

Abstract
This article briefly summarizes the research activities in the field of hydrogen storage in sorbent materials and reports our recent works and future directions for the design of such materials. Distinct features of sorption-based hydrogen storage methods are described compared with metal hydrides and complex chemical hydrides. We classify the studies of hydrogen sorbent materials in terms of two key technical issues: (i) constructing stable framework structures with high porosity, and (ii) increasing the binding affinity of hydrogen molecules to surfaces beyond the usual van der Waals interaction. The recent development of reticular chemistry is summarized as a means for addressing the first issue. Theoretical studies focus mainly on the second issue and can be grouped into three classes according to the underlying interaction mechanism: electrostatic interactions based on alkaline cations, Kubas interactions with open transition metals, and orbital interactions involving Ca and other nontransitional metals. Hierarchical computational methods to enable the theoretical predictions are explained, from ab initio studies to molecular dynamics simulations using force field parameters. We also discuss the actual delivery amount of stored hydrogen, which depends on the charging and discharging conditions. The usefulness and practical significance of the hydrogen spillover mechanism in increasing the storage capacity are presented as well.
Renewable Energy and the Significance of Hydrogen Storage
The prosperity of modern civilization is inextricably based on energy supply networks (on-grid or off-grid) that deliver petroleum, natural gas, and electricity to residential, public, commercial, and industrial facilities, as well as transport systems. Concerns are growing over the limited availability of natural resources and the environmentally hazardous byproducts of the energy conversion process, such as greenhouse gases. From this perspective, renewable energy harvesting technologies based on natural energy flows, including solar power, wind power, geothermal energy, and tidal energy, are increasingly gaining momentum. The storage of the harvested energy remains a fundamentally important factor for the utilization of the renewable energy because the sources of the renewable energy usually undergo significant spatial and temporal variations (1). The issue of hydrogen storage can be understood in this context. Among chemical fuels, pure hydrogen (i.e., in the gas form) has the highest mass density but the poorest volumetric density (2, 3). As a result, hydrogen storage research traces a long history of studies toward the reversible condensation of hydrogen into a limited volume using lightweight devices. In recent decades a globally recognized objective is the development of a stored hydrogen carrier that can power vehicles through fuel cells (or, perhaps, combustion engines). For example, the guideline published by the US Department of Energy states that, for a commercially competitive vehicle, hydrogen storage systems need to achieve an overall capacity of 5.5 wt% hydrogen with a volumetric ratio of 40 g/L within a few years (4).
However, the objectives of hydrogen storage need to be defined from a wider perspective of the energy storage and energy back-up system. Although supercapacitors and rechargeable batteries have shown an optimistic outlook in the field of hybrid electric vehicles and limited forms of zero-emission cars, larger-scale energy operations such as stationary back-up power systems and full commercial scale automotive applications are very difficult to realize through the pure electric power storage devices. Hydrogen storage systems have obvious advantages in certain important aspects (e.g., energy density and durability), and the search for safe and efficient hydrogen storage materials continues for such wide application purposes.
Hydrogen Storage Methods and Sorption-Based Storage Systems
Methods for hydrogen storage may be divided into two categories: physical confinement inside vessels and binding onto host materials. The physical confinement of hydrogen gas involves either high-pressure compression or cryogenic liquefaction and has been used for special purposes or on the laboratory scale. The safety (for high-pressure) and cost (for liquefaction) concerns associated with such methods suggest that large-scale commercial applications of stored hydrogen might be unlikely via such physical storage methods. Regarding the method to bind hydrogen onto an absorbent medium, a variety of host materials have been investigated (3, 5). These materials may be classified into three distinct groups depending on the microscopic mechanism related to the charging and discharging properties: metal hydrides that contain atomic hydrogen as a constituent of the bulk material, complex chemical hydrides that react chemically upon charging and discharging, and the adsorption of molecular hydrogen onto the surfaces of highly porous materials. Each method has its own advantages and disadvantages. For example, metal hydride systems (in particular the AB5-type heavy metal alloys) can provide greatly satisfactory volumetric storage density, easy kinetics, and good reversibility, but the gravimetric density is fundamentally limited (6). The reversibility and reaction kinetics present the largest hurdle to complex chemical hydride storage systems. In a sense, the complex chemical hydride can perhaps be used with a compartment system that is subject to off-board regeneration processes (4, 7). A marked drawback of sorption-based storage in porous materials is the weak strength with which hydrogen is physisorbed onto surfaces. Without a particular enhancement of the adsorption strength, sorption-based storage systems are practically operative only around the liquid nitrogen temperature (77 K).
Nevertheless, sorbent materials and sorption-based storage systems deserve a more intensive investigation because they present a variety of inherent advantages over other storage systems. Microscopically, adsorption does not require the dissociation of hydrogen molecules and thus does not involve energy barriers. Macroscopically, this approach permits the rapid charging and discharging of hydrogen, and the corresponding thermal management is expected to be easier than chemical or metal hydrides (8). A fundamental requirement for sorbent materials is the presence of a stable framework structure with sufficient porosity and surface area. In this respect, recent experimental advancement in the field of reticular chemistry, that is, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), is particularly noteworthy (9–11). On the other hand, many theory-based studies have addressed the issues of increasing binding affinity of hydrogen molecules onto surfaces by introducing various strategies, as will be described in more detail in later sections.
Computational Methods Applied to Hydrogen Storage Problems
Several hierarchical computational methods have been used in this field. An accurate evaluation of the binding strength between hydrogen molecules and the host material surface is central to this study. Among various ab initio theories, the density functional theory (DFT) has been most widely used. The Kohn–Sham equation, which is an effective single-particle equation, is usually solved with the local one-body operators that approximate the electron–electron many-body effects (12). Commonly made choices are the form of a local density approximation or a generalized gradient approximation (13, 14). Such local approximations of the exchange–correlation potentials can have remnant (surplus) electron self-interactions and occasionally require a cautious treatment. The over-delocalization problems of the local functionals can arise in some cases of adsorptions (15). In this regard, effects of the exact exchange and the explicit inclusion of the Hubbard U term deserve detailed theoretical considerations (16, 17). Accurate and practical calculations of van der Waals (vdW)-type dispersion interactions are important for calculating hydrogen–surface interactions (18). The direct treatment of such interactions using the Schrödinger equation is impractical; therefore, wavefunction theories based on the Hartree–Fock (HF) self-consistent field (SCF) approximation provide a reference for or complement to the DFT results. For example, the Moller–Plesset second-order perturbation applied to the HF-SCF wavefunctions provides a reasonably practical and accurate approach (19).
More realistic adsorption isotherms may be calculated using molecular dynamics simulations at given pressures and temperatures. Such studies require transferrable force parameters that account for the chemical environment, such as the reactive force field. If reliable force parameters are available, a grand canonical Monte Carlo (GCMC) simulation is particularly useful for calculating hydrogen adsorption isotherms associated with a porous material (20). Porous materials are characterized by the surface area and the free pore volume. Experimental storage measurements, either through gravimetric or volumetric methods, typically count only the so-called excess amount of hydrogen uptake. However, the hydrogen gas present in the free volume of the pores should be included in calculating the total amount of hydrogen. GCMC calculations provide a practical tool for a direct evaluation of the total as well as excess amounts of stored hydrogen in a porous material (21).
Porous Framework Structures
Solid materials intended for gas uptake must possess a high specific surface area and porosity. Microporous and mesoporous materials, including zeolites and silica derivatives, have been developed throughout the history of materials sciences with a variety of applications, such as gas sorbents, gas separators, and catalysts (22). Recent advances in the reticular chemistry of MOFs and related materials, including zeolitic imidazolate frameworks (ZIFs) and COFs, are relevant to the design and synthesis of materials with a tunable pore size and chemical functionality derived from selected building units (9). The reversible hydrogen storage, CO2 uptake, and methane uptake are used to benchmark the performances of various MOFs and ZIFs. An extensive review of them is provided in refs. 11 and 23. Furukawa et al. (10) described the inclusion of a long and slim linker as conveying ultrahigh porosity. They recently reported the preparation of an extremely porous structure, named MOF-210, which shows an extremely high specific surface area. In terms of the Brunauer–Emmett–Teller area, MOF-210 reaches the highest value so far (6,240 m2/g), which exceeds that of other members of the family of MOFs [e.g., MOF-5 (3,800 m2/g) and MOF-177 (5,640 m2/g)]. Its excess and total hydrogen uptakes (86 mg/g and 176 mg/g at 77 K and 80 bar, respectively) are higher than those of other porous crystals (10). These results showed that an increase in the specific surface area of a porous material directly benefits the uptake of hydrogen storage at low temperatures, and also the storage of heavier gases like CO2 or methane. The governing thermodynamics associated with the gas uptake by MOFs is derived from the physisorption of gas molecules onto a chemically inert MOF surfaces. The polarity of a building unit and the appropriate spatial separation between the organic units can partially increase the binding strength of hydrogen molecules to some degree (24). However, these binding strengths still fall in the regime of physisorption and are on the order of several kJ/mol (a few tens of meV). Thus, these binding strengths are not relevant for hydrogen storage at room temperatures.
Decoration of Metal Atoms onto Porous Structures
The success of low-temperature hydrogen storage methods using microporous materials, as stated in previous paragraphs, can lead to a certain utility (10). Nevertheless, higher operational temperatures for the hydrogen storage systems would unarguably convey a much wider impact, for example by permitting vehicular applications under ambient conditions. For such a purpose, a large specific surface area in a sorbent material must be accompanied by an enhanced binding affinity of the surface to hydrogen molecules. As discussed below, the interactions between open transition metal atoms or alkaline cations with hydrogen molecules can yield a desirable binding strength. In an effort to use such an advantage, a few postsynthetic treatments have been considered to incorporate alkali, alkaline earth, and transition metal atoms onto the organic linkers of MOFs or COFs (25–27). Transition metal atoms have a strong tendency to aggregate, whereas alkaline cations tend to oxidize. Therefore, the preparation of dispersed under-coordinated metal elements for the adsorption sites in the porous material accessible to gas molecules remains an extremely challenging task in synthetic chemistry (28). Instead of decoration with metal elements, as a somewhat different approach, people considered modification of porous structures with embedded metal nanoparticles (29–31). In such cases, chemisorption into the destabilized nanoparticles and adsorption onto surfaces can lead to a synergic increase of storage capacity.
Focusing on methods of surface treatment, we explored modifications of framework structures that help anchor additional metal atoms. In our previous publications, we demonstrated that boron substitution in the organic building units of COFs substantially improved the stabilization of Sc, Ti, and Ca coordinated to the linker moieties (32). We also searched for functional groups capable of preferential binding to each transition metal atom so as to prevent the aggregation. In particular, the presence of OH terminal groups on the MOF linkers was found to be efficient in stabilizing transition metal atoms (33). Our ab initio calculations showed that the under-coordinated Fe(II) on the OH terminals could bind up to four H2 molecules, and the transition from a high spin state to a low spin state took place upon adsorption of H2 molecules. Such transition in the spin state obviously indicates the orbital interaction between the 3d states and H2 molecular orbitals. Other groups suggested that the decoration of Li on the benzenyl rings of MOF carboxylate linkers could lead to a substantial increase in the H2 uptake capacity near room temperature (34–37). This effect was attributed to the strong electrostatic field centered at the cation site.
Delivery Amount
Before we examine the detailed interaction mechanism between hydrogen molecules and these surface-modified structures, we want to precisely define the target value for the binding energy in the context of the actual vehicle-operating conditions. In this situation, the volumetric and gravimetric storage capacity should be assessed in terms of the reversibly deliverable amount rather than the maximum hydrogen uptake. The delivery amount is defined as the difference in the amount of hydrogen present under the charging and discharging conditions. Therefore, the delivery amount, also called the usable amount, depends on the macroscopic charging and discharging conditions [pressure (P) and temperature (T)], as well as on the microscopic binding characteristics (such as available binding sites and energies). For safety reasons, the maximum delivery pressure is recommended to be lower than 100 bar (4). It is also strongly desirable that the discharging temperature (the condition for hydrogen desorption) is not more than 100 °C. From equilibrium thermodynamics, the expression for g (the average number of adsorbed hydrogen molecules per adsorption site) is (38):
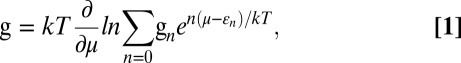 |
where µ is the chemical potential for H2 (common to both the gas and the adsorbed phase at the thermal equilibrium) at given P and T, k is the Boltzmann constant, εn is the adsorption energy per H2 molecule when the number of adsorbed molecules is n, and gn is the configurational degeneracy factor for given n. (In practice, the sum in the grand partition function is usually dominated by a single term. In such cases, the gn in the numerator and the denominator is effectively cancelled, and the effect of configurational degeneracy turns out to be minor.) Suppose that we use the isoreticular MOF 16 (IRMOF16) as the storage system. To increase the storage capacity, the linker is functionalized with OH and decorated with transitional metal atoms such as Fe or V. At the fuel charging (298 K and 100 atm) and discharging (373 K and 3 atm) conditions, μ = −0.196 eV and μ = −0.386 eV, respectively (39). If we choose Fe(II) for the decorating metal, the calculated binding energies of H2 to the Fe site are εn = −0.127, −0.246, −0.238, and −0.259eV for n = 1, 2, 3, and 4, respectively. Fig. 1A shows the configuration of the linker (terphenyl dicarboxylate) part of our system when the maximum number (nmax = 4) of H2 molecules are attached. (In reality, we considered four metal atom sites per linker to increase the storage capacity, but the case of only one metal site is shown here for visual clarity.) The expected adsorption number g from Eq. 1 is 3.987 and 0.001 at charging and discharging conditions, respectively, and the actual delivery amount is the difference (3.986 per adsorption site). In this case, the delivery amount (3.986) is close to nmax (4) because the binding energy is appropriate, namely, it falls within the range of two μ values for charging and discharging conditions. It should be as far below as possible from μ of charging (−0.196 eV) for efficient H2 adsorption and as far above as possible from μ of discharging (−0.386 eV) for efficient H2 release. Parenthetically, we note that εn is the binding energy level that is negative, whereas the binding energy is usually presented only with the magnitude (without the negative sign). Following this convention, a representative binding energy value for a maximized delivery amount under the aforementioned operation conditions can be said to be 0.29 eV (or 28 kJ/mol). It is obvious that, if the magnitude of binding energy is too small (such as the vdW interaction of 0.05 eV), the room temperature storage is almost zero. On the other hand, the magnitude of the binding energy should not be too large, either. The case of V, shown in Fig. 1B, constitutes a striking example. The configuration with the maximum number of H2 molecules adsorbed onto V (nmax = 4) looks similar to Fig. 1A. However, εns are out of the range of two μ values, namely, εn = −0.523, −0.517, −0.500, and −0.435 eV for n = 1, 2, 3, and 4, respectively. We have g = 3.861 and 2.930 for charging and discharging conditions, and the delivery amount is only 0.931 per site, mere 23% of Fe.
Fig. 1.

Atomic configurations of 4H2 molecules attached to the backbone structure. The backbone is (A) Fe-decorated, OH-functionalized terphenyl dicarboxylate (TPDC), and (B) V-decorated, OH-functionalized TPDC, respectively, where TPDC is the linker part of the IRMOF16. The blue, gray, red, green, and orange balls stand for hydrogen, carbon, oxygen, iron, and vanadium atoms, respectively.
The above results are the contribution from the relatively strong adsorption to transition metal sites. To include the contribution from the vdW interaction between H2 and the MOF, as well as that from the gas-phase storage in the void (pore) volume of the MOF, we have performed GCMC simulations for the Fe-decorated, OH-functionalized IRMOF16 with four Fe sites per linker (33). Including all of the contributions, we get the reversibly usable storage capacity of 6.0 wt% (4.3 wt% from the strong adsorption at Fe sites).
Some authors considered the delivery amount under different charging/discharging conditions. Han et al. (36) performed GCMC simulations using the force parameters obtained from ab initio works and considered the charging and discharging assuming hydrogen pressure as the single control parameter. They showed that the existence of a substantial free volume could improve the delivery amount (36). They suggested that not only the size but the shape of the pore should be important, and some ZIF structures could provide better adsorption isotherms than MOFs in terms of the delivery amount. On the other hand, we suggested that a mixture of H2 and a purging gas, such as NH3, would provide a control mechanism that could be an alternative or a complement to temperature as part of the discharging parameters (40).
Electrostatic Interactions and Alkaline Cations
In the present and following two sections, we elaborate on underlying interaction mechanisms between hydrogen molecules and adsorption sites. It has been widely perceived that the interaction mechanism of H2 with adsorption sites can be grouped into the vdW (dispersion), electrostatic, and orbital interactions, which are often intermixed in real physical systems. The vdW interaction is always present, but the strength by itself is too weak to stably confine the hydrogen gas molecules near room temperature (18). For a measurable amount of adsorption in noncryogenic conditions, additional treatments of surfaces are required to incorporate the electrostatic and orbital interactions. We focus on the former in the present section.
The electrostatic interaction involves the dipole and/or quadrupole moment of H2 induced by the charged species centered on the adsorption sites (41). There is an obvious contrast between the binding mode of H2 onto positively charged sites and that onto negatively charged ones: H2 molecules bind in the side-on fashion onto a positively charged center, whereas they are in the end-on shape onto negatively charged atoms. The driving mechanism of the end-on pattern is attributed to the dipole moment of the H2 induced by the negative charge, whereas the side-on adsorption is the outcome of the interaction between the electric field and quadrupole moment of H2. Lochan and Head-Gordon (42) published a comprehensive article on these issues, using hierarchical levels of theories. They showed that the negatively charged ligands (e.g., SO42- and F−) can attract H2 with some enhanced binding strength.
In terms of the adsorption strength and also the capability of gathering multiple H2 molecules onto a single adsorption site, the positively charged alkaline cations are superior to the negatively charged species. It has been shown that 6H2 molecules can be bound to a bare Li+ cation electrostatically (42). As stated in previous sections, however, because free ions by themselves cannot be used for the storage medium, they should be incorporated into porous framework structures in the solid-state form. Even though the experimental synthesis is yet to be proven, theoretical results for the alkaline-decorated organic porous structures deliver a promising message. We considered the substituted alkaline atoms for H atoms in the linker molecules before (43), whereas Goddard et al. explored the adsorbed alkaline atoms on the aromatic plane of the linker molecules (34–37). Both cases retained substantial cationic features and achieved the binding of a few H2 molecules with a reasonable strength.
Transition Metal and Kubas Interaction
Undercoordinated transition metal atoms can show a more salient binding affinity toward H2. This is described by the Dewar–Chatt–Duncanson coordination of dihydrogen to transition metal atoms and is called the Kubas interaction (44). Heavier metal atoms with 4d and 5d electrons can also have this quality, but they are not attractive for hydrogen storage because of the heavy weight. The Kubas interaction involves the orbital hybridization between the partially filled (and so partially empty) 3d orbitals of the transition metal atom and the σ (both bonding and antibonding) orbitals of H2. In another view, it is interpreted as the electron donation from the H2 σ-bonding orbital to empty metal 3d orbitals and the simultaneous back-donation from the filled metal d orbital to the H2 σ-antibonding orbital. Upon coordination, the H–H bond length increases by approximately 10% of the free H2 bond length (45, 46), mainly owing to some occupation of the H2 σ-antibonding orbital. Several H2 molecules can cluster onto a single metal atom through this type of orbital interactions. Numerous theorists explored the transition metal-decorated surface structures as a hydrogen storage medium (47–49), and experimentalists identified the Kubas-type hydrogen molecular adsorptions onto such sites of adsorption (50–53).
Despite positive predictions and anticipations, a practical manifestation of metal-coordinated open sites on the surfaces of porous materials, for use as hydrogen adsorption sites, still waits for a great breakthrough. One of the most serious obstacles is the strong tendency of under-coordinated metal atoms to aggregate or oxidize. The purpose of introducing functional groups such as OH mentioned before was to disperse metal atoms in the backbone matrix and suppress the metal aggregation. To disperse the metal atoms (or, more realistically, metal ions) using solution chemistry, it may be necessary to use organo-metallic precursors containing the metal element. The detailed route for the synthesis of such sorbent materials is a subject beyond the scope of the present article. Furthermore, even after the desired synthesis, the metal-coordinated structures require a sealing procedure to protect the inside from oxidation or other contamination. Therefore, the transition-metal decoration on porous structures seems to require more critical investigations and harder efforts on the synthetic chemistry side in the future.
Theoretical Issues in the Interaction Between Ca and H2
Other than alkaline and transition metals, calcium has attracted attention as decorating-metal atoms recently (54–56). Calcium has a lower aggregation tendency because of its small cohesive energy compared with transition metals. Besides the potential use of Ca-coordinated surface structures as hydrogen absorbents, the interaction between Ca and hydrogen molecules constitutes an interesting academic topic from a theoretical point of view. Although a free Ca atom does not have 3d electrons and the outermost electron is in the 4s state, the adsorption of a few H2 molecules and the accompanying orbital hybridization lowers the 3d energy levels below the Fermi level (54, 57). Thus a sharp discontinuous transition in the valence configuration of Ca atoms is observed from 4s to 3d upon H2 adsorption (58–60). Fig. 2 A and B illustrate such a change in the Ca valence states. This involves the multireference character of the electron wavefunction and provides a prototypical example of the failure of locally approximated density functionals. The exchange energy as a local functional of density (and the gradient of density) underestimates the energy costs associated with the transition from an s orbital to a higher angular momentum state and thus artificially favors the occupation of 3d orbitals over 4s orbitals (61).
Fig. 2.

Geometries of Ca atom with (A) attached and (B) far separated four hydrogen molecules. (C) Optimized geometry and (D) molecular orbital levels of the Ca adsorbed anthracene structure. (E) Same structure as in C, with four adsorbed hydrogen molecules. The isosurface plot of each HOMO state is also illustrated in A, B, and C. Large ivory balls represent Ca. Symbols of other atoms follow the same convention used in Fig. 1. Figure is adapted from the published data in Ref (58), copyrighted by the American Physical Society and Ref (64), with permission from Elsevier.
We tested various theories, including the DFT, hybrid functionals, and Hartree–Fock-based theories. We found that the hybrid functional could alleviate the errors if a somewhat larger-than-usual ratio of the exact exchange was used. Other groups also performed computations with various theories (62, 63). In particular, Bajdich et al. (62) reported that the exact exchange combined with a correlation density functional yielded a quite accurate energy curve similar to the quantum Monte Carlo calculations. Putting aside these theoretical issues, the Ca-decorated structures might be more useful than the transition metal structures if the adsorbed Ca stabilized the down-shifted 3d-derived orbitals after anchoring to the surface. We observed that the valence state of Ca on defective graphene or other reactive carbon surface structures readily possessed the 3d characters, and the abrupt orbital change induced by adsorption could be effectively eliminated (57, 64). As an example, adsorption configurations of Ca on anthracene and the electronic structure are shown in Fig. 2 C and D, respectively. The highest occupied molecular orbital (HOMO), as shown in Fig. 2C, much resembles the 3d orbital centered at Ca. In this case, the adsorption of hydrogen molecules, as depicted in Fig. 2E, does not induce a sharp orbital change in the valence states.
Search for Kubas-Type Orbital Interactions in Light Elements
In previous sections we discussed the mechanisms (electrostatics or orbital interactions) that can bind a few hydrogen molecules onto alkaline cations, transition metal atoms, and (the 3d orbital state of) Ca. In a broad sense, Ca can be included in the family of transition metals as far as the Kubas interaction via 3d orbitals is concerned. Although under-coordinated transition metal, Ca, and alkaline metals are all very attractive for hydrogen molecular adsorptions, the synthesis of the desired structure and its postsynthetic maintenance are not well established. Therefore, it is natural that the search for alternative methods continues. Recent DFT calculations revealed that Be 2p orbitals can interact with H2 molecular orbitals in a similar way (65, 66). Because of the light weight and the weaker tendency of aggregation, Be may bear a greater potential from the scientific viewpoint, but its toxicity brings about another problem for the realization. Others previously reported that B-doped fullerene could exhibit a similar Kubas-like H2 adsorption property (66). However, our refined calculations revealed that the suggested states were only locally stable, and the adsorbed H2 molecules were easily dissociated on B sites of the fullerene (67). We also tested a few other light elements but found no comparable orbital interactions. Therefore, the observed orbital interaction between H2 and Be seems quite exceptional among light elements.
Spillover of Hydrogen onto Inert Surfaces
A mechanism that is quite distinct from metal hydride, chemical hydride, and Kubas-enhanced adsorption mechanisms has been suggested for the enhancement of the hydrogen storage at elevated temperature. It has been proposed that accumulated hydrogen molecules on metal sites may be activated through catalysis and then spill over onto the otherwise inert surface, called the receptor (68–71). Platinum nanoparticles are commonly used as catalysts, and activated carbon materials combined with MOFs have been tested as receptors. Experimentalists report a high wt% hydrogen storage in metal–carbon complexes even at room temperature and suggest potential contributions from the spillover mechanism (68, 69). Theorists have suggested a variety of atomistic models related to the hydrogen movement from the catalyst to the receptor, along with the migration within the receptor surfaces (72). Despite a reported increase in the room-temperature storage capacity, the desorption kinetics and reversibility must be improved and the movement of hydrogen atoms and binding states on the receptor surfaces must be unambiguously identified.
Carbon materials have been considered as a good candidate for a cost-effective receptor. We present here the hydrogen migration characteristics of pure graphene, which is a generic sp2-bonded carbon surface. In Fig. 3 we show the hydrogen desorption barrier and the migration barrier on graphene. The stability of the hydrogenated domains and the barriers, either for the desorption or for the migration of hydrogen adatoms, largely depends on the adsorption configuration (73, 74). Fig. 3A, Left, is the case in which two adsorbed hydrogen atoms constitute the metadimer configuration. In this case the balance between the bipartite sublattice of the graphene is violated, and the adsorbed state (Fig. 3A, Left) is thermodynamically unstable relative to the desorbed state (Fig. 3B, Right). When the hydrogen adatoms in the hydrogenated domain are well paired by occupying each bipartite sublattice equally (Fig. 3 B, Left and C, Left), the desorption involves a large energy cost, and the desorption barrier increases substantially. The difference of desorption barriers between Fig. 3 A and B is remarkable. Of greater significance is the even larger migration barrier of two right-most hydrogen adatoms in Fig. 3C. This suggests that, on the pure graphene surface, the unpaired hydrogen adatoms tend to desorb to become molecular hydrogen gas rather than migrate within the plane. Note that the gaseous phase has obvious advantage in the entropy (75). On the other hand, once hydrogen adatoms are paired on the graphene plane, they are likely neither to desorb nor to migrate.
Fig. 3.

Desorption barrier of two hydrogen atoms from (A) the metadimer configuration and (B) the paired six adatoms configuration. (C) Migration barrier of two hydrogen adatoms from the paired 24 adatoms configurations into a separated paradimer configuration. IS, TS, and FS stand for initial, transition, and final states, respectively. Blue balls are hydrogen atoms, and the network of carbon atoms is represented by the wireframe. (C, Right) The two carbon atoms from which hydrogen atoms migrated are highlighted with gray balls. Dashed lines are only a guide for the eye. (C, Inset) A zoomed-in view depicting the direction of the migration. Parts of figure adapted from the published data in Ref (75). Copyrighted by the American Physical Society.
The most important necessary condition for an efficient hydrogen spillover is probably the existence of migrations channel along which hydrogen adatoms can travel in a deep inner surface of the material. Aforementioned features of hydrogen adatoms on graphene suggest that the pure sp2 carbon surfaces are not relevant as a receptor because of the poor kinetics. There must be some extrinsic sources so as to boost the hydrogen migration within the carbon surface. In this respect, we have explored shuttling molecular species that can exchange hydrogen atoms with carbon surfaces (76). In a sense, the spillover mechanism can be regarded as an intermediate storage mechanism between the chemical hydride and the surface adsorption. We stated in the previous sections that poor reversibility and slow kinetics are considered to be the limitation of chemical hydrides. It was also pointed out that under-coordinated transition metals and alkaline metal cations on the surfaces of porous materials have not been shown to be synthesizable yet. Taking all these limitations into consideration, an appropriate choice of a catalyst and a receptor to facilitate spillover-type hydrogen storage would be highly desirable and deserves more focused study in the future. A recent study that demonstrated hydrogen dissociation and ensuing migration on Pd-doped Cu surfaces suggests a clue for a choice of catalyst and receptor (77).
Summary and Future Directions
In this article we have reviewed recent progress on sorption-based hydrogen storage, emphasizing the materials design aspects. Experimental studies concerning the development of large surface porous framework structures were briefly sketched. Computation-based studies that mainly concentrated on enhancing the binding affinity of hydrogen molecules onto surfaces were closely examined and summarized. We classified the mechanisms of hydrogen molecular adsorption (beyond the vdW interaction) into three groups: electrostatic interactions based on alkaline cations, Kubas interactions with open transition metals, and orbital interactions involving Ca and other nontransitional metals. Various efforts were undertaken to take advantage of such metal elements to increase the H2 binding strength onto porous materials. Theoretical issues and methods of computations were also summed up.
Besides the recent focus on the automotive applications, the objectives of hydrogen storage can be defined in a wide perspective. As the global capacity for harvesting renewable (such as solar and wind) energy increases, large-scale energy back-up systems are attracting greater attention than before. To develop a hydrogen storage system for such wide spectra of applications, the volumetric storage density and reversibility can be a target of prime consideration, which imposes different limitations on the choice of storage materials than the vehicular applications. These features should be considered more seriously in future investigations. In any case, although we have searched for many different absorbent materials based on the idea of metal-decorated porous structures, we must concede that much more effort is still required for the synthesis of intended materials. The study should be further extended to postsynthetic management to avoid the problem of oxidation or other contaminations of the adsorption sites. Regarding the spillover mechanism, despite numerous suggestions and debates, the microscopic details of the hydrogen adatoms on receptors have not been fully explored and deserve intensive study. In particular, when the sp2 carbon materials are considered as the receptor, methods for an efficient hydrogen migration along the carbon surfaces need to be developed to establish the hydrogen spillover onto carbon materials.
Up to now the capacity of sorption-based hydrogen storage is acceptable only at very low temperatures, and room temperature reversible storage is yet to be achieved. However, in view of a very short history of this (sorbent-based storage) research area compared with others, the developments in the last decade may be regarded as impressive. We expect that a more thorough study of porous structures and surface adsorption mechanisms in the future will eventually lead to a successful development of efficient and safe storage materials usable under ambient conditions.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) through the Ministry of Education, Science and Technology (MEST) Grant 2006-0093853, the 2010 Korea-Sweden Research Cooperation Program of NRF, and the Core Competence Enhancement Program (2E22790) of the Korea Institute of Science and Technology. N.P. was supported by the Hydrogen Energy R&D Center, a 21st Century Frontier R&D Program, funded by MEST, Korea. Computations were performed through the support of the Korea Institute of Science and Technology Information.
Footnotes
The authors declare no conflict of interest.
References
- 1.Jacobson MZ, Delucchi MA. A path to sustainable energy by 2030. Sci Am. 2009;301(5):58–65. doi: 10.1038/scientificamerican1109-58. [DOI] [PubMed] [Google Scholar]
- 2. George Thomas (2000) Overview of Storage Development DOE Hydrogen Program. Available at http://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/storage.pdf. Accessed October 30, 2012.
- 3.van den Berg AWC, Areán CO. Materials for hydrogen storage: Current research trends and perspectives. Chem Commun (Camb) 2008;(6):668–681. doi: 10.1039/b712576n. [DOI] [PubMed] [Google Scholar]
- 4. US Department of Energy (2011) DOE Targets for Onboard Hydrogen Storage Systems for Light-Duty Vehicles. Available at http://www1.eere.energy.gov/hydrogenandfuelcells/storage/current_technology.html. Accessed October 30, 2012.
- 5.Lim KL, Kazemian H, Yaakob Z, Daud WRW. Solid-state materials and methods for hydrogen storage: A critical review. Chem Eng Technol. 2010;33(2):213–226. [Google Scholar]
- 6.Schlapbach L, Züttel A. Hydrogen-storage materials for mobile applications. Nature. 2001;414(6861):353–358. doi: 10.1038/35104634. [DOI] [PubMed] [Google Scholar]
- 7.Xiong Z, et al. High-capacity hydrogen storage in lithium and sodium amidoboranes. Nat Mater. 2008;7(2):138–141. doi: 10.1038/nmat2081. [DOI] [PubMed] [Google Scholar]
- 8.Zhao D, Yuan D, Zhou H-C. The current status of hydrogen storage in metal-organic frameworks. Energy Environ Sci. 2008;1(2):222–235. [Google Scholar]
- 9.Yaghi OM, et al. Reticular synthesis and the design of new materials. Nature. 2003;423(6941):705–714. doi: 10.1038/nature01650. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa H, et al. Ultrahigh porosity in metal-organic frameworks. Science. 2010;329(5990):424–428. doi: 10.1126/science.1192160. [DOI] [PubMed] [Google Scholar]
- 11.Suh MP, Park HJ, Prasad TK, Lim D-W. Hydrogen storage in metal-organic frameworks. Chem Rev. 2012;112(2):782–835. doi: 10.1021/cr200274s. [DOI] [PubMed] [Google Scholar]
- 12.Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev. 1964;136(3B):B864–B871. [Google Scholar]
- 13.Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133–A1138. [Google Scholar]
- 14.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38(6):3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 15.Baker TA, Head-Gordon M. Modeling the charge transfer between alkali metals and polycyclic aromatic hydrocarbons using electronic structure methods. J Phys Chem A. 2010;114(37):10326–10333. doi: 10.1021/jp105864v. [DOI] [PubMed] [Google Scholar]
- 16.Heyd J, Scuseria GE. Assessment and validation of a screened Coulomb hybrid density functional. J Chem Phys. 2004;120(16):7274–7280. doi: 10.1063/1.1668634. [DOI] [PubMed] [Google Scholar]
- 17.Anisimov VI, Zaanen J, Andersen OK. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys Rev B Condens Matter. 1991;44(3):943–954. doi: 10.1103/physrevb.44.943. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto Y, Miyamoto Y. Ab initio investigation of physisorption of molecular hydrogen on planar and curved graphenes. J Phys Chem B. 2001;105(17):3470–3474. [Google Scholar]
- 19.Møller C, Plesset MS. Note on an approximation treatment for many-electron systems. Phys Rev. 1934;46(7):618–622. [Google Scholar]
- 20.Adams DJ. Chemical potential of hard-sphere fluids by Monte Carlo methods. Mol Phys. 1974;28(5):1241–1252. [Google Scholar]
- 21.Han SS, Mendoza-Cortés JL, Goddard WA., 3rd Recent advances on simulation and theory of hydrogen storage in metal-organic frameworks and covalent organic frameworks. Chem Soc Rev. 2009;38(5):1460–1476. doi: 10.1039/b802430h. [DOI] [PubMed] [Google Scholar]
- 22.Venna SR, Carreon MA. Highly permeable zeolite imidazolate framework-8 membranes for CO2/CH4 separation. J Am Chem Soc. 2010;132(1):76–78. doi: 10.1021/ja909263x. [DOI] [PubMed] [Google Scholar]
- 23.Rosi NL, et al. Hydrogen storage in microporous metal-organic frameworks. Science. 2003;300(5622):1127–1129. doi: 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- 24.Jung DH, et al. Grand canonical Monte Carlo simulation study on the catenation effect on hydrogen adsorption onto the interpenetrating metal-organic frameworks. J Phys Chem B. 2006;110(46):22987–22990. doi: 10.1021/jp065819z. [DOI] [PubMed] [Google Scholar]
- 25.Dincă M, Long JR. Hydrogen storage in microporous metal-organic frameworks with exposed metal sites. Angew Chem Int Ed Engl. 2008;47(36):6766–6779. doi: 10.1002/anie.200801163. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Cohen SM. Postsynthetic modification of metal-organic frameworks. Chem Soc Rev. 2009;38(5):1315–1329. doi: 10.1039/b802258p. [DOI] [PubMed] [Google Scholar]
- 27.Mulfort KL, Farha OK, Stern CL, Sarjeant AA, Hupp JT. Post-synthesis alkoxide formation within metal-organic framework materials: A strategy for incorporating highly coordinatively unsaturated metal ions. J Am Chem Soc. 2009;131(11):3866–3868. doi: 10.1021/ja809954r. [DOI] [PubMed] [Google Scholar]
- 28.Park N, Hong S, Kim G, Jhi SH. Computational study of hydrogen storage characteristics of covalent-bonded graphenes. J Am Chem Soc. 2007;129(29):8999–9003. doi: 10.1021/ja0703527. [DOI] [PubMed] [Google Scholar]
- 29.Cheon YE, Suh MP. Enhanced hydrogen storage by palladium nanoparticles fabricated in a redox-active metal-organic framework. Angew Chem Int Ed Engl. 2009;48(16):2899–2903. doi: 10.1002/anie.200805494. [DOI] [PubMed] [Google Scholar]
- 30.Lim DW, Yoon JW, Ryu KY, Suh MP. Magnesium nanocrystals embedded in a metal–organic framework: Hybrid hydrogen storage with synergistic effect on physi- and chemisorption. Angew Chem. 2012;124(39):9952–9955. doi: 10.1002/anie.201206055. [DOI] [PubMed] [Google Scholar]
- 31.Jeon K-J, et al. Air-stable magnesium nanocomposites provide rapid and high-capacity hydrogen storage without using heavy-metal catalysts. Nat Mater. 2011;10(4):286–290. doi: 10.1038/nmat2978. [DOI] [PubMed] [Google Scholar]
- 32.Zou X, Zhou G, Duan W, Choi K, Ihm J. A chemical modification strategy for hydrogen storage in covalent organic frameworks. J Phys Chem C. 2010;114(31):13402–13407. [Google Scholar]
- 33.Cha MH, Nguyen MC, Lee YL, Im J, Ihm J. Iron-decorated, functionalized metal organic framework for high-capacity hydrogen storage: First-principles calculations. J Phys Chem C. 2010;114(33):14276–14280. [Google Scholar]
- 34.Han SS, William A G., 3rd Lithium-doped metal-organic frameworks for reversible H2 storage at ambient temperature. J Am Chem Soc. 2007;129(27):8422–8423. doi: 10.1021/ja072599+. [DOI] [PubMed] [Google Scholar]
- 35.Blomqvist A, Araújo CM, Srepusharawoot P, Ahuja R. Li-decorated metal-organic framework 5: A route to achieving a suitable hydrogen storage medium. Proc Natl Acad Sci USA. 2007;104(51):20173–20176. doi: 10.1073/pnas.0708603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han SS, Choi SH, Goddard WA. Improved H2 storage in zeolitic imidazolate frameworks using Li+, Na+, and K+ dopants, with an emphasis on delivery H2 uptake. J Phys Chem C. 2011;115(8):3507–3512. [Google Scholar]
- 37.Mendoza-Cortés JL, Han SS, Goddard WA., 3rd High H2 uptake in Li-, Na-, K-metalated covalent organic frameworks and metal organic frameworks at 298 K. J Phys Chem A. 2012;116(6):1621–1631. doi: 10.1021/jp206981d. [DOI] [PubMed] [Google Scholar]
- 38.Lee H, Choi WI, Ihm J. Combinatorial search for optimal hydrogen-storage nanomaterials based on polymers. Phys Rev Lett. 2006;97(5):056104. doi: 10.1103/PhysRevLett.97.056104. [DOI] [PubMed] [Google Scholar]
- 39.Lee H, et al. Ab initio study of dihydrogen binding in metal-decorated polyacetylene for hydrogen storage. Phys Rev B. 2007;76(19):195110. [Google Scholar]
- 40.Lee H, Huang B, Duan W, Ihm J. Releasing H_{2} molecules with a partial pressure difference without the use of temperature. Phys Rev B. 2010;82(8):085439. [Google Scholar]
- 41.Yoon M, Yang S, Wang E, Zhang Z. Charged fullerenes as high-capacity hydrogen storage media. Nano Lett. 2007;7(9):2578–2583. doi: 10.1021/nl070809a. [DOI] [PubMed] [Google Scholar]
- 42.Lochan RC, Head-Gordon M. Computational studies of molecular hydrogen binding affinities: The role of dispersion forces, electrostatics, and orbital interactions. Phys Chem Chem Phys. 2006;8(12):1357–1370. doi: 10.1039/b515409j. [DOI] [PubMed] [Google Scholar]
- 43.Huang B, Lee H, Duan W, Ihm J. Hydrogen storage in alkali-metal-decorated organic molecules. Appl Phys Lett. 2008;93(6):063107–063103. [Google Scholar]
- 44.Kubas GJ. Metal–dihydrogen and σ-bond coordination: The consummate extension of the Dewar–Chatt–Duncanson model for metal–olefin π bonding. J Organomet Chem. 2001;635(1–2):37–68. [Google Scholar]
- 45.Niu J, Rao BK, Jena P. Binding of hydrogen molecules by a transition-metal ion. Phys Rev Lett. 1992;68(15):2277–2280. doi: 10.1103/PhysRevLett.68.2277. [DOI] [PubMed] [Google Scholar]
- 46.Gagliardi L, Pyykkö P. How many hydrogen atoms can be bound to a metal? Predicted MH12 species. J Am Chem Soc. 2004;126(46):15014–15015. doi: 10.1021/ja045991l. [DOI] [PubMed] [Google Scholar]
- 47.Yildirim T, Ciraci S. Titanium-decorated carbon nanotubes as a potential high-capacity hydrogen storage medium. Phys Rev Lett. 2005;94(17):175501. doi: 10.1103/PhysRevLett.94.175501. [DOI] [PubMed] [Google Scholar]
- 48.Sun YY, Kim YH, Zhang SB. Effect of spin state on the dihydrogen binding strength to transition metal centers in metal-organic frameworks. J Am Chem Soc. 2007;129(42):12606–12607. doi: 10.1021/ja0740061. [DOI] [PubMed] [Google Scholar]
- 49.Bhattacharya A, Bhattacharya S, Majumder C, Das GP. Transition-metal decoration enhanced room-temperature hydrogen storage in a defect-modulated graphene Sheet. J Phys Chem C. 2010;114(22):10297–10301. [Google Scholar]
- 50.Hamaed A, Trudeau M, Antonelli DM. H2 storage materials (22 KJ/mol) using organometallic Ti fragments as σ-H2 binding sites. J Am Chem Soc. 2008;130(22):6992–6999. doi: 10.1021/ja710288g. [DOI] [PubMed] [Google Scholar]
- 51.Hamaed A, Mai HV, Hoang TKA, Trudeau M, Antonelli D. Functionalized porous silicas with unsaturated early transition metal moieties as hydrogen storage materials: Comparison of metal and oxidation state. J Phys Chem C. 2010;114(18):8651–8660. [Google Scholar]
- 52.Kim TS, Kim KJ, Jo SK, Lee J. Interaction of D2 with Ti-adsorbed polyaniline and implication for hydrogen storage. J Phys Chem B. 2008;112(51):16431–16436. doi: 10.1021/jp8033243. [DOI] [PubMed] [Google Scholar]
- 53.Phillips AB, Shivaram BS. High capacity hydrogen absorption in transition metal-ethylene complexes observed via nanogravimetry. Phys Rev Lett. 2008;100(10):105505. doi: 10.1103/PhysRevLett.100.105505. [DOI] [PubMed] [Google Scholar]
- 54.Yoon M, et al. Calcium as the superior coating metal in functionalization of carbon fullerenes for high-capacity hydrogen storage. Phys Rev Lett. 2008;100(20):206806. doi: 10.1103/PhysRevLett.100.206806. [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Sun Q, Jena P, Kawazoe Y. Theoretical study of hydrogen storage in Ca-coated fullerenes. J Chem Theory Comput. 2009;5(2):374–379. doi: 10.1021/ct800373g. [DOI] [PubMed] [Google Scholar]
- 56.Lee H, Ihm J, Cohen ML, Louie SG. Calcium-decorated graphene-based nanostructures for hydrogen storage. Nano Lett. 2010;10(3):793–798. doi: 10.1021/nl902822s. [DOI] [PubMed] [Google Scholar]
- 57.Kim G, Jhi SH, Lim S, Park N. Crossover between multipole Coulomb and Kubas interactions in hydrogen adsorption on metal-graphene complexes. Phys Rev B. 2009;79(15):155437. [Google Scholar]
- 58.Cha J, Lim S, Choi CH, Cha MH, Park N. Inaccuracy of density functional theory calculations for dihydrogen binding energetics onto Ca cation centers. Phys Rev Lett. 2009;103(21):216102. doi: 10.1103/PhysRevLett.103.216102. [DOI] [PubMed] [Google Scholar]
- 59.Ohk Y, Kim Y-H, Jung Y. Comment on “Inaccuracy of density functional theory calculations for dihydrogen binding energetics onto Ca cation centers”. Phys Rev Lett. 2010;104(17) doi: 10.1103/PhysRevLett.104.179601. 179601, author reply 179602. [DOI] [PubMed] [Google Scholar]
- 60.Cha J, Choi CH, Park N. Cha, Choi, and Park reply. Phys Rev Lett. 2010;104(17):179602. [Google Scholar]
- 61.Gunnarsson O, Jones RO. Total-energy differences: Sources of error in local-density approximations. Phys Rev B Condens Matter. 1985;31(12):7588–7602. doi: 10.1103/physrevb.31.7588. [DOI] [PubMed] [Google Scholar]
- 62.Bajdich M, Reboredo FA, Kent PRC. Quantum Monte Carlo calculations of dihydrogen binding energetics on Ca cations: An assessment of errors in density functionals for weakly bonded systems. Phys Rev B. 2010;82(8):081405. [Google Scholar]
- 63.Purwanto W, Krakauer H, Virgus Y, Zhang S. Assessing weak hydrogen binding on Ca+ centers: An accurate many-body study with large basis sets. J Chem Phys. 2011;135(16):164105–164111. doi: 10.1063/1.3654002. [DOI] [PubMed] [Google Scholar]
- 64.Cha J, Choi CH, Park N. Ab initio study of Kubas-type dihydrogen fixation onto d-orbital states of Ca adatoms. Chem Phys Lett. 2011;513(4–6):256–260. http://www.sciencedirect.com/science/journal/00092614. [Google Scholar]
- 65.Lee H, Huang B, Duan W, Ihm J. Beryllium-dihydrogen complexes on nanostructures. Appl Phys Lett. 2010;96(14):143120–143123. [Google Scholar]
- 66.Kim YH, Zhao Y, Williamson A, Heben MJ, Zhang SB. Nondissociative adsorption of H2 molecules in light-element-doped fullerenes. Phys Rev Lett. 2006;96(1):016102. doi: 10.1103/PhysRevLett.96.016102. [DOI] [PubMed] [Google Scholar]
- 67.Lee H, et al. Room-temperature dissociative hydrogen chemisorption on boron-doped fullerenes. Phys Rev B. 2008;77(23):235101. [Google Scholar]
- 68.Li Y, Yang RT. Hydrogen storage in metal-organic frameworks by bridged hydrogen spillover. J Am Chem Soc. 2006;128(25):8136–8137. doi: 10.1021/ja061681m. [DOI] [PubMed] [Google Scholar]
- 69.Tsao CS, et al. Effect of catalyst size on hydrogen storage capacity of Pt-impregnated active carbon via spillover. J Phys Chem Lett. 2010;1(7):1060–1063. [Google Scholar]
- 70.Psofogiannakis GM, Froudakis GE. DFT study of hydrogen storage by spillover on graphite with oxygen surface groups. J Am Chem Soc. 2009;131(42):15133–15135. doi: 10.1021/ja906159p. [DOI] [PubMed] [Google Scholar]
- 71.Psofogiannakis GM, Froudakis GE. Fundamental studies and perceptions on the spillover mechanism for hydrogen storage. Chem Commun (Camb) 2011;47(28):7933–7943. doi: 10.1039/c1cc11389e. [DOI] [PubMed] [Google Scholar]
- 72.Lin Y, Ding F, Yakobson BI. Hydrogen storage by spillover on graphene as a phase nucleation process. Phys Rev B. 2008;78(4):041402. [Google Scholar]
- 73.Stojkovic D, Zhang P, Lammert PE, Crespi VH. Collective stabilization of hydrogen chemisorption on graphenic surfaces. Phys Rev B. 2003;68(19):195406. [Google Scholar]
- 74.Sofo JO, Chaudhari AS, Barber GD. Graphane: A two-dimensional hydrocarbon. Phys Rev B. 2007;75(15):153401. [Google Scholar]
- 75.Han SS, Jung H, Jung DH, Choi SH, Park N. Stability of hydrogenation states of graphene and conditions for hydrogen spillover. Phys Rev B. 2012;85(15):155408. [Google Scholar]
- 76.Han SS, Kim H, Park N. Effect of shuttling catalyst on the migration of hydrogen adatoms: A strategy for the facile hydrogenation of graphene. J Phys Chem C. 2011;115(50):24696–24701. [Google Scholar]
- 77.Kyriakou G, et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science. 2012;335(6073):1209–1212. doi: 10.1126/science.1215864. [DOI] [PubMed] [Google Scholar]