

Abstract
The highly conserved first 23 residues of the influenza hemagglutinin HA2 subunit constitute the fusion domain, which plays a pivotal role in fusing viral and host-cell membranes. At neutral pH, this peptide adopts a tight helical hairpin wedge structure, stabilized by aliphatic hydrogen bonding and charge–dipole interactions. We demonstrate that at low pH, where the fusion process is triggered, the native peptide transiently visits activated states that are very similar to those sampled by a G8A mutant. This mutant retains a small fraction of helical hairpin conformation, in rapid equilibrium with at least two open structures. The exchange rate between the closed and open conformations of the wild-type fusion peptide is ∼40 kHz, with a total open-state population of ∼20%. Transitions to these activated states are likely to play a crucial role in formation of the fusion pore, an essential structure required in the final stage of membrane fusion.
Keywords: conformational ensemble, dynamics, membrane proteins, RDC, relaxation dispersion
Proteins play a central role in stabilizing the high-energy intermediates of membrane fusion (1), with critical roles in biology as diverse as synaptic vesicle fusion (2), membrane remodeling (3), and cellular entry of enveloped viruses (4). The influenza glycoprotein hemagglutinin is responsible for receptor binding and membrane fusion upon viral infection, and has been the subject of numerous detailed studies (5). Hemagglutinin is a homotrimer of two subunits, the product of a posttranslational cleavage of HA0 into HA1 and HA2, whose C-terminal helix traverses the outer membrane of the virus. A “spring-loaded” conformational change driven by a pH decrease in the endosome activates the HA2 domain of hemagglutinin for fusion (5, 6). The N-terminal 23 residues of HA2, which are quite hydrophobic and highly conserved across all serotypes, are often referred to as the fusion peptide or fusion domain. In the fusion-active state, the fusion peptide is projected from a coiled-coil stem to become anchored in the host-cell membrane. The importance of the fusion peptide to membrane fusion has been underscored by mutational studies, which have shown an arrest in fusion activity upon even the most conservative mutations in this domain (7).
As was demonstrated in the 1980s by Lear and DeGrado (8), the 20-residue N-terminal peptide of HA2 suffices to promote fusion of unilamellar vesicles. Whereas this 20-residue peptide was reported to adopt a static open boomerang-shaped kinked helix structure, based on 1H-only NMR spectroscopy (9, 10), 15N NMR revealed a high degree of dynamic disorder (11, 12). By contrast, the 23-residue N-terminal peptide of HA2 adopts a highly ordered, tight helical hairpin structure (11) with a ridge of conserved Gly residues lining the inner face of the two adjacent helices. The hairpin is stabilized by a charge–dipole interaction between the positive charge of the Gly1 α-amino group and the C terminus of the second helix (13), as well as by four interhelical aliphatic H-bonds (13) of a type commonly found in transmembrane proteins (14). The side chains of the structure are segregated in an amphiphilic pattern such that the bulky hydrophobic groups are embedded in the lipophilic core of the detergent micelles, or of membrane-mimicking bicelles, and the hydrophilic surface interacts with the aqueous solvent (11, 15). The helical hairpin structure points to the structural basis of many of the mutations that decrease fusion activity for the peptide. For example, mutations at the N terminus (G1Δ, L2Δ) disrupt the charge–dipole interaction, and mutations at the packing interface between the two helices (A5V, G8A) destabilize the hairpin structure (16–18).
In dodecylphosphocholine (DPC) micelles, the 23-residue fusion domain, HAfp23, retains a high population of the hairpin structure when the pH is lowered from 7 to 4 (11). However, it is believed that the fusion peptide structure must reorganize to enable formation of the fusion pore required at the later stages of fusion (19). Previous experimental evidence also points to transient structural changes taking place in the fusion peptide upon acidification: the backbone 15N transverse relaxation rates at pH 4 become elevated for a number of its residues, consistent with exchange to a minor, activated species on a submillisecond timescale (11).
In this report, we identify the multiple conformations associated with the activated states of the hemagglutinin fusion peptide at pH 4 by studying the conformational ensemble resulting from a G8A mutation (HAfp23-G8A). The G8A mutation produces a steric clash at the interface between the two helices when the Gly8 pro-S hydrogen is replaced by a methyl group, thereby opening the closed helical hairpin structure. The chemical shift changes induced by the G8A mutation relative to HAfp23 closely correlate with the exchange broadening seen in the wild-type structure, making it an appropriate model for the activated state. The G8A mutation is shown to interchange dynamically between at least three different conformations: a lowly populated helical hairpin closed structure and two opened structures. Here, we characterize these structures at high resolution using residual dipolar couplings (RDCs) and nuclear Overhauser effect (NOE) restraints. The opened, activated states are likely intermediates on the pathway to formation of the fusion pore when multiple hemagglutinin trimers assemble in the later stages of fusion (20).
Results
Activated States of the Wild-Type Peptide Can Be Modeled by the G8A Mutant.
The wild-type HAfp23 fusion peptide adopts a helical hairpin structure at pH 7 and predominantly maintains this structure at pH 4, as demonstrated by the close agreement between the pH 4 backbone RDCs and the helical hairpin structure determined at neutral pH (11). However, backbone relaxation measurements at pH 4 (Fig. 1) reveal a broadening contribution that is indicative of an exchange process to one or more lowly populated activated states.
Fig. 1.

HAfp23 in DPC micelles at pH 4 shows evidence for conformational exchange to an activated state populated by the open conformers of HAfp23-G8A. (A) Backbone 15N R2 relaxation rates at 900 MHz measured from a 15N R1ρ experiment at 32 °C for HAfp23 at pH 7 (●) and pH 4 (○), reproduced from ref. 11. (B) The pH 4 15N R2 rates at 900 MHz are plotted against the 15N chemical shift difference squared, Δω2, between the wild-type and HAfp23-G8A mutant at pH 7. Excluded from the plot are the chemical shifts of Gly8 and Phe9, which are dominated by the covalent modification introduced by the G8A mutation, and residue 11, which becomes protonated below pH 5.3 (13). 15N R2 rates are derived in the standard manner (21) from 15N R1ρ measurements, carried out under conditions where ωSLτex « 1, i.e. without suppressing the exchange contribution.
We hypothesized that such an activated state likely would involve a disruption of the tightly packed helical hairpin structure. By generating the G8A mutant (HAfp23-G8A), where the steric clash of Ala8 with the backbone of the second helix destabilizes the hairpin, an experimental model for the activated state of the wild-type peptide is obtained, and we set out to prove that the mutant is a good working model for the activated state.
The impact of the G8A mutation on the helical hairpin structure is clearly revealed by comparing its backbone amide chemical shifts (Fig. 2) and its diminished fusion activity (Fig. S1) to that of wild-type HAfp23. Chemical shifts are a sensitive measure of local structure, and a number of residues show chemical shift changes as large as 0.5 ppm and 4 ppm for 1HN and 15N nuclei, respectively, with the largest changes occurring for residues involved in the interhelical CαHα—O hydrogen bonds (Ala5, Gly13, Met17, Trp21) and the turn region (Gly12–Gly16) of the helical hairpin.
Fig. 2.

Transverse relaxation-optimized spectroscopy-HSQC spectra showing the backbone 1H and 15N chemical shifts, δ, at 900 MHz for the wild-type HAfp23 (black), mutant HAfp23-G8A (red), and truncated HAfp14 (blue) peptides in DPC micelles at pH 7. Black dotted lines are drawn to show the changes in resonance positions among HAfp23, HAfp23-G8A, and HAfp14. Peaks without assignment labels correspond to residues in the C-terminal SGK4D tag; the Gly1 and Leu2 amides are not observed because of elevated hydrogen exchange rates with water.
With these chemical shift differences, we tested whether the mutant HAfp23-G8A peptide could be used to model the relaxation broadening seen in Fig. 1, therefore testing its suitability as an accurate model for the activated state of the wild-type peptide at pH 4. The relaxation rate from a T1ρ spin-lock experiment can be written to include exchange between two sets of states (21):
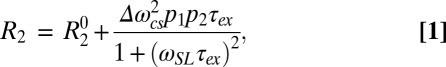 |
where Δωcs (in radians per second) is the chemical shift difference between the native and activated states; p1 and p2 are their populations; ωSL is the strength of the spin-lock field used for the T1ρ measurement; and τex is the time constant of the exchange process, defined as the inverse of the sum of the forward and backward exchange rates. The first term, R20, represents the natural line width of the peptide, and it is governed by the overall tumbling rate of the peptide–DPC complex and by picosecond-to-nanosecond timescale internal motions. The second term represents the exchange broadening contribution to the R2 rate, originating from the microsecond-to-millisecond dynamic processes that change chemical shift frequencies on the timescale of the chemical shift evolution. For simplicity, the off-resonance effect of the spin-lock field is not shown in Eq. 1 but can be easily included (21), as was done for all analyses in our study.
The 15N chemical-shift differences squared (Δωcs2), taken between the HAfp23-G8A and the wild-type HAfp23 peptides, show a strong linear correlation with the wild-type HAfp23 15N R2 rates at pH 4 (Fig. 1B). This result illustrates that the 15N chemical shifts of the G8A mutant closely correlate with those of the HAfp23 activated state at low pH, and therefore substantiates the use of HAfp23-G8A as representative for the activated state of the wild-type HAfp23 peptide at pH 4.
G8A Mutant Is a Dynamic Tertiary-Level Conformational Mixture.
In the heteronuclear single-quantum coherence (HSQC) spectrum of the HAfp23-G8A mutant, the amide resonances fall close to positions seen in the 14-residue N-terminal peptide, HAfp14, but are slightly shifted toward those seen in the closed helical hairpin of HAfp23 (Fig. 2). Because only a single set of peaks is observed for HAfp23-G8A, and these lie on average between HAfp23 and HAfp14, the HAfp23-G8A chemical shifts represent a population-weighted average between these two end-states, with an exchange rate that is much faster than their chemical shift difference (21), i.e., on a time scale much faster than milliseconds. The HAfp14 peptide, by its very nature, lacks the interhelical hairpin contacts of the full-length peptide; however, it remains helical from residues 2–14, as judged by its secondary chemical shifts (12), and is therefore representative of an “open” structure that lacks interhelical contacts. Clearly, the observation that the HAfp23-G8A resonances fall much closer to HAfp14 than to the hairpin structure of the wild-type sequence indicates that the mutant structure is predominantly open, analogous to the truncated HAfp20, which also shows reduced fusion activity compared with the full-length HAfp23 (15). Most of the HAfp23-G8A resonances fall close to but not directly on the trace between the HAfp23 and HAfp14 resonances, further suggesting that more than two states are appreciably populated in the HAfp23-G8A peptide (see below).
RDCs also represent the conformation time-averaged over the entire timescale from picoseconds to milliseconds (22, 23), and internal motions that reorient internuclear vectors will result in an attenuation of the corresponding dipolar couplings. The 1DNH and 1DCαHα RDCs for the N-terminal helices (Leu2–Glu11) of HAfp23 and HAfp23-G8A immersed in stressed acrylamide gels (24, 25) are highly similar to one another (Fig. 3 A and B), confirming that this region remains α-helical in the mutant, and also indicating that the N-terminal helices in the wild-type and mutant peptides are subject to similar molecular alignments. However, RDCs for the C-terminal helix (Trp14–Gly23) are significantly smaller for the HAfp23-G8A mutant, corresponding to an approximately threefold-weaker alignment strength (Fig. 3 C and D), but fit very well to the coordinates of the second helix in the HAfp23 structure. Considering that the mutant is predominantly in the open state (∼85%; see below), this observation indicates that the second helix retains its secondary structure while sampling multiple orientations relative to the N-terminal helix, which remains tightly anchored in the micelle. In other words, the open state itself is a dynamic ensemble of multiple structures, in which both the N- and C-terminal helices remain largely intact.
Fig. 3.

Residual dipolar coupling analysis of wild-type HAfp23 (filled symbols) and mutant HAfp23-G8A. (open symbols). Experimental RDC values were measured in stretched acrylamide gel at pH 7 for (A) 1DNH, circles, and (B) 1DCαHα, triangles. Singular-value decomposition fits of the HAfp23-G8A RDCs to the (C) N-terminal helix (residues 2–11) and (D) C-terminal helix (residues 14–23) of the hairpin structure (PDB ID code 2KXA) show good agreement, but nearly threefold-smaller alignment strength for the C-terminal helix. The 1DCαHα RDCs in C and D have been scaled by 0.485 to account for the difference in the static-limit dipolar coupling constant between 1H-13C and 1H-15N dipolar interactions.
Evaluation of the backbone chemical shifts in terms of secondary structure, using the TALOS+ program (26), confirms that both Leu2–Glu11 and Trp14–Tyr22 remain α-helical in HAfp23-G8A, although a random coil index order parameter (RCI-S2) analysis (27) indicates a decrease in local order for the last few residues of the second helix (Fig. S2 A and B). Characteristic α-helical NOE patterns between residues “i” and “i + 3” are seen in both the N- and C-terminal helices (Fig. S2C), and stable hydrogen bonds in the two helices are responsible for the more than 20-fold protection from solvent exchange observed for the backbone amide protons (Fig. S2D). Together with the RDC data, the chemical shift, hydrogen exchange, and NOE measurements confirm that both the N- and C-terminal helices remain largely intact for HAfp23-G8A, and that the reduced alignment strength seen for this C-terminal helix represents its tertiary-level reorientation with respect to the N-terminal helix/micelle complex.
The backbone 15N R1 and R2 relaxation rates and 15N-{1H} NOE values probe the dynamic behavior of the molecule on the picosecond-to-nanosecond timescale. For the N-terminal helix of HAfp23 and HAfp23-G8A, the similar rates (Fig. 4) indicate comparable tumbling times for the two peptides, which for HAfp23 is 10.9 ns (15). For the C-terminal helix, a modest increase in R1 and decrease in R2 point to an effective correlation time, τc,eff, that is slightly shorter in the mutant. Presumably this lower τc,eff value reflects the above-noted motion of the second helix relative to the N-terminal helix–micelle complex. A detailed motional analysis remains difficult due to the overlap in time scales between the overall tumbling of the micelle–peptide complex, motion of the peptide relative to the micelle (15, 28), and the reorientation of the second helix. However, a reduced spectral density mapping analysis (29) (Fig. S3) suggests a motional time scale in the ∼100-ns range for the second helix, which is too slow to contribute significantly to the 15N-{1H} NOE, but has a nonnegligible impact on R1 and R2.
Fig. 4.

The backbone 15N relaxation rates at 600 (circle) and 900 MHz (triangle) for HAfp23 (11) (solid symbols) and HAfp23-G8A (open symbols) in DPC micelles at pH 7.
Populations of the Closed and Open States for the G8A Mutant.
The chemical shift and NOE data can be used as separate and independent parameters to quantify the populations of the open and closed states of HAfp23-G8A. To estimate the population from chemical shifts, we used the wild-type HAfp23 peptide as a reference for the closed helical hairpin state, and the HAfp14 peptide as a reference for the open states. The chemical shifts of these two peptides therefore serve as endpoints between the fully closed and open structures of the first helix (residues 2–12). The population-weighted average chemical shifts of Fig. 2 then can be used to estimate the populations for the individual conformations (closed, HAfp23; open, HAfp14) in the HAfp23-G8A sample. Using the relation
the chemical shifts, δ*, and their differences can be plotted together to yield the population of the open conformers, popen, from the slope. The range in chemical shifts, δ, varies for different types of nuclei, and therefore appropriately scaled chemical shifts, δ*, were used to generate the plot (see SI Materials and Methods for details). The linear correlation in chemical shifts (Fig. 5A) confirms that, on average, the HAfp23-G8A chemical shifts fall between those of HAfp23 and HAfp14, with a population of 85 ± 4% for the open conformations in HAfp23-G8A.
Fig. 5.

Population of the open conformations for HAfp23-G8A from (A) chemical shift and (B) 1H-1H NOE data. (A) The differences in the 1HN (black), 1Hα (green), 15N (red), and 13Cα (blue) backbone chemical shifts among HAfp23 (completely closed conformation), HAfp14 (completely open conformation), and HAfp23-G8A (open/closed mixture) at pH 7 show a high degree of correlation with a slope of 85 ± 4%, corresponding to the population of the open conformation(s) for HAfp23-G8A. The chemical shift differences have been scaled (δ*) relative to 1HN by the following factors derived from database SDs in secondary chemical shifts: 15N = 0.181, 13Cα = 0.315, 1Hα = 1.373. Chemical shifts from the site of mutation, residue 8, and the C-terminal residues of HAfp14, Gly13, and Trp14 were not included in the fit. (B) Intensities of interhelical NOEs in HAfp23 and HAfp23-G8A show a similar correlation with a slope of 14 ± 4%, corresponding to the population of the closed helical hairpin conformation. Open symbols correspond to NOEs between Met17 and the aromatic side chain of Phe9 and have been excluded from the fit, because they show evidence for close proximity in the open structures.
Very weak interhelical NOE interactions (e.g., between Ala5 and Trp21) remain present in HAfp23-G8A, indicating that there still exists a minor population of the closed helical hairpin structure in this mutant, and their intensities relative to those seen in the native HAfp23 structure provide a second, independent measure for the residual population of the closed hairpin state. As discussed previously, the timescale for exchange is much faster than the evolution timescale of the chemical shifts, yet much slower than the overall rotational correlation time. Therefore, the NOE simply represents the population-weighted average of the NOE for the different conformers. If we assume, at first, that the interhelical NOEs can build up only in the closed conformation, where the interproton distances are within ∼6 Å, the population-weighted NOE rate is expected to be weaker by a factor pclosed, representing the population of the closed state. This relation uses the fact that the NOE datasets for HAfp23 and HAfp23-G8A were collected with the same mixing times and that both peptides have approximately the same overall tumbling times, a condition validated by the 15N relaxation rates.
A comparison of the long-range NOEs between HAfp23 and HAfp23-G8A, after referencing to an internal cross-peak of fixed distance (Ala5Hα–Hβ) to account for differences in absolute peptide concentration in the two samples, shows that most of the interhelical NOEs in the HAfp23-G8A spectrum are about sevenfold attenuated compared with wild-type HAfp23 (Fig. 5B). This NOE-derived population, pclosed = 14 ± 4% or popen = 86 ± 4%, agrees well with the 85 ± 4% open population derived independently from chemical shifts (Fig. 5A), and therefore, populations of 15% and 85% for the closed and open states, respectively, were used in all subsequent analyses. It is interesting to note, however, that several long-range NOEs, in particular those between protons from residues Phe9 and Met17, have intensities in HAfp23-G8A that are higher than expected from only closed-state contributions (Fig. 5B), indicating that these protons must be within ∼6 Å of one another in at least some of the open structures.
Structure of the HAfp23-G8A Conformers.
To identify the conformations sampled by the ∼85% population of HAfp23-G8A open states, we subtracted the RDCs corresponding to the ∼15% helical hairpin population, measured previously for the wild-type structure, thereby generating an “open-only” RDC dataset. Direct subtraction of the HAfp23 RDCs (after scaling by 0.15) is possible because the closed-to-open exchange rate is slower than the overall rotational correlation time (30), and aligned HAfp23 and HAfp23-G8A samples were created under matching conditions with the same alignment strength. The open-state RDCs show the large amplitude helical oscillatory pattern for the residues in the N-terminal helix, but much smaller RDCs for the C-terminal helix (Fig. S4).
At a minimum, two open conformations of the C-terminal helix relative to that of the N-terminal helix–micelle aggregate are needed to accurately model the smaller RDCs of the C-terminal helix. Including the ∼15% population of the closed state, HAfp23-G8A spectral data therefore represent the average over at least three conformers. The limited number of experimental NMR observables relative to the degrees of freedom needed to uniquely define the individual conformers contributing to even a two-state equilibrium has proven challenging in the past, and a number of special approaches have been developed to solve such problems (31, 32). These approaches include the measurement of paramagnetic relaxation enhancement (33) and extraction of the chemical shifts of a lowly populated conformer to derive its structure (34). For HAfp23-G8A, the experimental data correspond to an even-more-challenging three-conformer equilibrium, but the structure of one of these conformers, the closed hairpin state, is already known accurately. We therefore modeled our three conformers by including a 15% population for a closed helical hairpin conformation that must closely mimic the structure of the wild-type, and two open-state conformations. The structures of all three conformers were then determined by using the knowledge that both the N- and C-terminal helix remain largely intact in all three conformers, thereby limiting the degrees of freedom required to define these structures; this allows these HAfp23-G8A structures to be determined from the NOEs and RDCs by using the protocol described in SI Materials and Methods, Structure Calculations.
Next to the closed hairpin structure, the ensemble (Fig. 6; Table S1) includes two conformations for the open state: an extended and an l-shaped structure. The contribution of each open conformer to the observed RDCs is such that the helical oscillations in the N-terminal residues add constructively, whereas the helical oscillations in the C-terminal residues are out of phase and largely cancel one another. Note that due to the manner in which RDCs are used in the structure determination process, the conformations of the extended and l-shaped structures are rather insensitive to their relative populations (SI Materials and Methods).
Fig. 6.

Structures for the 33 lowest-energy three-conformer ensembles of HAfp23-G8A, and ribbon model representations of the three conformers taken from the ensemble with lowest total energy. The structures have interhelical angles of 159° ± 1°, 110° ± 6°, and 73° ± 11° for the helical hairpin (black), the l-shaped (blue), and the extended (red) conformations, respectively. The structures have been aligned to yield minimal coordinate rmsd for the backbone heavy atoms of residues 3–10 for each conformer. The ribbon models include side chains for the bulky hydrophobic residues, highlighting the amphiphilic character of all three structures, with hydrophobic residues all located on one face of each conformer.
The closed and two open structures each have a natural amphiphilic profile such that the hydrophobic and hydrophilic side chains are segregated on opposite sides of the peptide (Fig. 6). Structures of these conformers were calculated in the absence of lipids, and at no stage during the structure determination process was there any explicit or implicit restraint to invoke the segregation of side chains by polarity. However, from a physical perspective, this outcome appears plausible because it strongly suggests that for all three conformers the two helices remain located at the lipid/water interface of the detergent micelle. For both open conformers, the close proximity of the Phe9 and Met17 side chains is responsible for the elevated NOE intensities observed between these residues (Fig. 5B).
Time Scale of Exchange to the Activated States for Wild-Type HAfp23 at pH 4.
The time scale for the opening and closing transition of the wild-type peptide at pH 4 can be probed by a relaxation dispersion NMR experiment (21, 35), where the magnetization decay is probed as a function of the strength of an applied spin-lock field, ωSL (Eq. 1). However, exchange processes on a time scale much faster than the spin-lock nutation frequency remain invisible in such measurements. For this reason, attempts to measure the exchange rates by the commonly used 15N relaxation dispersion experiment proved unsuccessful. However, carrying out relaxation dispersion measurements on the amide protons, where much stronger spin-lock fields are available (36), and cooling the sample temperature to 12 °C, instead of the 32 °C sample temperature used in all other experiments, allowed probing of the exchange process for the wild-type peptide (Fig. 7).
Fig. 7.

1H relaxation dispersion data from a 1H T1ρ experiment for 2H, 13C, 15N-labeled HAfp23 at pH 4 and 12 °C. Relaxation dispersion curves are shown for R2unlike (36) of several representative residues. Curves were fit to Eq. 1 with a spin-lock field offset correction (21), and a common τex value of 28 ± 1 μs was fit for all residues.
Two sets of parameters are obtained from fitting the 1H relaxation dispersion curves: p1p2Δωcs2, and the exchange time constant, τex, which represents the inverse sum of the opening and closing rates. For a concerted conformational change, a common exchange time constant is expected for all residues, and indeed a common τex = 28 ± 1 μs sufficed to fit all of the relaxation dispersion curves at 12 °C. The best-fit values for p1p2Δωcs2, again using the chemical shift differences between HAfp23 and HAfp23-G8A at 12 °C and pH 7, gives an open-state population of 20–28% (Fig. S5). This estimate agrees well with the open population of 21 ± 4% determined using the chemical shifts from the HAfp14 peptide (Fig. S6), as was done in the analysis of Fig. 5.
Discussion
Acidification of the endosome triggers a key step in the fusion of the viral and endosomal membranes, which ultimately results in formation of a pore connecting the interior of the virus to the cytosol of the host cell. After proteolytic cleavage of hemagglutinin into its two subunits, HA2 becomes the physical link between the viral envelope and the endosomal membrane. Though the HA2 C-terminal transmembrane helix anchors to the virus, its N-terminal fusion peptide becomes embedded in the surface of the endosomal membrane. At neutral pH, this fusion peptide adopts a helical hairpin shape, and prior NMR data indicate this shape is predominantly maintained even when lowering the pH below the pKa value of its acidic side chains (11). In its hairpin conformation, this fusion peptide is too small to span a bilayer and to participate in the formation of the previously postulated transmembrane pore (19). However, at low pH, our data show clear evidence for transient population of a minor, activated state, with relaxation dispersion experiments providing a tight link between the conformation of this activated state and that of a G8A mutant, which destabilizes the hairpin structure of the fusion peptide. This activated state itself is shown to consist of two stable helices switching between L-shape and extended arrangements, where the L-shape provides an intermediate step in transitioning from the hairpin to the extended structure. Although a minimum of two open structures suffices to fit the experimental NMR data, a much wider array of interhelical angles presumably is sampled, because both the L-shape and extended shape lack any prominent features that stabilize their relative helix orientations. Attempting to generate a single model that fits the experimental restraints using the standard NMR protocol, as was done for the truncated HAfp20 peptide, which is ∼90% open (12), results in a structure that bears similarity to the previously reported static boomerang shape (9, 10), but cannot fully satisfy the NOE and RDC data.
The conformational change between closed and open structures appears to be driven by the changes in protonation states of the acidic side chains, somewhat analogous to that seen in subunit c of ATP synthase (37). Neutralization of the Glu11 and Asp19 side chains in HAfp23 upon acidification of the endosome favors deeper burial of the peptide in the apolar region of the phospholipids (11), thereby driving the partial transition to the activated open states. In its final stage, fusion is then believed to progress through a mechanism where the N- and C-terminal helices of multiple hemagglutinin trimers zipper together in an antiparallel fashion to form a competent fusion pore (20). Opening of the fusion peptide therefore appears a requisite step for adopting a helical structure that is sufficiently long to traverse the lipid bilayer, which is needed to allow formation of the stipulated oligomeric fusion pore structure, assembled from fusion peptides and HA2 C-terminal transmembrane helices (38).
Materials and Methods
The HAfp23-G8A and HAfp14 peptides of the H1 serotype were expressed and purified as fusion proteins with the IgG-binding domain B1 of streptococcal protein G (GB1; PDB ID code 3GB1) at the N terminus and a polyionic solubilization sequence, SGKKKKD, at the C terminus to aid in the purification. The GB1 sequence was removed by Factor Xa proteolytic cleavage. Details on the amino acid sequence and purification protocol are presented in SI Materials and Methods.
Uniformly, 13C,15N-, and 15N-enriched samples were prepared with a peptide concentration of 0.3–0.6 mM and 130–150 mM DPC. NMR experiments were conducted at 600 and 900 MHz. NMR ensembles were calculated using NOE distance restraints, RDCs, and TALOS+ dihedral restraints using XPLOR-NIH v2.31. For details, see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Annie Aniana for help with protein expression and purification, and acknowledge support from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) mass spectrometry facility. This work was funded by the National Institutes of Health (NIH) Intramural Research Programs of the NIDDK and the Center for Information Technology, and by the Intramural AIDS-Targeted Antiviral Program of the Office of the Director, NIH.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and restraints have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2LWA); and NMR chemical shifts have been deposited in the BioMagResBank, www.bmrb.wisc.edu (accession no. 18617).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213801109/-/DCSupplemental.
References
- 1.Chernomordik LV, Kozlov MM. Mechanics of membrane fusion. Nat Struct Mol Biol. 2008;15(7):675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15(7):665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNeil PL, Steinhardt RA. Plasma membrane disruption: Repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 4.Lamb RA, Jardetzky TS. Structural basis of viral invasion: Lessons from paramyxovirus F. Curr Opin Struct Biol. 2007;17(4):427–436. doi: 10.1016/j.sbi.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 6.Carr CM, Chaudhry C, Kim PS. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci USA. 1997;94(26):14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross KJ, Langley WA, Russell RJ, Skehel JJ, Steinhauer DA. Composition and functions of the influenza fusion peptide. Protein Pept Lett. 2009;16(7):766–778. doi: 10.2174/092986609788681715. [DOI] [PubMed] [Google Scholar]
- 8.Lear JD, DeGrado WF. Membrane binding and conformational properties of peptides representing the NH2 terminus of influenza HA-2. J Biol Chem. 1987;262(14):6500–6505. [PubMed] [Google Scholar]
- 9.Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat Struct Biol. 2001;8(8):715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 10.Lai AL, Tamm LK. Locking the kink in the influenza hemagglutinin fusion domain structure. J Biol Chem. 2007;282(33):23946–23956. doi: 10.1074/jbc.M704008200. [DOI] [PubMed] [Google Scholar]
- 11.Lorieau JL, Louis JM, Bax A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc Natl Acad Sci USA. 2010;107(25):11341–11346. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorieau JL, Louis JM, Bax A. 2013. The impact of influenza hemagglutinin fusion peptide length and viral subtype on its structure and dynamics. Biopolymers, 10.1002/bip.22102/pdf.
- 13.Lorieau JL, Louis JM, Bax A. Helical hairpin structure of influenza hemagglutinin fusion peptide stabilized by charge-dipole interactions between the N-terminal amino group and the second helix. J Am Chem Soc. 2011;133(9):2824–2827. doi: 10.1021/ja1099775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senes A, Ubarretxena-Belandia I, Engelman DM. The Calpha-H2O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorieau JL, Louis JM, Bax A. Whole-body rocking motion of a fusion peptide in lipid bilayers from size-dispersed 15N NMR relaxation. J Am Chem Soc. 2011;133(36):14184–14187. doi: 10.1021/ja2045309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinhauer DA, Wharton SA, Skehel JJ, Wiley DC. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus hemagglutinin. J Virol. 1995;69(11):6643–6651. doi: 10.1128/jvi.69.11.6643-6651.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross KJ, Wharton SA, Skehel JJ, Wiley DC, Steinhauer DA. Studies on influenza haemagglutinin fusion peptide mutants generated by reverse genetics. EMBO J. 2001;20(16):4432–4442. doi: 10.1093/emboj/20.16.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han X, Steinhauer DA, Wharton SA, Tamm LK. Interaction of mutant influenza virus hemagglutinin fusion peptides with lipid bilayers: Probing the role of hydrophobic residue size in the central region of the fusion peptide. Biochemistry. 1999;38(45):15052–15059. doi: 10.1021/bi991232h. [DOI] [PubMed] [Google Scholar]
- 19.Blumenthal R, Sarkar DP, Durell S, Howard DE, Morris SJ. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J Cell Biol. 1996;135(1):63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133(3):559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton N. Protein NMR Spectroscopy: Principles and Practice. Burlington, MA: Elsevier; 2007. [Google Scholar]
- 22.Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. NMR evidence for slow collective motions in cyanometmyoglobin. Nat Struct Biol. 1997;4(4):292–297. doi: 10.1038/nsb0497-292. [DOI] [PubMed] [Google Scholar]
- 23.Peti W, Meiler J, Brüschweiler R, Griesinger C. Model-free analysis of protein backbone motion from residual dipolar couplings. J Am Chem Soc. 2002;124(20):5822–5833. doi: 10.1021/ja011883c. [DOI] [PubMed] [Google Scholar]
- 24.Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. Solution NMR of proteins within polyacrylamide gels: Diffusional properties and residual alignment by mechanical stress or embedding of oriented purple membranes. J Biomol NMR. 2000;18(4):303–309. doi: 10.1023/a:1026703605147. [DOI] [PubMed] [Google Scholar]
- 25.Tycko R, Blanco FJ, Ishii Y. Alignment of biopolymers in strained gels: A new way to create detectable dipole-dipole couplings in high-resolution biomolecular NMR. J Am Chem Soc. 2000;122:9340–9341. [Google Scholar]
- 26.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44(4):213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berjanskii MV, Wishart DS. A simple method to predict protein flexibility using secondary chemical shifts. J Am Chem Soc. 2005;127(43):14970–14971. doi: 10.1021/ja054842f. [DOI] [PubMed] [Google Scholar]
- 28.Prosser RS, Davis JH. Dynamics of an integral membrane peptide: A deuterium NMR relaxation study of gramicidin. Biophys J. 1994;66(5):1429–1440. doi: 10.1016/S0006-3495(94)80933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng JW, Wagner G. Mapping of spectral density functions using heteronuclear NMR relaxation measurements. J Magn Reson. 1992;98:308–332. [Google Scholar]
- 30.Clore GM, Schwieters CD. How much backbone motion in ubiquitin is required to account for dipolar coupling data measured in multiple alignment media as assessed by independent cross-validation? J Am Chem Soc. 2004;126(9):2923–2938. doi: 10.1021/ja0386804. [DOI] [PubMed] [Google Scholar]
- 31.Kessler H, et al. Conformational dynamics detected by nuclear magnetic resonance NOE values and J coupling constants. J Am Chem Soc. 1988;110(11):3393–3396. [Google Scholar]
- 32.Schwieters CD, Clore GM. Reweighted atomic densities to represent ensembles of NMR structures. J Biomol NMR. 2002;23(3):221–225. doi: 10.1023/a:1019875223132. [DOI] [PubMed] [Google Scholar]
- 33.Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449(7165):1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- 34.Bouvignies G, et al. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature. 2011;477(7362):111–114. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulder FAA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: Application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2001;123(5):967–975. doi: 10.1021/ja003447g. [DOI] [PubMed] [Google Scholar]
- 36.Ishima R, Wingfield PT, Stahl SJ, Kaufman JD, Torchia DA. Using amide H-1 and N-15 transverse relaxation to detect millisecond time-scale motions in perdeuterated proteins: Application to HIV-1 protease. J Am Chem Soc. 1998;120:10534–10542. [Google Scholar]
- 37.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402(6759):263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 38.Donald JE, et al. Transmembrane orientation and possible role of the fusogenic peptide from parainfluenza virus 5 (PIV5) in promoting fusion. Proc Natl Acad Sci USA. 2011;108(10):3958–3963. doi: 10.1073/pnas.1019668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.