 μM) and nearly identical to the positive control Amphotericin B (IC50= 0.79 μM).
In vitro cytotoxicity
1 and its analogs 2 and 4 were selected and evaluated for in vitro anticancer drug screening in a panel of 60 human cancer cell lines (the NCI-60) as a part of the Developmental Therapeutics Program at the National Cancer Institute (supplementary information). These 1 analogs exhibited significant activity against selected cell lines, in particular NSCL, colon, ovarian, and breast cancers, and especially for prostate cancer (Table 4, Figure S1).
Table 4.
In vitro activity data of 1 and its analogs 2 and 4 (μM) against various cell lines.
| Cancer | Cell line | Paclitaxel a | 1a | 2 | 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| GI50 | TGI | GI50 | TGI | GI50 | TGI | GI50 | TGI | ||
| NSCL | A549/ATCC | 0.004 | 25.11 | 0.135 | 0.302 | 0.167 | - | 0.175 | - |
| NCI-H322M | 0.013 | 6.310 | 0.191 | 0.372 | 0.131 | 0.244 | 0.133 | 0.264 | |
| Leukemia | RPMI-8226 | 0.002 | 5.012 | 1.738 | 6.918 | 0.885 | 2.260 | 1.130 | 2.510 |
| Colon | COLO 205 | 0.003 | 0.316 | - | - | 0.135 | - | 0.152 | - |
| HCC-2998 | 0.003 | 0.126 | 0.288 | 0.616 | 0.172 | 0.367 | 0.250 | 0.877 | |
| HCT-15 | 0.158 | 15.85 | 0.269 | 0.741 | 0.181 | 0.419 | 0.264 | 0.814 | |
| HT29 | 0.002 | 0.251 | 0.162 | 0.316 | 0.164 | - | 0.224 | - | |
| KM12 | 0.004 | 15.85 | 0.182 | 0.363 | 0.155 | 0.328 | 0.201 | - | |
| CNS | SNB-75 | 0.004 | 0.126 | 0.224 | 1.905 | 0.370 | 1.250 | 0.808 | 1.900 |
| Melanoma | UACC-257 | 0.040 | 25.12 | 1.023 | 2.818 | 1.280 | 2.550 | 1.340 | 2.650 |
| Ovarian | SK-OV-3 | 0.008 | 15.85 | 0.191 | 0.355 | 0.173 | 0.370 | 0.178 | 0.415 |
| Renal | ACHN | 0.398 | 12.59 | 1.659 | 3.236 | 1.120 | 2.140 | 1.270 | 2.470 |
| Prostate | PC-3 | 0.004 | 10.00 | 0.170 | 0.324 | 0.165 | 0.342 | 0.156 | 0.340 |
| DU-145 | 0.005 | 0.794 | - | - | 0.123 | 0.240 | 0.128 | 0.257 | |
| Breast | T-47D | - | - | - | - | 0.144 | 0.327 | 0.167 | 0.466 |
| HS 578T | 0.003 | 0.100 | 0.162 | 0.479 | 0.217 | 0.665 | 0.453 | 0.444 | |
Developmental Therapeutics Program NCI/NIH (http://dtp.nci.nih.gov), “ - = No data”
GI50 (50% inhibition of cell growth: the concentration needed to reduce the growth of treated cells to half that of untreated (i.e., control) cells. TGI (100% (total) growth inhibition): the concentration required to completely halt the growth of treated cells.
The total growth inhibition (TGI) of 1 and its analogs exhibited higher cytotoxic potency than paclitaxel against most of the human cell lines and in particular colon. A possible future goal maybe to scale these analogs and investigate the drug delivery so that the dose delivered to colon cancer patients will further enhance the selectivity that shown here. Analogs 2 and 4 showed better activity than natural product 1 against NCI-H322M, RPMI-8226, HCC-2998, HT29, SK-OV-3, ACHN and PC3 cell lines. These findings revealed the highly promising improvements associated with the modification of 1. Both of the analogs 2 and 4 demonstrate significant in vitro cytotoxicity against all human cancer cell lines in the panel while some of them display improved activity than parent molecule. Regarding the activity against NSCL, analogs 2 and 4 caused 50% growth inhibition at 0.131 and 0.133 μM, respectively, for NCI-H322M while parent molecule 1 provided an inhibition at relatively higher concentration (GI50 = 0.191 μM). Analogs 2 and 4 showed potent in vitro cytotoxic activity against a panel of human prostate and breast cancer cell lines, with a GI50 ranging from 0.123 (DU-145) to 0.453 (HS 578T) μM.
Maximum tolerated assay (MTD)
It is reported that the MTD for 1 in female mice was 280 μg/kg after a single bolus intravenous injection.15 The MTD of compounds 2 and 4 was determined by Nontumored Animal Toxicity Assay (Tables S1 and S2). Intraperitoneal (IP) injections of 400, 200 and 100 mg/kg/dose were given to the female athymic nude mice and monitored their weight loss. In the case of analog 2, there was no survival at 400 and 100 mg/kg/dose after 19 days, while the animal survived at 200 mg/kg/dose. The experiment was repeated with compounds 2 and 4, this time three doses of 50, 25 and 12.5 mg/kg/dose were given through IP injection to three mice. There was no observed toxicity after 24 days in the lowest concentration indicating 12.5 mg/kg as the MTD of analogs of 2 and 4.
Hollow fiber assay (HFA)
The preliminary in vivo activity of 1 analogs was demonstrated by a hollow fiber assay, which provided quantitative indices of drug efficacy of these compounds. A panel of 12 tumor cell lines viz. NCI-H23, NCI-H522, MDA-MB-231, MDA-MB-435, SW-620, COLO 205, LOX, UACC-62, OVCAR-3, OVCAR-5, U251 and SF-295 was used (supplementary information, Table S2). Based on the MTD, each mice was administered by IP injection at 2 dose levels. The fibers were collected from the mice on the day following the fourth compound treatment and subjected to the stable endpoint MTT (3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide) assay. The optical density of each sample was determined spectrophotometrically at 540 nm and the mean of each treatment group was calculated. Out of the maximum possible score of 96 for an agent (12 cell lines × 2 sites × 2 dose levels × 2 [score]), compound 2 showed IP score 4 (out of 48), SC score 2 (out of 48) and it did not cause cell killing. Interestingly, compound 4 displayed higher drug activity indices than analog 2. Compound 4 exhibited IP score 12 (out of 48), SC score 4 (out of 48) while no cell killing was observed. In summary, these results reveal that the rational design and semisynthetic modification of 1 to generate new active analogs can be achieved. These limited lead exploration studies led to the discovery of new kahalalide F analogs with in vitro improvements against selected cancer cell lines and fungi compared to the parent molecule. These new analogs demonstrated sufficient in vitro and in vivo activity to suggest further investigations are warranted.
Experimental
General Experimental Procedures
The 1H and 13C NMR spectra were recorded in DMSO-d6 and MeOD on a Bruker DRX NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C NMR. Chemical shift (δ) values are expressed in parts per million (ppm) and are referenced to the residual solvent signals of DMSO-d6 and MeOD at δH/δC 2.50/39.5 and 3.31,4.78/49.1, respectively. UV and IR spectra were respectively obtained using a Perkin-Elmer Lambda 3B UV/Vis spectrophotometer and an AATI Mattson, Genesis Series FTIR. Optical rotations were measured with a JASCO DIP-310 digital polarimeter. The High Resolution ESI-MS spectra were measured using a Bruker Daltonic (GmbH, Germany) microTOF series with electrospray ionization. TLC analysis was carried out on precoated silica gel G254 aluminium plates.
Chemicals
1/isoKF was prepared according to the previously reported methods with some modifications.1a Elysia rufescens and Bryopsis pennata were collected by snorkeling at low tide near Black Point, O’ahu in Hawaii. The ethanolic extract of freeze-dried plant/animal material was subjected to flash column chromatography on silica gel (EtOAc/MeOH). Preparative HPLC using Phenomenex 100 mm RP C8 column (250 × 100 mm) and a gradient MeCN (0.05% TFA)/H2O, followed by further HPLC purification on an amino column (250 × 22 mm) using gradient EtOAc/MeOH afforded 1 as a white amorphous powder. All reagents and solvents were obtained from commercial vendors and were utilized without further purification.
General Preparation of Compounds 2–4
To a solution of 1/isoKF (29.5 mg, 20 μmol) and aldehyde in anhydrous methanol (5 mL) was added 3 Å molecular sieve (2 g) and stirred for 30 min at room temperature under argon followed by portionwise addition of sodium triacetoxyborohydride (8.5 mg, 40 μmol) over a 20 min period. The reaction mixture was stirred for period of time described below, quenched with water (20 mL) and extracted with IPA/CHCl3 (1:2) (2×10 mL). The combined organic layers were dried over anhydrous Na2SO4 and the solvent was removed under vacuum. The resulting residue was purified with preparative HPLC using Phenomenex Luna RP C8 column (250 × 22 mm) and eluted with gradient MeCN (0.05% TFA)/water to yield corresponding monoalkyl-KF (major) and dialkyl-KF (minor) products as colorless powders.
4-Fluoro-3-methyl-benzylamino-KF (2)
Starting material 4-fluoro-3-methyl-benzaldehyde (10.4 μL, 100 μmol) was used and the reaction mixture was stirred for 16 h. Yield 68.7 %; [α]25D – 7.8 (c = 0.22, MeOH); UV λmax (MeOH) 197 nm; IR neat (NaCl) 3283 (s, br), 2965 (s), 2936 (s), 1736 (s), 1639 (s), 1560 (s), 1528 (s), 1460 (s), 1206 (s) cm−1; HRESIMS m/z calcd for C83H132FN14O16 [M+H]+ 1599.9924, found 1599.9945.
Bis(4-fluoro-3-methyl-benzyl)amino-KF (3)
Yield 6.6 %; [α]25D – 4.2 (c = 0.10, MeOH); UV λmax (MeOH) 197 nm; IR neat (NaCl) 3281 (s, br), 2961 (s), 2925 (s), 1732 (s), 1644 (s), 1566 (s), 1538 (s), 1470 (s), 1206 (s) cm−1; HRESIMS m/z calcd for C91H139F2N14O16 [M+H]+ 1723.0487, found 1723.0488.
Morpholin-4-yl-benzylamino-KF (4)
Starting material morpholin-4-yl-benzaldehyde (8.5 mg, 40 μmol) was used and the reaction mixture was stirred for 3 days.
Yield 69.7 %; [α]25D – 7.5 (c = 0.25, MeOH); UV λmax (MeOH) 194 nm; IR neat (NaCl) 3288 (s, br), 2964 (s), 2937 (s), 1728 (s), 1644 (s), 1530 (s), 1467 (s), 1205 (s), 1137 (s) cm−1; HRESIMS m/z calcd for C86H138N15O17 [M+H]+ 1653.0389, found 1653.0366.
Assay for antimicrobial activity
All organisms are obtained from the American Type Culture Collection (Manassas, VA) and include the fungi Candida albicans ATCC 90028, Cryptococcus neoformans ATCC 90113, and Aspergillus fumigatus ATCC 90906 and the bacteria methicillin-resistant Staphylococcus aureus ATCC 43300 (MRS), Escherichia coli ATCC 35218, Pseudomonas aeruginosa ATCC 27853, and Mycobacterium intracellulare ATCC 23068. Susceptibility testing is performed using a modified version of the CLSI (formerly NCCLS) methods. M. intracellulare is tested using a modified method of Franzblau, et al.16 Samples are serially-diluted in 20% DMSO/saline and transferred in duplicate to 96 well flat bottom microplates. Microbial inocula are prepared by correcting the OD630 of microbe suspensions in incubation broth to afford final target inocula. Drug controls [Ciprofloxacin (ICN Biomedicals, Ohio) for bacteria and Amphotericin B (ICN Biomedicals, Ohio) for fungi] are included in each assay. All organisms are read at either 630 nm using the EL-340 Biokinetics Reader (Bio-Tek Instruments, Vermont) or 544ex/590em, (M. intracellulare, A. fumigatus) using the Polarstar Galaxy Plate Reader (BMG LabTechnologies, Germany) prior to and after incubation. Minimum fungicidal or bactericidal concentrations are determined by removing 5ul from each clear well, transferring to agar and incubating. The MFC/MBC is defined as the lowest test concentration that kills the organism (allows no growth on agar).
In vitro tumor growth inhibitory activities
In-vitro tumor growth inhibitory activities of these compounds were investigated at NCI, Bethesda on 60 cell line panel of human cancer cells using standard procedure. Compounds were first tested at 10×5 M for the growth inhibitory activities at entire 60 cell line panel. Compounds with considerable activities at 10×5 M concentration (as per the standard of NCI) were subjected to detailed tumor growth inhibitory studies at five concentrations viz. 10×4 M, 10×5 M, 10×6 M, 10×7 M and 10×8 M.
Maximum tolerable dose test (acute toxicity determination)
A single mouse was given a single injection of 400 mg/kg; a second mouse received a dose of 200 mg/kg and a third mouse received a single dose of 100 mg/kg. Dose volumes were generally 0.1 ml/10 gm body weight. The mice were observed for a period of 2 weeks. They were sacrificed if they lost more than 20% of their body weight or if there were other signs of significant toxicity. If all 3 mice were sacrificed, then the next 3 dose levels (50, 25, 12.5 mg/kg) were tested in a similar way. The process was repeated until a tolerated dose was found. The maximum tolerated dose was used to calculate the amount of material given to experimental mice during antitumor testing.
In vivo anticancer activities
A panel of 12 tumor cell lines viz. NCI-H23, NCI-H522, MDA-MB-231, MDA-MB-435, SW-620, COLO 205, LOX, UACC-62, OVCAR-3, OVCAR-5, U251 and SF-295, cultivated in RPMI-1640 containing 10% FBS and 2 mM glutamine was used. The cell suspension (2–10 × 106 cells/ml) was flushed into 1 mm (internal diameter) polyvinylidene fluoride hollow fibers with molecular weight exclusion of 500,000 Da. The hollow fibers were heat-sealed at 2 cm intervals and the samples generated from these seals were placed into tissue culture medium and incubated at 37 °C in 5% CO2 for 24–48 h prior to implantation. Samples of each tumor cell line preparation were quantitated for viable cell mass by a stable endpoint MTT (3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide) assay before and after the administration of test agent. The optical density of each sample was determined spectrophotometrically at 540 nm and the mean of each treatment group was calculated. A 50% or greater reduction in percent net growth in the treated samples compared to the vehicle control samples was considered a positive result.
Supplementary Material
Acknowledgments
Division of Cancer Treatment and Diagnosis, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, Maryland 21701 is gratefully acknowledged for biological testing under the developmental therapeutics program. This research is supported in part by grants from the national institute of health of the United States of America R01AI36596 antimicrobial testing was supported by the NIH, NIAID, division of aids, grant no. AI27094, and the USDA agricultural research service cooperative agreement no. 58-6408-2-009. The government of the United States has certain rights in this invention.
Footnotes
Supplementary data (In vitro activity data of 60 NCI cell lines, HFA data, and MTB results for compounds 2 and 4) associated with this article can be found, in the online version, at doi:xxx/j.bmc.xxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Hamann MT, Scheuer PJ. J Am Chem Soc. 1993;115:5825–5826. [Google Scholar]; (b) Hamann MT, Otto CS, Scheuer PJ, Dunbar DC. J Org Chem. 1996;61:6594–6600. doi: 10.1021/jo960877+. [DOI] [PubMed] [Google Scholar]; (c) Hamann MT, Otto CS, Scheuer PJ, Dunbar DC. J Org Chem. 1996;61:6594–6600. doi: 10.1021/jo960877+. [DOI] [PubMed] [Google Scholar]; (d) Goetz G, Nakao Y, Scheuer PJ. J Nat Prod. 1997;60:562–567. [Google Scholar]; (e) Horgen FD, de los Santos DB, Goetz G, Sakamoto B, Kan Y, Nagai H, Scheuer PJ. J Nat Prod. 2000;63:152–154. doi: 10.1021/np990402o. [DOI] [PubMed] [Google Scholar]; (f) Hill RT, Hamann MT, Enticknap J, Rao KV. WO 2005/042720 PCT Int Appl. 2005; (g) Ashour M, Edrada R, Ebel R, Wray V, Wätjen W, Padmakumar K, Müller WEG, Lin WH, Proksch PJ. Nat Prod. 2006;69:1547–1553. doi: 10.1021/np060172v. [DOI] [PubMed] [Google Scholar]; (h) Tilvi S, Naik CG. J Mass Spectrom. 2006;42:70–80. doi: 10.1002/jms.1140. [DOI] [PubMed] [Google Scholar]
- 2.Dmitrenok A, Iwashita T, Nakajima T, Sakamoto B, Namikoshi M, Nagai H. Tetrahedron. 2006;62:1301–1308. [Google Scholar]
- 3.Gao J, Hamann MT. Chemical Reviews. 2011 (in press) [Google Scholar]
- 4.Gao J, Caballero-George C, Wang B, Rao KV, Shilabin AG, Hamann MT. J Nat Prod. 2009;72:2172–2176. doi: 10.1021/np900287e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Faircloth G, Sheuer P, Avila J, Hendricks H, Drees M, Jimeno J. Ann Oncol. 1996;7:33. [Google Scholar]; (b) Rademaker-Lakhai JM, Horenblas S, Meinhardt W, Stokvis E, de Reijke TM, Jimeno JM, Lopez-Lazaro L, Martin JAL, Beijnen JH, Schellens JHM. Clin Cancer Res. 2005;11:1854. doi: 10.1158/1078-0432.CCR-04-1534. [DOI] [PubMed] [Google Scholar]; (c) Janmaat M, Kruyt F, Jimeno J, Rodriguez JA, Giaccone G. Proceedings of the 2nd International Symposium on Signal Transduction Modulators in Cancer Therapy; Amsterdam. 2003. p. 60. [Google Scholar]
- 6.Jimenez JC, Lopez-Macia A, Gracia C, Varon S, Carrascal M, Caba JM, Royo M, Francesch AM, Cuevas C, Giralt E, Albericio F. J Med Chem. 2008;51:4920. doi: 10.1021/jm8000828. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hamann MT. Curr Opin Mol Ther. 2004;6:657. [PMC free article] [PubMed] [Google Scholar]; (b) Izquierdo Delso MA. 20070032412. US Patent. 2007; Chem Abstr. 2004;141:236712. [Google Scholar]
- 8.Sewell JM, Langdon SP, Smyth JF, Jodrell DI, Guichard S. Proc Am Assoc Cancer Res. 2004;45:1509. [Google Scholar]
- 9.Suarez Y, Gonzalez L, Cuadrado A, Berciano M, Lafarga M, Munoz A. Mol Cancer Ther. 2003;2:863. [PubMed] [Google Scholar]
- 10.Janmaat ML, Rodriguez JA, Jimeno J, Kruyt FAE, Giaccone G. Mol Pharmacol. 2005;68:502. doi: 10.1124/mol.105.011361. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Macia A, Jimenez JC, Royo M, Giralt E, Albericio F. J Am Chem Soc. 2001;123:11398–11401. doi: 10.1021/ja0116728. [DOI] [PubMed] [Google Scholar]
- 12.(a) Albericio PF, Giralt LE, Jiménez GJ-C, Lopez MA, Manzanares I, Rodrigues I, Royo EM. WO 01/58934 PCT Int Appl. 2001; (b) Albericio PF, Fernandez DA, Giralt LE, Gracia CC, Lopez RP, varon CS, Cuevas MC, Lopez MA, Francesca SA, Jiménez GJ-C, Royo EM. WO 2005/023846 PCT Int Appl. 2005; (c) Gracia C, Isidro-Llobet A, Cruz LJ, Acosta GA, Alvarez M, Cuevas C, Giralt E, Albericio F. J Org Chem. 2006;71:7196–7204. doi: 10.1021/jo060976f. [DOI] [PubMed] [Google Scholar]; (d) Faircloth GT, Elices M, Sasak H, Aviles Marin PM, Cuevas Marchante MDC. WO 2004/035613. PCT Int Appl. 2004
- 13.Shilabin AG, Kasanah N, Wedge DE, Hamann MT. J Med Chem. 2007;50:4340–4350. doi: 10.1021/jm061288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Gribble GW, Ferguson DC. J Chem Soc, Chem Commun. 1975:535–536. [Google Scholar]; (b) Nutaitis CF, Gribble GW. Tetrahedron Lett. 1983;24:4287. [Google Scholar]
- 15.Faircloth GT, Grant W, Smith B, Supko J, Brown A, Geldof A, Jimeno J. Proc Am Assoc Cancer Res. 2000;41:600. [Google Scholar]
- 16.Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. J Clin Microbiol. 1998;36(2):362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.