

Graphical abstract
Highlights
► Detailed molecular evolution of metalloenzymes that catalyse the dismutation of hydrogen peroxide. ► Three protein families of differing structure, catalytic mechanism, distribution and evolutionary age. ► Catalatic enzymes in pathogenic organisms are promising targets for drug design. ► Occurrence of biotechnological interesting representatives in extremophiles.
Keywords: Catalase, Catalase–peroxidase, Manganese catalase, Molecular evolution, Pathogen, Horizontal gene transfer
Abstract
For efficient removal of intra- and/or extracellular hydrogen peroxide by dismutation to harmless dioxygen and water (2H2O2 → O2 + 2H2O), nature designed three metalloenzyme families that differ in oligomeric organization, monomer architecture as well as active site geometry and catalytic residues. Here we report on the updated reconstruction of the molecular phylogeny of these three gene families. Ubiquitous typical (monofunctional) heme catalases are found in all domains of life showing a high structural conservation. Their evolution was directed from large subunit towards small subunit proteins and further to fused proteins where the catalase fold was retained but lost its original functionality. Bifunctional catalase–peroxidases were at the origin of one of the two main heme peroxidase superfamilies (i.e. peroxidase–catalase superfamily) and constitute a protein family predominantly present among eubacteria and archaea, but two evolutionary branches are also found in the eukaryotic world. Non-heme manganese catalases are a relatively small protein family with very old roots only present among bacteria and archaea. Phylogenetic analyses of the three protein families reveal features typical (i) for the evolution of whole genomes as well as (ii) for specific evolutionary events including horizontal gene transfer, paralog formation and gene fusion. As catalases have reached a striking diversity among prokaryotic and eukaryotic pathogens, understanding their phylogenetic and molecular relationship and function will contribute to drug design for prevention of diseases of humans, animals and plants.
Introduction
Hydrogen peroxide represents the most abundant reactive oxygen species (ROS) 1 and is an inevitable by-product of aerobic metabolism. It is formed by oxidoreductases by two-electron reduction of dioxygen or by dismutation of superoxide, in respiratory or photosynthetic electron transport chains or simply by autoxidation of biomolecules. However, H2O2 also acts as messenger in cell signaling pathways in eukaryotic organisms or as (antimicrobial) weapon in unspecific (innate) immune defence reactions [1,2]. Its cellular steady-state concentration is the consequence of continuos formation and degradation reactions and organisms differ enormously in their tolerance for H2O2.
Degradation of hydrogen peroxide can be done enzymatically by its reduction to water with the help of endogenous electron donors (mediated by metal-containing peroxidases or thiol-containing peroxiredoxins) but more efficiently by metalloenzyme-mediated dismutation to harmless O2 and water according to Reaction (1). In daily life its efficient degradation is of essential importance for both prokaryotic and eukaryotic cells, even for some anaerobic bacteria [3].
There are indications that the evolutionary design of catalatic enzymes could have started in Archaean already some 3.5 billion years ago – at the time when the ancestral planctonic bacteria acquired the potential of aerobic respiration [4]. This was (later) associated with accelerated evolution of prehistoric catalases (and other ROS degrading enzymes) in the primordial cyanobacteria [4,5] by about 2.7 billion years ago. Cyanobacteria had succeeded in the development of tandem operation of two photosystems (namely a high-potential water-oxidizing photosystem II and a low-potential ferredoxin reducing photosystem I) resulting in oxygenic photosynthesis [6]. This most decisive evolutionary step marked a turning point in evolution on Earth, opening up the era of an aerobic, oxygen – containing biosphere and atmosphere. Primordial cyanobacteria performed both oxygenic photosynthesis and (mitochondria-like) respiration within a single prokaryotic cell [7] with a high demand on ROS detoxification. The catalase evolutionary process increased further in intensity mostly in Proterozoic (2.45–2.32 billion years ago) in accordance with the beginning of an (slow) increase in atmospheric dioxygen [8]. Another qualitative step of the evolution of ROS degrading enzymes was the occurrence of Eukaryotes around 2 billion years ago [4].
In any case, the evolution of H2O2 dismutating (catalatically active) enzymes was a fundamental process in evolution of aerobic life [1] and independently led to the appearance of three metalloenzyme gene families, namely typical (monofunctional) heme catalases (KatEs), (bifunctional) heme catalase–peroxidases (KatGs) and (non-heme) manganese catalases (MnCats) [1]. This review focuses on the phylogeny and distribution of these oxidoreductases, whereas other contributions of this special issue report the relation between their differing structures and reaction mechanisms [9–11].
| (Reaction 1) |
The molecular phylogeny of catalatically active enzymes has already been the focus of several previous analyses and reviews with various scopes [1,12,13]. Here, we present updates and new aspects for all three gene families based on newly available data from recent genome sequencing projects of numerous organisms. Additionally, we also report on rare fusion events of catalase genes during evolution and the intense evolution and high diversity of H2O2 dismutating enzymes in prokaryotic and eukaryotic pathogens enabling survival during oxidative burst induced by the attacked hosts.
Materials and methods
Sequence data mining
Protein sequences of all three catalatically active enzyme families (KatEs, KatGs and MnCats) were collected from the public databases GenBank and UniProt. They were classified and analyzed in PeroxiBase (http://peroxibase.toulouse.inra.fr), where each collected sequence got its abbreviation and identification number. The latter is used throughout the present work.
Phylogenetic analysis
Phylogenetic analyses were performed with the MEGA package Version 5.05 [14]. First, protein sequences of each catalase family were aligned with the Muscle program implemented in the MEGA package with up to 100 iterations. Obtained alignment was subjected to Neighbor-Joining (NJ), Minimum-Evolution (ME) or Maximum Likelihood (ML) method of phylogeny reconstruction available in the MEGA 5.05 package. For NJ and ME 1000 bootstraps and for ML 100 bootstraps were applied. Obtained phylogenetic trees were depicted with the Tree Explorer program of the MEGA package. Conserved regions of obtained multiple sequence alignments were presented with GeneDoc [15].
Results and discussion
Evolution of typical (monofunctional) heme catalases
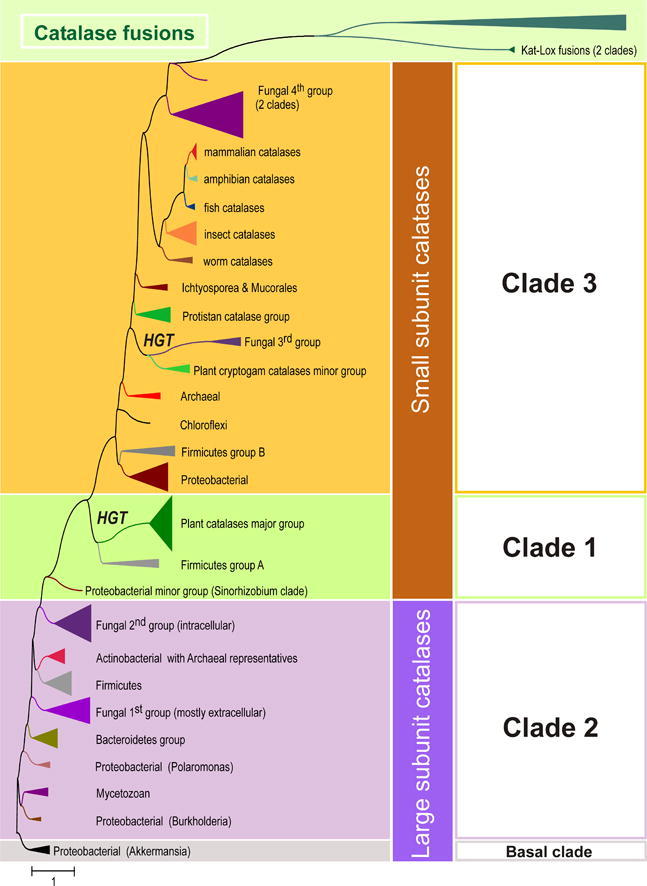
Typical (monofunctional) heme catalases are widely distributed among bacteria, archaea and eukarya. Reconstructed phylogeny of 200 representatives (out of 346 currently available in PeroxiBase; January 2012) and related gene fusions belonging to the catalase-like superfamily is presented in Fig. 1 . The three main evolutionary clades of the catalase (KatE) superfamily depicted in Fig. 1 were already defined in previous works [1,12,16] with Clade 2 comprising large subunit catalases (∼750 residues per subunit) and Clades 1 and 3 small subunit catalases (∼500 residues per subunit). The oligomeric organization and the architecture of the typical catalase fold that includes about 460 residues is highly conserved in large- and small subunit catalases and described in detail by Diaz et al. in this special issue [9]. This holds also for the high conservation and organization of catalytic residues in the heme cavity as well as differences between small- and large-subunit enzymes regarding heme orientation and type (heme b versus d) as well as posttranslational modifications of amino acids at the active site.
Fig. 1.

Reconstructed unrooted tree of 200 typical (monofunctional) catalases obtained with the ML method of the MEGA package [14]. Almost identical trees were obtained also with NJ and ME methods.
Evolution of typical catalases started with large-subunit catalases of Clade 2 that contains bacterial and fungal enzymes. Maximum likelihood analysis revealed that the ancestral representatives are located in the basal branch among Negibacteria, i.e. predecessors of modern Proteobacteria. Detailed reconstruction of this basal clade is presented in Supplemental Fig. 1 with representatives from γ-Proteobacteria and Verrucomicrobia being evolutionarily the closest neighbors of the proposed ancestor of the whole family. A minor mycetozoan group of catalases is closely related with a Gammaproteobacterial branch that is further connected with the basal clade. As this rare Dictyostelium branch also contains large subunit catalases with (yet) unknown function, most probably a horizontal gene transfer (HGT) from Negibacteria into ancestral single cell eukaryots occurred. HGT events to Dictyostelium from bacteria living in the same environment were found to be frequent [17].
Further steps of catalase evolution (middle of Fig. 1) led through proteobacterial large subunit enzymes (including typical representatives from the aquatic gram-negative cocci of the genus Polaromonas) towards Bacteroidetes and fungal large subunit groups. Bacteroidetes, with Flavobacteria as typical representatives, are aerobic rods possessing genes for both monofunctional catalases and bifunctional catalase–peroxidases (see below). Although most of extant Bacteroidetes are pathogenic, their predecessors living in soils and water apparently developed two different genes for highly efficient removal of both external and internal ROS.
The peculiar fungal group 1 of large subunit catalases is probably the oldest line of KatE evolution in fungal genomes and as it has representatives among Zygomycota and Mucorales (Fig. 2 ) it must have evolved already in the first fungal ancestor. Most sequences of this group have a predicted signal sequence for secretion, thus group 1 evolved in fungal genomes as extracellular KatE paralog with many representatives in (phyto)pathogenic fungi [18]. Further branches of large subunit catalases are constituted by closely related Firmicutes and actinobacterial KatEs that evolved through gene duplications from their common ancestor. Also few Euryarchaeota large subunit representatives are situated in this clade (Fig. 2), probably deriving through a single HGT event from Gram-positive bacteria. Clade 2 is completed with the second fungal KatE group containing mainly sequences from Basidiomycota and Ascomycota that, although intracellular, are slightly longer than secreted KatEs from the first fungal group. Similar to the first group, many of them also originate from pathogenic fungi.
Fig. 2.

Reconstroncted unrooted tree of Clade 2 of typical catalases. Details about phylogeny of large-subunit catalases from Bacteriodetes, Archaea, Actinobacteria, Firmicutes and Euryarchaeota as well as fungi are shown. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given.
From Fig. 1 and previous analysis [12] it is obvious that Clade 1 and Clade 3 typical catalases (as well as catalase gene fusions) evolved from ancestral Clade 2 catalases. This can be interpreted by the means of adaptation of KatE evolution, as frequently two distinct paralogs in the same genome code for functional catalase variants that differ in their behavior. Phenotypic diversity was already verified experimentally for 16 eubacterial and ascomycetous catalases [19]. All known representatives from clades 1 and 3 have lost the extra C-terminal “flavodoxin-like” domain [9,12]. In some further evolved proteins the catalase domain is fused to a lipoxygenase domain. In Clade 1 catalases (Fig. 3 ) there are several Firmicutes representatives at the phylogenetic base of this clade but the most abundant group is represented by plant catalases (analyzed in detail in the work of Scandalios et al. [20]). New in this reconstruction is the introduction of Cryptogam catalase genes that evolved from the corresponding Chlorophyta genes during speciation events (Fig. 3).
Fig. 3.

Reconstructed unrooted tree of Clade 1 of typical catalases. Details about phylogeny of small-subunit catalases from plants and fungi are shown. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given.
Clade 3 (Fig. 4 ) is the most versatile and abundant evolutionary lineage of typical (monofunctional) catalases. It has its roots among small-subunit catalases from Proteobacteria and Firmicutes. Further evolution of Clade 3 enzymes occurred through short katE genes of the Chloroflexi group towards small subunit archaeal representatives (Fig. 4) and in another direction towards various eukaryotic groups. Among them the close relationship between the short subunit fungal 3rd group and a rather small paralog group of plant – Cryptogam catalases is very interesting (Fig. 4). The wood-degrading fungi Phanerochaete and Coprinopsis might have acquired these katE genes via HGT from simplest plants (Cryptogams) and, finally, phytopathogenic fungi, e.g. Phaeosphaeria, made use of this gene as an evolutionary advantage in attacking higher plants.
Fig. 4.

Reconstructed unrooted tree of Clade 3 of typical catalases. Details about phylogeny of small-subunit bacterial, archaeal, fungal, plant and animal catalases are shown. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given.
Further evolution of Clade 3 catalases occurred mainly among unicellular protistan genomes. There is a distinct branch of catalases from ciliates (Ciliophora) that is closely related with the second Mycetozoan branch, containing KatEs shorter than 500 amino acids. Phylogeny of Clade 3 continues with katE genes found in multicellular metazoan genomes (lower part of Fig. 4) following the general animal evolution line as reconstructed by other sequence markers [21]. An unexpected turn in later steps of catalase gene evolution is the node between the fourth fungal group (Supplemental Fig. 2) which is connected with branches of catalase-lipoxygenase fusion proteins. The fourth group of fungal catalases consists of intracellular proteins mainly located in peroxisomes as is evident from the presence of the conserved PTS1 signal sequence [22]. During evolution of the catalase-lipoxygenase fusion gene, the 5′-region coding for the catalase fold was subjected to a more rapid evolution as is evident from the comparison of the length of branches (Fig. 1 and Supplemental Fig. 2). Today this part of the protein has lost its catalase function but still retained a high structural similarity to the catalase fold including a complete β-barrel and part of the α-helical domain. The modified domain has now the functionality of an allene oxide synthase (E.C. 4.2.1.92) as was proven for a fused protein from the coral Plexaura homomalla [23]. Supplemental Fig. 2 depicts that similar fusion proteins are also found in cyanobacteria, whereas in some plants they are part of putative multidomain complexes (so far not investigated). Nevertheless, this late events in catalase phylogeny may suggest that the stable catalase fold can be used as a scaffold for introduction of new functionalities by using directed evolution methods.
Summing up, typical heme catalases are found in all kingdoms of life and originally nature designed large-subunit proteins that lost at least around 150 C-terminal amino acids during evolution. Surprisingly, in cyanobacteria, the pacemaker of aerobic evolution, these metalloenzymes are found only in very few representatives in a branch of Clade 3, clearly suggesting that monofunctional catalase does not represent the oldest H2O2 scavenging enzyme designed immediately after development of oxygenic photosynthesis.
Evolution of (bifunctional) catalase–peroxidases
The phylogeny of catalase–peroxidases (KatGs) was reconstructed recently in the context of the evolution of the whole Class I of the peroxidase–catalase superfamily that also contains homologous cytochrome c peroxidases, ascorbate peroxidases and hybrid-type peroxidases [24]. In analogy to the phylogeny of typical catalases, also the evolution of the peroxidase–catalase superfamily started with more complex proteins, i.e. predecessors of KatGs, being comprised of two domains per subunit, namely a N-terminal heme-containing domain and a C-terminal domain without cofactor [9,24]. Extant KatGs retained as the sole subfamily a real bifunctionality, i.e. a peroxidase activity according to Reaction 2 (with yet unknown physiological electron donors) and a significant (pseudo-)catalase activity that differs mechanistically from the classical catalatic mechanism of typical catalases [25] but also follows Reaction 1. During further evolution of the peroxidase–catalase superfamily the (KatG-typical) C-terminal domain was lost as was the functionality of H2O2 dismutation [24]. All other representatives of this heme peroxidase superfamily are typical (monofunctional) peroxidases with homology to the N-terminal domain of KatG.
| (Reaction 2) |
Here, we have updated the amount of full-length KatG sequences for phylogenetic reconstruction to 204 (out of 471 currently deposited in PeroxiBase; January 2012) by covering representatives from archaea, bacteria and lower eukaryotes including Protista, stramenopiles and fungi. The resulting robust tree clearly shows division into two basic paralog clades, namely into main Clade 1 and minor Clade 2 that diverged at the beginning of evolution of KatGs (Fig. 5 ) [24].
Fig. 5.

Reconstructed unrooted tree of 204 catalase–peroxidases (KatGs) obtained with the ML method of the MEGA package [14]. Very similar trees were obtained with NJ and ME methods of the same package. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given.
Maximum likelihood analysis located the origin of katG gene evolution in the clade of marine heterotrophic bacteria from the phylum Planctomycetes (Figs. 5 and 6) with the closest phylogenetic neighbor being a KatG from the cyanobacterium Gloeobacter violaceus. It is important to note that Gloeobacteria might represent the closest extant relatives of primordial cyanobacteria that did not possess thylakoid membranes as specific site for oxygenic photosynthesis [26]. Probably early katG acquisition by Gloeobacter (or its predecessor) was important to cope with photooxidative stress. Fig. 6 suggests that KatGs that first emerged in aquatic heterotrophs were very soon transferred to marine autotrophs probably inhabitating the same environment. The branch of ancestral planctomycetes clade is closely connected to a homogenous basal branch containing solely sequences from Bacteroidetes that are obligate chemoheterotrophs comprising a significant portion of prokaryotic communities in the oceans [27].
Fig. 6.

The origin of the catalase–peroxidase evolution among bacteria of Planctomycetes and closely related groups.
In (main) Clade 1 the branches of catalase–peroxidases from Cyanobacteria [including also representatives from Deltaproteobacteria and Gammaproteobacteria] and Firmicutes segregated quite early in evolution (Fig. 6). Among these sequences (putative) KatGs from the anaerobic organisms are found, e.g. from the alkaliphilic and thermophilic bacterium Anoxybacillus flavithermus [28] or Geobacter sulfurreducens, suggesting the occurrence of catalase–peroxidases in extant anaerobic bacteria. A single eukaryotic representative from an Ecdysozoan parasite of fish (“sea louse”), Lepeophtherius salmonis, present among early branches of cyanobacterial KatGs (Fig. 6), needs further analysis as it has sequence features typical for eubacterial KatG.
In Clade 1 further steps of evolution led to segregations of a KatG branch containing proteins from Actinobacteria as well as three main proteobacterial branches designated major groups A, B and C (Figs. 6 and 7). The few Euryarchaeal representatives may be the result of HGTs between ancient Proteobacteria and Archaea. In the proteobacterial group A mainly representatives from Gammaproteobacteria but also several Verrucomicrobia/Chlamydiae are found that are considered as deeply branching phylum in bacterial phylogeny [29]. This large group includes also KatGs from phytopathogens (Xanthomonas) as well as endophytes (e.g. Burkholderia phytofirmans) in neighbored branches.
Fig. 7.

Branching of the Proteobacterial A group of catalase–peroxidases from the main paralogs Clade 1 and major HGT event of katG genes between bacteroidetes and sac fungi.
Besides bacterial and archaeal proteins recent sequencing projects showed the occurrence of KatGs in fungi including Sordariomycetes, Eurotiomycetes, Dothideomycetes and Basidiomycetes. All known fungal KatGs are descendants of a major HGT event between Bacteroidetes (here represented mainly by strictly aerobic Sphingobacteria and Flavobacteria) and an ancestor of the sac fungi (Fig. 7 ). This situation is different to monofunctional catalases where corresponding fungal genes are also found in older lineages. The phylogeny of KatGs suggests a HGT from Bacteroidetes to Ascomycetes with a high bootstrap support (Fig. 7). There is no indication of a close relationship of Actinobacterial KatGs with fungal counterparts, although Actinobacteria are considered as close relatives with Neomura (i.e. the very ancestral eukaryotic branch [30]).
Soon after KatG acquisition in ancestral ascomycetous genomes, a gene duplication – supported by high bootstrap values in our analysis – led to the divergence of KatG1 (intracellular) and KatG2 (secreted variant) groups (Fig. 7). The intracellular enzymes are more abundant than the extracellular proteins. Finally, the KatG1 group diversified into subgroups of Eurotiomycetes and Sordariomycetes catalase–peroxidases that are most probably located in peroxisomes, as is suggested by the presence of the PTS1 signal [31]. Among Eurotiomycetes a further HGT of katG towards basidiomycetes (class of Ustilaginomycetes) occurred. The presence of a katG gene in the arthropod Nilaparvata lugens needs further investigation since it could derive from a (yet unknown) fungal pathogen of the brown planthopper.
In contrast to KatG1, all representatives of the fungal KatG2 group are extracellular heme proteins, since they possess a N-terminal signal sequence for secretion [32]. All katG2 genes originate in genomes of pathogenic fungi from the class Sordariomycetes being primarily phytopathogens (and few mycoparasites of the order Hypocreales). One species, Fusarium oxysporum, has two KatG2 orthologs and is a transkingdom pathogen. Extracellular catalase–peroxidase seems to be closely related with the pathogenicity of these organisms as has been demonstrated for Magnaporthe oryzae. In this rice blast fungus the extracellular KatG2 is essential for overcoming the oxidative burst and release of H2O2 by the attacked plant thereby enabling penetration of the host by the fungal cells [18,33].
Clade 2 catalase–peroxidases segregated from the (main) Clade 1 proteins quite early in evolution (Fig. 5) [24] but contained eukaryotic lineages from its very beginning (Fig. 8 ). One early branch of this minor Clade 2 is represented by intracellular (partially peroxisomal) KatGs from single-cell protozoa. In detail, a protein from a marine flagellated heterotroph from the phylum Apusozoa [34] is closely related to a protein from unicellular photosynthetic algae belonging to the phylum Bacillariophyta [35] pointing to eukaryotic HGT between (ancient) marine heterotrophs and autotrophs. The branch of KatGs from Proteobacteria in minor Clade 2 contains mostly sequences from various pathogens of human, animal, plants, and even protists. Examples are KatGs from Francisella tularensis, which is a multi-species intracellular pathogen with a demand on efficient H2O2 decomposition [13] as well as the plasmid-born KatG (named “KatP”) from the enterohaemorrhagic strain of Escherichia coli with unique periplasmic location [36]. Another branch of Clade 2 contains KatGs from Euryarchae (Fig. 8). It is closely related with the protistan and the proteobacterial branches thus pointing to evolutionary connections between Archaean and unicellular eukaryotic phylogeny [37]. This group includes KatGs from extremophiles, e.g. Archaeoglobus fulgidus [38] or (related and anaerobic) Ferroglobus placidus with temperature optima around 85 °C [38,39]. Further interesting katG-containing extremophiles include Halogeometricum borinquense that exists in hypersaline environments with a salinity range between 1.4 and 5.2 M [40]. In any case, (putative) bifunctional catalase–peroxidase from these extremophiles could be an attractive starting point for future applications.
Fig. 8.

Details on eukaryotic lineages of catalase–peroxidases in the minor Clade 2 and also related bacterial and archaeal representatives.
The second known KatG from an eukaryotic Bacillariophyt (diatom), namely from Phaeodactylum tricornutum is related to the quite abundant group of algal/stramenopiles catalase–peroxidases. Among them, several representatives from green and brown algae are present, but the dominant group here is the branch of phytopathogenic oomycetes (Fig. 8). These (lower) eukaryotes are phylogenetically related to protists of the Stramenopile group, which is also supported by present analysis of KatG evolution (Fig. 5 and 8). Similar to fungal KatG2 group also these pathogenic oomycete KatGs are secreted enzymes [41] but sequence similarity with (already investigated) ascomycetous KatG2 group is low (around 46%).
To sum up, bifunctional catalase–peroxidase are at the origin of evolution of one major heme peroxidase superfamily and are distributed among archaea and bacteria. These hydrogen peroxide dismutating metalloenzymes seem to be older than typical catalases and most probably were already present in ancient cyanobacteria at the time of development of oxygenic photosynthesis. In contrast to KatEs, KatGs are not found in higher plants, or animals but in protists and fungi. Normally, catalase–peroxidase is an intracellular oxidoreductase, which in eukaryotes is located in the peroxisomes. However, exclusively in phytopathogenic fungi a second secreted KatG is synthesized that seems to be involved in host attack, thereby being an interesting future target for specific drug design. Similarly to KatEs, also KatGs reveal during their evolutionary divergence adaptation to environmental conditions. This was demonstrated mainly for mycobacterial KatGs [42] although for cyanobacterial KatG variants there can also be a neutral drift [5]. Recently, the adaptation of two distinct fungal KatG groups was described also on the phenotypic level to a good extent [43,44].
Evolution of manganese (non-heme) catalases
Manganese catalases (MnCats, also known as T-catalases or N-catalases or non-heme catalases) evolved among the domains of Eubacteria and Archaea. However, as there are already known representatives in Actinobacteria, expansion into Eukaryotes cannot be ruled out due to the proposed common origin of Neomura [30]. Manganese catalases belong to a complex (ferritin-like) superfamily, comprising up to 12 distinct families with over 11,500 known sequences [45]. In contrast to KatEs and KatGs, MnCats are single domain proteins with a typical ferritin-like four helical bundle [10,46]. The robust tree depicted in Fig. 9 is based on 100 full length protein sequences. Besides the five distinct clades defined previously [1], a new basal clade and a connecting clade were found in the updated phylogenetic reconstruction presented herein. Maximum likelihood analysis revealed that MnCat from Halothermothrix orenii is at the origin of the whole MnCat family. This MnCat gene comes from a strictly anaerobic thermohalophilic Gram-negative bacterium with optimal growth conditions around 60 °C and 1.7 M NaCl [47]. This bacterial lineage is thought to be closely related to (thermophilic) LUCA [48] suggesting that manganese catalases were present in anaerobic thermohalophilic bacteria long before the proposed increase of atmospheric oxygen by oxygenic photosynthesis. This moves the origin of the MnCat family evolution to a former date than previously supposed [12].
Fig. 9.

Reconstructed unrooted tree for 100 manganese catalases obtained with the ML method of the MEGA package. Nearly identical trees were obtained also with NJ and ME methods of the same package. Numbers in the nodes indicate bootstrap values for 1000/100/100 replications in NJ/ME/ML, respectively.
The sequence from the soil Acidobacterium Solibacter usitatus [49] resides in the outgroup clade (Figs. 9 and 10) and – although annotated as MnCat – it is closely related with the family of YciF proteins, known as bacterial stress proteins [50] from the ferritin-like superfamily, with no manganese ions bound and consequently without catalase activity. The closely related basal clade of manganese catalases (Fig. 10 ) contains only sequences from Firmicutes (including Clostridia) with anaerobic metabolism reflecting slow evolution via speciation events.
Fig. 10.

Details on the reconstructed tree covering the basal manganese catalase clade connected with an outgroup as well as Clades 1–3.
An important point in MnCat evolution was an early gene duplication (mentioned already in [12]) visible in the node separating Clades 1–3 from Clades 4–5 (Fig. 10). Firmicutes sequences are present also in the roots of Clade 3, represented by MnCat from Lactobacillus plantarum with known 3D structure [46] but further steps of this evolution led to the acquisition of MnCat in the Phyla Cyanobacteria and Actinobacteria. In contrast with the roots of katG evolution, in this clade only sequences from specifically evolved Cyanobacteria, like diazothrophic (N2-fixing) Cyanothece sp. and Nostoc punctiforme (Fig. 10) point to a possible HGT event from Firmicutes. A big group of Alphaproteobacterial MnCats exists in Clade 3 with MnCat from Rhodopirellula baltica as a typical example. This planctomycete contains in addition a bifunctional KatG (at the hypothetical root of that family). As the RbaMnCat is located in later steps of manganese catalase gene evolution, it is reasonable to assume that KatG started to evolve later in genomic evolution compared to MnCat.
Further steps of evolution in this direction lead to a mixed Actinobacteria and Firmicutes Clade 2 and a Deinococci and Crenoarchaeota Clade 1 that diversified by a late gene duplication event. In Clade 1 the extremophile (aerobic) Thermus thermophilus with temperature optimum above 62 °C [51] is represented by two related MnCats. Neither KatE nor KatG is found in this organism [51]. The Crenarchaeota representatives of Clade 1 are represented by MnCat sequences from the phylum Pyrobaculum. These putative manganese catalases are very interesting, since these microorganisms are also aerobic extremophiles living around 100 °C. Manganese catalases from such boundaries of life as probably sole catalysts for H2O2 decomposition are of exceptional biotechnological interest.
Clades 4 and 5 separated themselves by a series of later gene duplication events as apparent from Fig. 11 . In Clade 4 a second group of cyanobacterial sequences is present, among them also a MnCat from the primordial G. violaceus. Presence of planctobacterial sequences mixed with proteobacterial further support the hypothesis that MnCats are older than KatGs. Clade 5 is dominated by numerous Gamma- and Betaproteobacterial sequences but also a mixed group containing MnCats from Actinobacteria and Bacteroidetes are found. In some cases, like for the erythromycin producer Saccharopolyspora erythraea [52] it is interesting to see, that all three hydrogen peroxide dismutating protein families occur, namely two typical catalases (a small and a large subunit protein), a catalase–peroxidase (KatG) as well as a manganese catalase (labeled in bold in the corresponding phylogenetic trees). Coming from different evolutionary routes they constitute the armory of a typical actinobacterial cell against oxidative stress but details of their different physiological regulation and function need to be elucidated in the future.
Fig. 11.

Details on the reconstructed tree covering manganese catalase Clades 4–5.
In conclusion, manganese catalases are the oldest catalysts designed by nature for H2O2 dismutation. So far no eukaryotic representative is found suggesting that this protein family is very old and probably stayed restricted to bacteria and archaea where indications for an adaptation to various environments could be observed [42] but the phenotypic details on this adaptation still need more proofs. The physiological limitations could be that heme catalases (KatG and KatE) have a better catalytic efficiency [9–11] than manganese variants. Nevertheless, the occurrence of non-heme catalases in extremophils make these proteins interesting for biotechnological purposes.
Conclusion
Catalatic enzymes are ubiquitous in nature and found in all kingdoms of life. Manganese catalase is the oldest H2O2 dismutating catalyst and its occurrence is restricted to the prokaryotic world. Heme b containing catalase–peroxidases were designed later in evolution – besides prokaryotic proteins – also enzymes from protists and fungi are found. Finally, typical catalases evolved that are widely distributed in eukaryotes including plants and animals. The present phylogenetic analyses demonstrate the distribution of these oxidoreductases that is closely correlated with the actual physiological demand. Most interestingly, especially (prokaryotic and eukaryotic) pathogens have a great armory of catalatic enzymes that play distinct roles in pathogenesis. Understanding their specific involvement and relations between function and catalase structure may help to design specific inhibitors and prevent diseases of humans, animals and plants.
Acknowledgments
The work was supported by the Austrian science Fund (FWF Project P20996-B11) as well as the Doctoral Program Biomolecular Technology of Proteins – BioToP (FWF W1224).
Footnotes
Abbreviations used: HGT, horizontal gene transfer; KatE, typical (monofunctional) catalase; KatG, (bifunctional) catalase–peroxidase; MnCat, manganese catalase; LUCA, last universal common ancestor; ROS, reactive oxygen species; PTS, peroxisomal targeting signal.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.abb.2012.01.017.
Appendix A. Supplementary data
(Fig. 1) The evolutionary origin of large-subunit catalases. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given. (Fig. 2) Details on relationships between 4th fungal small subunit group of catalases with branches of catalase-lipoxygenase fusion proteins. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given.
References
- 1.Zamocky M., Furtmüller P.G., Obinger C. Antioxid. Redox Signal. 2008;10:1527–1548. doi: 10.1089/ars.2008.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peus D., Meves A., Vasa R.A., Beyerle A., O’Brien T., Pittelkow M.R. Free Radic. Biol. Med. 1999;27:1197–1202. doi: 10.1016/s0891-5849(99)00198-7. [DOI] [PubMed] [Google Scholar]
- 3.Brioukhanov A.L., Netrusov A.I. Biochemistry (Moscow) 2004;69:949–962. doi: 10.1023/b:biry.0000043537.04115.d9. [DOI] [PubMed] [Google Scholar]
- 4.Lenton T.M. In: Evolution of Planet Earth. Rothschild L., Lister A., editors. Elsevier; 2003. pp. 35–53. [Google Scholar]
- 5.Bernroitner M., Zamocky M., Furtmüller P.G., Peschek G.A., Obinger C. J. Exp. Bot. 2009;60:423–440. doi: 10.1093/jxb/ern309. [DOI] [PubMed] [Google Scholar]
- 6.Drews G. In: Bioenergetic Processes of Cyanobacteria. Peschek G.A., Obinger C., Renger G., editors. Springer; Dordrecht: 2011. pp. 265–284. [Google Scholar]
- 7.Peschek G.A., Bernroitner M., Sari S., Pairer M., Obinger C. In: Bioenergetic Processes of Cyanobacteria. Peschek G.A., Obinger C., Renger G., editors. Springer; Dordrecht: 2011. pp. 3–68. [Google Scholar]
- 8.Bekker A., Holland H.D., Wang P.L., Rumble D., Stein H.J., Hannah J.L., Coetzee L.L., Beukes N.J. Nature. 2004;427:117–220. doi: 10.1038/nature02260. [DOI] [PubMed] [Google Scholar]
- 9.Diaz A., Loewen P.C., Fita I., Carpena X. Arch. Biochem. Biophys. 2012;525:102–110. doi: 10.1016/j.abb.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Whittaker J.W. Arch. Biochem. Biophys. 2012;525:111–120. doi: 10.1016/j.abb.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholls P. Arch. Biochem. Biophys. 2012;519:1–7. [Google Scholar]
- 12.Klotz M., Loewen P.C. Mol. Biol. Evol. 2003;20:1098–1112. doi: 10.1093/molbev/msg129. [DOI] [PubMed] [Google Scholar]
- 13.Passardi F., Favet J., Zamocky M., Jakopitsch C., Penel C., Obinger C., Dunand C. Gene. 2007;397:101–113. doi: 10.1016/j.gene.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 14.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.K.B. Nicholas, H.B. Nicholas, Genedoc, 1997, distributed by the author.
- 16.Klotz M., Klassen G., Loewen P. Mol. Biol. Evol. 1997;14:951–958. doi: 10.1093/oxfordjournals.molbev.a025838. [DOI] [PubMed] [Google Scholar]
- 17.Eichinger L., Pachebat J.A., Glöckner G., Rajandream M.A., Sucgang R., Berriman M., Song J., Olsen R., Szafranski K., Xu Q., Tunggal B., Kummerfeld S., Madera M., Konfortov B.A., Rivero F., Bankier A.T., Lehmann R., Hamlin N., Davies R., Gaudet P., Fey P., Pilcher K., Chen G., Saunders D., Sodergren E., Davis P., Kerhornou A., Nie X., Hall N., Anjard C., Hemphill L., Bason N., Farbrother P., Desany B., Just E., Morio T., Rost R., Churcher C., Cooper J., Haydock S., van Driessche N., Cronin A., Goodhead I., Muzny D., Mourier T., Pain A., Lu M., Harper D., Lindsay R., Hauser H., James K., Quiles M., Madan Babu M., Saito T., Buchrieser C., Wardroper A., Felder M., Thangavelu M., Johnson D., Knights A., Loulseged H., Mungall K., Oliver K., Price C., Quail M.A., Urushihara H., Hernandez J., Rabbinowitsch E., Steffen D., Sanders M., Ma J., Kohara Y., Sharp S., Simmonds M., Spiegler S., Tivey A., Sugano S., White B., Walker D., Woodward J., Winckler T., Tanaka Y., Shaulsky G., Schleicher M., Weinstock G., Rosenthal A., Cox E.C., Chisholm R.L., Gibbs R., Loomis W.F., Platzer M., Kay R.R., Williams J., Dear P.H., Noegel A.A., Barrell B., Kuspa A. Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanabe S., Ishii-Minami N., Saitoh K.I., Otake Y., Kaku H., Shibuya N., Nishizawa Y., Minami E. Mol. Plant–Microb. Interact. 2011;24:163–171. doi: 10.1094/MPMI-07-10-0175. [DOI] [PubMed] [Google Scholar]
- 19.Switala J., Loewen P.C. Arch. Biochem. Biophys. 2002;401:145–154. doi: 10.1016/S0003-9861(02)00049-8. [DOI] [PubMed] [Google Scholar]
- 20.Scandalios J.G., Guan L., Polidoros A.N. Oxidative Stress and the Molecular Biology of Antioxidant Defenses. Cold Spring Harbor Laboratory Press; 1997. pp. 343–406. [Google Scholar]
- 21.Perelman P., Johnson W.E., Roos C. Plos Genet. 2011;7:21001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurvitz A., Wabnegger L., Langer S., Hamilton B., Ruis H., Hartig A. Mol. Genet. Genom. 2001;265:276–286. doi: 10.1007/s004380000412. [DOI] [PubMed] [Google Scholar]
- 23.Oldham M.L., Brash A.R., Newcomer M.E. Proc. Natl. Acad. Sci. USA. 2005;102:297–302. doi: 10.1073/pnas.0406352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamocky M., Furtmüller P.G., Obinger C. Arch. Biochem. Biophys. 2010;500:45–57. doi: 10.1016/j.abb.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 25.Vlasits J., Jakopitsch C., Bernroitner M., Zamocky M., Furtmüller P.G. Arch. Biochem. Biophys. 2010;500:45–57. doi: 10.1016/j.abb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 26.Mimuro M., Tsuchiya T., Koyama K., Peschek G.A. In: Bioenergetic Processes of Cyanobacteria. Peschek G.A., Obinger C., Renger G., editors. Springer; Dordrecht: 2011. pp. 211–238. [Google Scholar]
- 27.Oh H.-M., Kang I., Ferriera S., Giovannoni S.J., Cho J.C. J. Bacteriol. 2010;192:4796–4797. doi: 10.1128/JB.00733-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pikuta E., Lysenko A., Chuvilskaya N., Mendrock U., Hippe H., Suzina N., Nikitin D., Osipov G., Laurinavichius K. Int. J. Syst. Evol. Microbiol. 2000;50:2109–2117. doi: 10.1099/00207713-50-6-2109. [DOI] [PubMed] [Google Scholar]
- 29.Kant R., Van Passel M.W.J., Palva A., Lucas S., Lapidus A., Glavina del Rio T., Dalin E., Tice H., Bruce D., Goodwin L., Pitluck S., Larimer F.W., Land M.L. J. Bacteriol. 2011;193:2902–2903. doi: 10.1128/JB.00295-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavalier-Smith T. Biol. Direct. 2006;1:19. doi: 10.1186/1745-6150-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brocard C., Hartig A. Biochim. Biophys. Acta. 2006;1763:1565–1573. doi: 10.1016/j.bbamcr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 32.Zamocky M., Furtmüller P.G., Obinger C. Biochem. Soc. Trans. 2009;37:772–777. doi: 10.1042/BST0370772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.S. Singh, S.A. Braus-Stromeyer, C. Timpner, O. Valerius, A. von Tiedemann, P. Karlovsky, C. Druebert, A. Polle, G.H. Braus, Mol. Plant Microb. Interact., in press, doi:10.1094/MPMI-08-11-0217. [DOI] [PubMed]
- 34.Cavalier-Smith T., Chao E.E. Protist. 2010;161:549–576. doi: 10.1016/j.protis.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Armbrust E.V., Berges J.A., Bowler C., Green B.R., Martinez D., Putnam N.H., Zhou S., Allen A.E., Apt K.E., Bechner M., Brzezinski M.A., Chaa B.K., Chiovitti A., Davis A.K., Demarest M.S., Detter J.C., Glavina T., Goodstein D., Hadi M.Z., Hellsten U., Hildebrand M., Jenkins B.D., Jurka J., Kapitonov V.V., Kröger N., Lau W.W., Larimer T.W.Lane.F.W., Lippmeier J.C., Lucas S., Medina M., Montsant A., Obornik M., Parker M.S., Palenik B., Pazour G.J., Richardson P.M., Rynearson T.A., Saito M.A., Schwartz D.C., Thamatrakoln K., Valentin K., Vardi A., Wilkerson F.P., Rokhsar D.S. Science. 2004;306:79–86. doi: 10.1126/science.1101156. [DOI] [PubMed] [Google Scholar]
- 36.Uhlich G.A. Microbiology. 2009;155:3589–3598. doi: 10.1099/mic.0.031435-0. [DOI] [PubMed] [Google Scholar]
- 37.Roberts E., Sethi A., Montoya J., Woese C.R., Luthey-Schulten Z. Proc. Natl. Acad. Sci. USA. 2008;105:13953–13958. doi: 10.1073/pnas.0804861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartzell P., Reed D.W. In: The Prokaryotes. Dworkin M., Falkow S., Schleifer K.-H., editors. Springer; Singapore: 2006. pp. 82–100. [Google Scholar]
- 39.Holmes D.E., Risso C., Smith J.A., Lovley D.R. Appl. Environ. Microbiol. 2011;77:5926–5933. doi: 10.1128/AEM.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oren A. In: The Prokaryotes. Dworkin M., Falkow S., Schleifer K.-H., editors. Springer; Singapore: 2006. pp. 113–164. [Google Scholar]
- 41.Blackman L.M., Hardham A.R. Plant Pathol. 2008;9:495–510. doi: 10.1111/j.1364-3703.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chelikani P., Fita I., Loewen P.C. Cell. Mol. Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zamocky M., Furtmüller P.G., Bellei M., Battistuzzi G., Stadlmann J., Vlasits J., Obinger C. Biochem. J. 2009;418:443–451. doi: 10.1042/BJ20081478. [DOI] [PubMed] [Google Scholar]
- 44.M. Zamocky, E. Droghetti, M. Bellei, B. Gasselhuber, M. Pabst, P.G. Furtmüller, G. Battistuzzi, G. Smulevich, C. Obinger, Biochimie, in press, doi: 10.1016/j.biochi.2011.09.020. [DOI] [PMC free article] [PubMed]
- 45.Andrews S.C. Biochim. Biophys. Acta. 2010;1800:691–705. doi: 10.1016/j.bbagen.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Barynin V.V., Whittaker M.M., Antonyuk S.V., Lamzin V.S., Harrison P.M., Artymiuk P.J., Whittaker J.W. Structure. 2001;9:725–738. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 47.Mavromatis K., Ivanova N., Anderson I. PloS One. 2009;4:e41192. doi: 10.1371/journal.pone.0004192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciccarelli F.D., Doerks T., Von Mering C., Creevey C.J., Snel B., Bork P. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 49.Challacombe J.F., Eichorst S.A., Hauser L. Plos One. 2011;6:e24882. doi: 10.1371/journal.pone.0024882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hindupur A., Liu D., Zhao Y., Bellamy H.D., White M.A., Fox R.O. Prot. Sci. 2006;15:2605–2611. doi: 10.1110/ps.062307706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cava F., Hidalgo A., Berenguer J. Extremophiles. 2009;13:213–231. doi: 10.1007/s00792-009-0226-6. [DOI] [PubMed] [Google Scholar]
- 52.Oliynyk M., Samborsky J.B., Lester T., Mironenko N., Scott S., Dickens S.F., Haydock P.F. Nat. Biotechnol. 2007;25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Fig. 1) The evolutionary origin of large-subunit catalases. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given. (Fig. 2) Details on relationships between 4th fungal small subunit group of catalases with branches of catalase-lipoxygenase fusion proteins. Numbers in the nodes represent bootstrap values for 1000/1000/100 replications in NJ/ME/ML, respectively. Additionally, the number of amino acids per subunit is given.