

Abstract
The nematode Caenorhabditis elegans is hypersusceptible to Burkholderia pseudomallei infection. However, the virulence mechanisms underlying rapid lethality of C. elegans upon B. pseudomallei infection remain poorly defined. To probe the host-pathogen interaction, we constructed GFP-tagged B. pseudomallei and followed bacterial accumulation within the C. elegans intestinal lumen. Contrary to slow-killing by most bacterial pathogens, B. pseudomallei caused fairly limited intestinal lumen colonization throughout the period of observation. Using grinder-defective mutant worms that allow the entry of intact bacteria also did not result in full intestinal lumen colonization. In addition, we observed a significant decline in C. elegans defecation and pharyngeal pumping rates upon B. pseudomallei infection. The decline in defecation rates ruled out the contribution of defecation to the limited B. pseudomallei colonization. We also demonstrated that the limited intestinal lumen colonization was not attributed to slowed host feeding as bacterial loads did not change significantly when feeding was stimulated by exogenous serotonin. Both these observations confirm that B. pseudomallei is a poor colonizer of the C. elegans intestine. To explore the possibility of toxin-mediated killing, we examined the transcription of the C. elegans ABC transporter gene, pgp-5, upon B. pseudomallei infection of the ppgp-5::gfp reporter strain. Expression of pgp-5 was highly induced, notably in the pharynx and intestine, compared with Escherichia coli-fed worms, suggesting that the host actively thwarted the pathogenic assaults during infection. Collectively, our findings propose that B. pseudomallei specifically and continuously secretes toxins to overcome C. elegans immune responses.
Keywords: Burkholderia pseudomallei, Caenorhabditis elegans, host-pathogen interaction, intestinal lumen colonization, toxin
Introduction
Burkholderia pseudomallei is a Gram-negative saprophyte that typically inhabits muddy soil and stagnant water throughout Southeast Asia and northern Australia.1 When acquired by humans and animals, B. pseudomallei can cause melioidosis, a life-threatening disease that, to this day, still presents a danger to most parts of the tropics.2 Decades of research on B. pseudomallei has only revealed how versatile this pathogen is, for example, it can (1) infect a multitude of organisms and invade a wide range of cell types,3,4 (2) result in a broad spectrum of clinical manifestations,5 (3) resist many clinical antimicrobials6 and (4) survive extremely harsh environmental conditions.7 Yet, the molecular mechanisms by which B. pseudomallei evades or modulates host immune responses remain elusive. Of the various forms of melioidosis, acute melioidosis raises the greatest medical concern owing to its high mortality rate regardless of appropriate antibiotic treatments.8 It is well documented that acute melioidosis tends to affect humans with risk factors such as diabetes mellitus but rarely immunocompetent individuals.9 For this reason, host models with clinically relevant predisposing backgrounds or sensitivities are particularly attractive in melioidosis research. To this end, several groups have recently begun to exploit host models engineered to mimic the risk factors for melioidosis, such as type 1 and 2 diabetic mice, in an effort to elucidate the attributes of B. pseudomallei virulence in the corresponding predisposed individuals.10-12
Over the last decade, there has been a growing appreciation that Caenorhabditis elegans can serve as a simple surrogate host for modeling bacterial diseases.13 C. elegans is also deemed a relevant host model for studying acute melioidosis. Diabetic patients prone to acute melioidosis have impaired innate immune responses such as macrophage phagocytosis and migration.14 C. elegans lacks circulating phagocytes and some innate immune system components essential for fighting an acute B. pseudomallei infection;15,16 however, it is protected by an innate immune system conserved with that in humans.15 Akin to acute melioidosis individuals, C. elegans is highly susceptible to B. pseudomallei infection,17-19 which strongly suggests that B. pseudomallei executes its pathogenicity by suppressing or breaching the host innate immune system. In addition, it has been shown that B. pseudomallei does not persist within C. elegans,19 similar to observations in acute melioidosis cases.20 These infection characteristics together suggest that C. elegans is an excellent model to simultaneously dissect the evolutionarily conserved determinants of B. pseudomallei virulence as well as host innate immune defense mechanisms.
A myriad of bacterial virulence mechanisms has been unraveled using C. elegans.21,22 One of the strategies commonly employed by bacterial pathogens in establishing infection in mammals is adhesion and colonization of host tissues.23 Likewise, virtually all Gram-positive and Gram-negative pathogens studied to date can colonize the brush border microvilli of C. elegans after they successfully escape the grinder and resist antimicrobial peptides in the pharynx, ultimately leading to colonization and distension of the intestinal lumen.13,21,22 In general, this active infectious process takes place only when the pathogens are cultured on a minimal or “slow-killing” medium, and the extent of colonization often correlates with host killing. Nevertheless, subtle differences have been noticed in terms of the intestinal lumen colonization by these pathogens. For example, Pseudomonas aeruginosa and Staphylococcus aureus grossly colonize and distend the worm intestinal lumen but do not persist within the host, unlike other pathogens such as Enterococcus faecalis, enteropathogenic Escherichia coli (EPEC), Salmonella Typhimurium, Serratia marcescens and Yersinia enterocolitica.22,24,25
Previously, we have established a C. elegans-B. pseudomallei model system and demonstrated that a clinical isolate, Human R15 (referred to as Bp R15 henceforth), was able to rapidly kill BALB/c mice and C. elegans.19 However, how Bp R15 interacts with C. elegans to elicit extreme symptoms and death is not completely understood. In the present study, we introduced a gfp construct into Bp R15 to visualize the passage of the pathogen across the host. Our results revealed a novel host-pathogen interaction in which the virulent Bp R15 failed to fully colonize C. elegans intestinal lumen under “slow killing” conditions, even though facilitated with host grinder dysfunction and accelerated feeding. With the use of a C. elegans detoxification gene reporter strain, we provided evidence that the rapid host killing may be mediated by bacterial toxin secretion.
Results
Construction and evaluation of GFP-expressing Bp R15
To visualize the events following Bp R15 infection of C. elegans, we first labeled Bp R15 with GFP. To prevent overexpression of GFP that may contribute to reduced bacterial fitness and infectivity, we fused a moderate-strength gene promoter, PompA,26 with a gfp coding sequence and cloned the DNA construct into a low-copy-number pBBR-derived vector (~20–30 copies/cell)27 to minimize potential burden imposed on the bacteria. Electroporation of the resultant recombinant plasmid, pSMompAgfp (Fig. S1A), into Bp R15 successfully generated R15-GFP with observable fluorescence under both in vitro (Fig. S1B) and in vivo (Fig. 1A–C) conditions.

Figure 1. R15-GFP displayed limited intestinal lumen colonization during C. elegans N2 infection. (A) A low number of R15-GFP cells were present along the intestinal lumen after 4 h of infection. (B) At 28 h post-infection, the major portion of the worm intestinal lumen was void of R15-GFP except for the anterior intestine which was grossly colonized and distended. Note that the worm body shrunk and the nose was contracted at this time point. (C) Three representative worms here depict an increase in R15-GFP colonization within the anterior intestine upon prolonged pathogen exposure. After 22 h post-infection, both pharyngeal bulbs of the worm were distorted and contained proliferating bacteria. (D) Shown are the three categories of colonization criteria that were used to classify the infected worms. (E) Stacked bars correspond to mean proportion of the infected N2 population encompassing all three colonization categories (n = 75–100). (F) The graph shows the number of R15-GFP colonizing N2 worm intestines throughout the infection. (G) Stacked bars represent mean proportion of infected tnt-3(aj3) population within the three colonization categories (n = 75–100). (H) The graph shows the number of R15-GFP colonizing tnt-3(aj3) worms up to 11 h infection. (F and H) Each marker corresponds to the average bacterial CFU extracted from 10 infected worms; horizontal lines represent geometric means. Light blue arrows in (A–C) point either to individual R15-GFP cells or colonization as well as distension of the intestinal lumen. Asterisks in (A) and (B) mark the terminal pharyngeal bulb. Dotted circles in (C) mark the anterior and terminal pharyngeal bulbs. Images (A and B) were taken at 100× magnification while (C) was captured at 400x magnification. Scale bars in (B?D) represent 200 µm, 50 µm and 30 µm, respectively.
Next, we tested the extrachromosomal stability of pSMompAgfp in R15-GFP under non-selective conditions. Loss of fluorescence was observed in a subpopulation of the bacteria after just one passage without chloramphenicol (Cm) (Fig. S2A), suggesting that pSMompAgfp was unstable and therefore Cm was required to select for R15-GFP. To verify that addition of Cm would not confound analyses of downstream infection assays, the C. elegans lifespan on E. coli OP50 plates supplemented with Cm was determined. The results showed that 100 µg/ml of Cm did not compromise worm lifespan and could be included in the assay media (Fig. S2B). The viability of R15-GFP could not be compared with that of Bp R15 as 100 µg/ml Cm is considerably bacteriostatic in the latter (data not shown).
As R15-GFP was to be used as a surrogate for Bp R15, we assessed the virulence of R15-GFP in C. elegans N2 to confirm that constitutive GFP expression did not interfere with the bacteria’s virulence. No significant difference was observed between Bp R15 and R15-GFP virulence (p > 0.01, Log-rank test) (Fig. S2C). In addition, both R15-GFP and Bp R15 were equally capable of causing intestinal discoloration, body shrinkage and contracted nose in C. elegans throughout the infection.
Bp R15 does not heavily colonize the C. elegans intestinal lumen
It has been suggested that B. pseudomallei, like other Gram-negative human pathogens such as P. aeruginosa, also gains entry into and colonizes the worm intestine.22 We followed bacterial accumulation within the C. elegans alimentary tract upon feeding on a R15-GFP lawn cultured on NG agar. As the presence of eggs in the C. elegans uterus complicates visualization and imaging of intestinal bacterial colonization, we used sterile (Glp) worms whose cdc-25.1 gene has been silenced by RNAi. We previously noted that susceptibility of Glp worms to Bp R15 infection was similar to wild-type N2 worms.19 After 4 h of infection, we did not observe any colonization in the infected worms but merely single R15-GFP cells irregularly interspersed along the intestinal lumen (Fig. 1A). As the infection proceeded, a progressive increase in the colonization and distension of the worm anterior intestinal lumen was observed. Nevertheless, even up to 28 h post-infection, the majority of infected worms were not fully colonized. Instead, only the anterior intestinal lumen was, to a great extent, colonized and distended with a clump of R15-GFP (Fig. 1B). In addition, most infected worms contained R15-GFP cells in their pharyngeal tracts, and the pharynx was noticeably plugged with bacteria during late infection, with R15-GFP spanning the buccal cavity through the terminal bulb (Fig. 1C). It is worth noting that R15-GFP barely colonized the mid- and posterior intestine. We also noticed that the greatly distended anterior intestinal lumen was not always densely filled with R15-GFP cells, suggesting that the distension was due to secretion of an unknown extracellular matrix similar to that previously observed in a P. aeruginosa infection.28 In all the worms examined, R15-GFP colonization was confined to the pharyngeal tract and intestinal lumen, suggesting that the infection was extracellular. Since we did not observe bacterial entry via other parts of the worm body such as the cuticle, anus or vulva, we conclude that B. pseudomallei is an intestinal pathogen of C. elegans.
Detailed examination of the infected worms revealed that not every single worm displayed similar degrees of intestinal lumen colonization. To ensure a representative microscopic observation, we conducted a quantitative infection assay whereby 75–100 infected worms were scored at the same time points and classified based on the severity of intestinal lumen colonization. As depicted in Figure 1D, worms were scored as “full” when a tract of R15-GFP was seen from the anterior to posterior intestinal lumen, regardless of the density of bacteria. Visible but not full colonization of R15-GFP anywhere along the intestinal lumen was categorized as “partial” whereas when none or only scattered R15-GFP cells were present in the lumen observed at 400× magnification, this was classified as “undetectable.” As noted above, only limited R15-GFP colonization was observed in the infected worm population. We found that less than 10% of the population were fully colonized even up to 28 h post-exposure (Fig. 1E), a time point at which a majority of the worms were severely ill. This implies that Bp R15 is unable to substantially colonize the C. elegans intestinal lumen on minimal media over the course of infection.
To the best of our knowledge, this observation of limited intestinal lumen colonization has not yet been reported for “slow killing” of C. elegans. To further validate our observation, we enumerated the number of colonizing bacteria by performing a colony forming unit (CFU) assay under the same infection conditions. By using the method described by Shapira and Tan,29 we were able to achieve CFU counts of 103–105 bacteria/worm for worms exposed to P. aeruginosa PA14 (PA14) (data not shown). On the other hand, for the R15-GFP infection, fairly low numbers of bacteria were enumerated within the infected worms throughout the infection period (in the range of 10–102 CFU/worm; Fig. 1F), reinforcing the belief that Bp R15 did not substantially colonize the host intestinal lumen.
It is thought that the C. elegans pharyngeal grinder that crushes ingested bacteria before thrusting the bacterial corpses into the intestine30 might hinder Bp R15 from efficiently colonizing its intestinal lumen. In worms infected by P. aeruginosa and S. Typhimurium, abrogating the grinder function accelerated the intestinal lumen colonization, leading to faster host killing.31,32 Hence, we tested this hypothesis by exposing a grinder-defective mutant strain, tnt-3(aj3), to R15-GFP and monitored the extent of intestinal lumen colonization at 4, 6, 8 and 11 h post-exposure. The tnt-3(aj3) mutant bears a recessive mutation in the troponin T gene which disrupts the grinder function; consequently, more intact bacteria can enter its intestine.32,33 We selected the aforementioned time points because a significant portion of the infected mutant population was severely sick beyond 12 h of infection (Yee and Nathan, in preparation). As expected, we noted a marked increase in R15-GFP colonization within the tnt-3(aj3) mutant intestinal lumen, but the hindgut was devoid of R15-GFP cells (data not shown). To quantify tnt-3(aj3) colonization, we scored 75–100 worms based on the same set of colonization criteria depicted in Figure 1D. Indeed, there was a significant increase in the number of fully colonized worms compared with N2 worms; however, the category percentage was only less than 70% at 11 h post-infection (Fig. 1G). We also conducted the CFU assay to quantitatively measure R15-GFP colonization within infected tnt-3(aj3) mutant worms. Consistent with the visualization assay, the number of R15-GFP in infected tnt-3(aj3) worms did not significantly increase over the course of infection (Fig. 1H). Therefore, Bp R15 is inferior to other well-studied intestinal pathogens in terms of host colonization.
C. elegans defecation and feeding do not appear to limit Bp R15 colonization of the intestinal lumen
In our previous study, we demonstrated that C. elegans was able to eliminate ingested Bp R15 from its intestinal lumen via defecation when the infected worms were shifted to an innocuous food source.19 Hence, if the infected worms were actively defecating during Bp R15 infection, this would explain the lack of intestinal lumen colonization by R15-GFP. To address this issue, an assay was performed to compare the defecation rate between infected and uninfected worms. Defecation is a rhythmic behavior that consists of three consecutive muscle contractions—posterior body contraction (pBoc), anterior body contraction (aBoc) and expulsion (Exp).34 Owing to the consistency of the defecation motor program, measurement of defecation rate typically involves determining the frequency of pBoc.35 However, we persistently observed that most Bp R15-infected worms were highly lethargic and failed to expulse following every pBoc. In lieu of this observation, we chose to enumerate the expulsion rate instead as a representation of defecation rates. In infected worms, defecation rates were significantly lower compared with E. coli OP50-fed worms throughout the infection, except at 8 h post-infection (Fig. 2A). Suffice to say, the inadequate Bp R15 colonization of the C. elegans intestinal lumen is not a result of routine worm defecation.
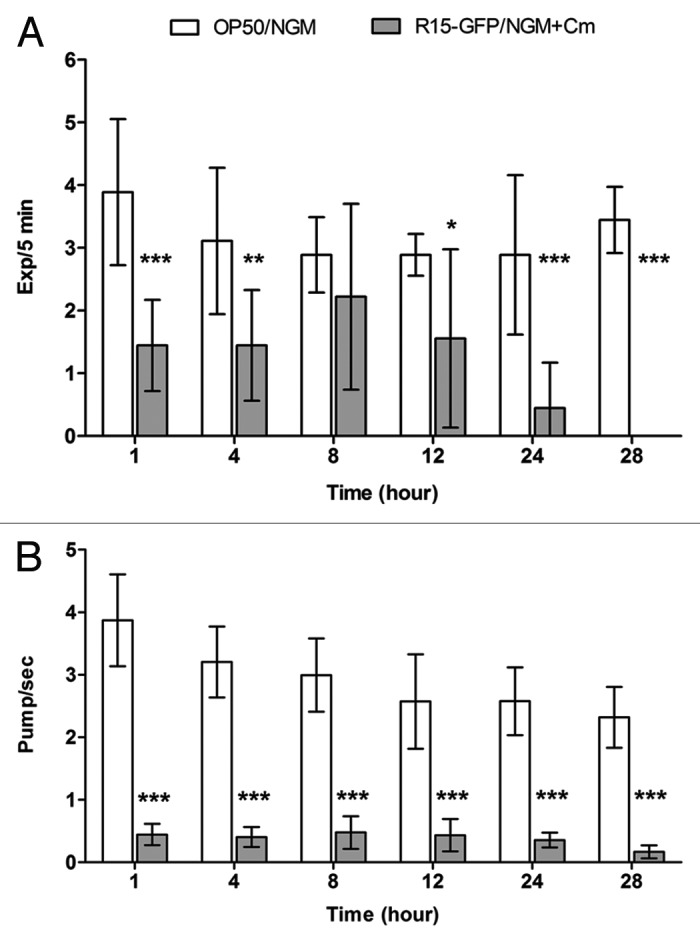
Figure 2.Bp R15 infection caused a significant reduction in both C. elegans defecation and feeding rates. (A) Bars correspond to mean ± SD of the frequency of Exp over 5 min per worm (n = 3). *p < 0.05,**p < 0.01, ***p < 0.001 (Mann-Whitney U-test). (B) Bars correspond to mean ± SEM of pharyngeal pumping per second for each worm (n = 30). ***p < 0.001 (Student’s t-test). Graphs (A and B) share the same labels, which are shown above (A).
In response to pathogen infection, C. elegans can orchestrate its behavior, such as limiting its ingestion of bacteria, in order to promote survival.13 Therefore, it was possible that the infected worms adjusted their feeding to restrict the uptake of bacteria, hence limiting Bp R15 colonization. To clarify this, we measured host pharyngeal pumping rates during Bp R15 infection. Surprisingly, we observed an almost immediate decline in worm pharyngeal pumping rates (average < 1 pump/sec) upon infection (Fig. 2B). Concomitantly, we also observed convulsion-like head contractions and relaxation in the infected worms similar to but slower than that described by Williams et al.36 With continuous monitoring of the pharyngeal pumping of infected worms, it was observed that the worms did not completely cease feeding but instead, extended the intervals between pumps, i.e., the infected worms fed on Bp R15 only intermittently.
The dramatic decline in Bp R15-infected C. elegans pharyngeal pumping rates raised the question of the possible impact of host feeding on the number of bacteria colonizing the intestine.30 To test whether the decrease in feeding rate was a host strategy to mitigate Bp R15 colonization, we supplemented the assay medium with 5 mM serotonin, a neurotransmitter that can stimulate C. elegans feeding, to reverse the sluggish worm pharyngeal pumping. This drug concentration has been shown to increase feeding rates to 3–4 pumps/sec in E. coli-fed worms.37 We first assessed the drug efficacy by demonstrating that 5 mM serotonin enhanced pharyngeal pumping rates in worms fed on R15-GFP (Fig. 3A, p < 0.01, Student’s t-test), although the stimulatory effect weakened over time. To ensure that the exogenous serotonin did not significantly affect defecation, we measured the worm defecation rate in the presence of serotonin. A higher rate of defecation was indeed apparent in the treated worms, but the increase was not significant (Fig. 3B, p > 0.05, Mann-Whitney U-test). Next, we performed a CFU assay on the serotonin-stimulated infected worms, with untreated infected worms as a control. However, we failed to observe any significant difference in the number of colonizing bacteria in the serotonin-induced worms (Fig. 3C, p > 0.05, Mann-Whitney U-test). While this result suggests that the reduction in host feeding rates did not contribute to the limited intestinal lumen colonization, it also implies that the primary virulence mechanism employed by Bp R15 in C. elegans killing is distinct from intestinal lumen colonization.
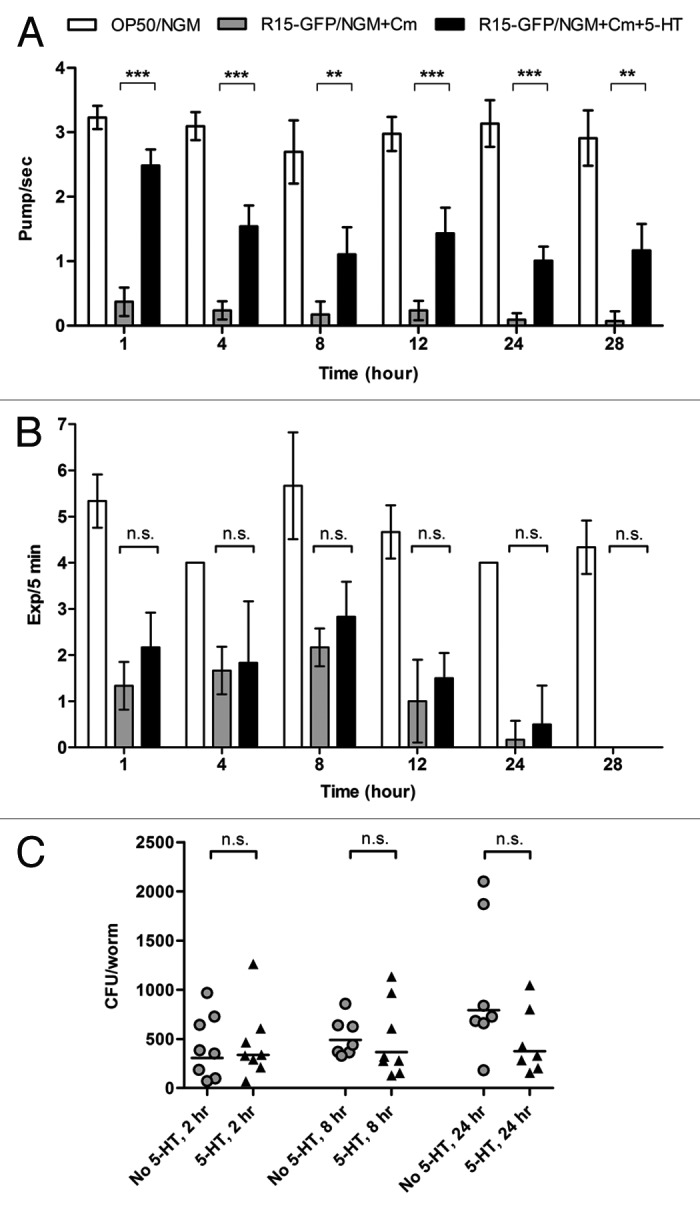
Figure 3. Serotonin-induced feeding did not improve intestinal lumen colonization by Bp R15. (A) The efficacy of 5 mM serotonin toward feeding stimulation is shown by the significant increase in pharyngeal pumping rates in worms exposed to R15-GFP. Bars correspond to pharyngeal pumping mean ± SEM (pump/sec for each worm) (n = 30). **p < 0.01, ***p < 0.001 (Student’s t-test). (B) Defecation rates of infected worms were not significantly affected by the addition of 5 mM serotonin into the assay medium. Bars correspond to the mean ± SD of Exp/5 min for each worm (n = 3). (C) Despite increased feeding rates, the number of colonizing R15-GFP did not change significantly within the serotonin-induced infected worms. Each marker corresponds to the average bacterial CFU of 10 infected worms; horizontal lines represent the geometric means of CFU. Graphs (B and C) were analyzed using the Mann-Whitney U-test. Graphs (A–C) share the same labels: white bar, N2 worms fed on E. coli OP50; gray bars/circles, worms infected with R15-GFP; black bars/triangles, serotonin-induced worms exposed to R15-GFP. Abbreviations: 5-HT, 5-hydroxytryptamine or serotonin; n.s., not significant.
C. elegans strongly induces the pgp-5 gene in response to Bp R15 infection
As the degree of intestinal lumen colonization was not commensurate with the level of Bp R15 virulence and its ability to kill C. elegans, we asked whether rapid host killing was mediated by secreted bacterial toxins. A previous study has demonstrated that C. elegans pgp-5 plays a significant role in the worm’s defense against P. aeruginosa and S. Typhimurium infections as well as cadmium and copper intoxication.38 C. elegans pgp-5 encodes a transmembrane P-glycoprotein, a conserved member of the ATP-binding cassette (ABC) transporter superfamily, which is predicted to mediate detoxification of exogenous toxins by the host.38 To determine changes in host pgp-5 expression upon Bp R15 infection, we measured pgp-5 transcript levels in Bp R15-infected worms using quantitative RT-PCR (qRT-PCR). We found that the transcription of pgp-5 was highly induced at 4 and 12 h post-infection (140- and 290-fold, respectively; Fig. 4A).

Figure 4. Transcription of host pgp-5 encoding the detoxification ABC-transporter was strongly induced in C. elegans infected by Bp R15. (A) qRT-PCR analysis of Bp R15-infected worms shows significant fold change of host pgp-5 transcription relative to E. coli-fed worms at 4 and 12 h of infection. Bars represent mean values ± SD. (B) Green fluorescence reflecting pgp-5 gene induction was weakly visible in uninfected ppgp-5::gfp transgenic worms. (C) However, at 28 h post-infection, the transgenic worms exhibited bright green fluorescence in the pharyngeal muscle, more prominently from the metacorpus to terminal bulb. Other loci of pgp-5 expression observed included (C--E) the entire intestine and (D) gonad. For (B--E), DIC images are displayed as insets either on the lower left or on upper right of the corresponding images. (F) Shown in the upper panel are the criteria used for scoring ppgp-5::gfp transgene expression upon Bp R15 infection. Bars in the lower panel correspond to the mean ± SD of pgp-5 induction scores (expressed in arbitrary units) for each worm (n = 20). (G) C. elegans exhibited increased susceptibility to Bp R15 infection upon RNAi knock-down of pgp-5. Graph shows the mean ± SD of the live worm fraction (n = 120). Images (B–E) are overlaid images captured using I3 and N2.1 filter cubes, therefore the green fluorescence region appears slightly yellowish while the non-specific gut autofluorescence is in orange. Asterisks in (B and C) mark the terminal pharyngeal bulb whereas those in (F) mark the worm nose. The red asterisk in (D) points to the vulva. Images (B–E) were taken at 400× magnification while images in (F) were captured at 100× magnification. Scale bars in (E and F) represent 50 µm and 100 µm, respectively.
To determine the spatio-temporal tissue distribution of pgp-5 expression, we infected ppgp-5::gfp transgenic worms with Bp R15 and monitored changes in the transgene expression using E. coli OP50-fed worms as a control. These transgenic worms harbor an integrated pgp-5::gfp construct made by fusing the 300-bp pgp-5 promoter region to a gfp coding sequence, thus any green fluorescence observed would reflect the transcription of pgp-5. Consistent with the findings of Kurz et al.,38 worms fed on E. coli exhibited very weak green fluorescence in the intestine throughout the observation period (Fig. 4B). In contrast, bright green fluorescence was visible in the pharynx (from isthmus to terminal bulb) and intestine of Bp R15-infected worms as early as 4 h post-exposure (data not shown). We also noted that the green fluorescence gradually increased in intensity over time. At 28 h post-infection, intense green fluorescence was apparent in the pharynx (from the isthmus through the terminal bulb), intestine as well as gonad (Fig. 4C–E). To confirm the steady increment of pgp-5 induction in the worm population upon Bp R15 infection, we also visually quantified the green fluorescence during the assay. Twenty live worms were randomly picked at 4, 12 and 28 h post-infection, observed under 100× magnification and scored based on defined criteria (Fig. 4F, upper panel). Briefly, a score of “1” was assigned to worms with very dim green fluorescence. Worms with detectable green fluorescence in the pharynx and intestine were given a score of “2” while a score of “3” was assigned to worms displaying intense fluorescence in the pharynx, intestine and gonad. This visual quantification confirmed that the pgp-5 induction level was significantly higher in Bp R15-infected worms (Fig. 4F, lower panel, p < 0.001, Mann-Whitney U-test), thus underscoring a strong yet specific detoxification process that occurred in response to Bp R15 infection.
In light of the high and specific induction of pgp-5 expression, we next asked if disrupting gene expression would render C. elegans more susceptible to Bp R15 infection. To answer this question, we fed rrf-3(pk1426);glp-4(bn2) double mutant worms with E. coli expressing dsRNA directed against pgp-5 and assessed survival upon Bp R15 infection at 25°C. RNAi knock-down of pgp-5 expression indeed resulted in greater susceptibility of C. elegans toward Bp R15; however, the difference in survival was not significant compared with worms treated with the L4440 vector (TDmean: 40.19 ± 0.69 and 41.86 ± 0.69 h, respectively; p = 0.115, Log-rank test). To better resolve the subtle difference in survival observed at 25°C, we repeated the assay at 16°C. We demonstrated that C. elegans was significantly more susceptible to Bp R15 killing upon RNAi silencing of pgp-5; TDmean values for pgp-5 RNAi-treated and control worms were 99.77 ± 1.47 h and 108.28 ± 1.68 h, respectively (Fig. 4G, p < 0.0001, Log-rank test), thus highlighting a protective role for pgp-5 during Bp R15 infection. This increased resolution of host susceptibility at lower temperatures was also observed in P. aeruginosa and S. Typhimurium infections.38
Discussion
Although C. elegans has long been established as a model organism for investigating B. pseudomallei pathogenicity, much remains to be learnt about how this pathogen severely damages the host. Previous studies have asserted that B. pseudomallei infection of C. elegans is an active process involving both live bacteria and continuous intoxication.17-19 However, at the organismal level, whether the interaction between B. pseudomallei and C. elegans is similar to other bacterial species, has yet to be clarified. Our earlier study had demonstrated that Bp R15 did not persistently colonize the C. elegans intestinal lumen.19 In this study, we sought to investigate the host-pathogen interaction in greater detail, hoping to shed light on the possible complex mechanism(s) by which B. pseudomallei kills C. elegans.
As B. pseudomallei is phylogenetically related to P. aeruginosa and they share a number of features in terms of infection (both are opportunistic pathogens that infect human lungs), we initially anticipated that R15-GFP would colonize the worm intestinal lumen to an extent comparable to that of PA14 but with faster kinetics owing to the lower TDmean of Bp R15-infected worms (~36 h, Fig. S2C) compared with PA14-infected worms (TDmean of ~75 h).38 However, we demonstrated that Bp R15 failed to extensively colonize the C. elegans intestinal lumen even during late infection. Furthermore, the bacterial CFU counts of Bp R15-infected worms are much lower (in the magnitude of 102 CFU/worm) compared with other bacterial infections such as Y. enterocolitica, which can reach up to ~106 CFU/worm.25 A similar lack of intestinal lumen colonization was also observed when Bp R15 was replaced with the Bp K96243 reference strain.39 These findings collectively suggest that virulent B. pseudomallei isolates are universally incapable of heavily colonizing the C. elegans intestinal lumen. This suggestion is further supported by the finding that Bp R15 does not fully colonize the tnt-3(aj3) mutant worm population over the course of infection.
The relatively poor intestinal lumen colonization ability of B. pseudomallei could also be a consequence of other host barriers or factors that interfere or prevent Bp R15 from efficient colonization. For example, the hostile intestinal lumen microenvironment may impede B. pseudomallei intraluminal growth or replication. On the other hand, unfavorable host factors may include suboptimal pH (~4.1),40 temperature (~25°C, depending on the assay temperature) and oxygen concentration. Moreover, the C. elegans intestinal lumen may not be conducive to colonization owing to continual gut peristalsis41 as well as circulation of antimicrobial peptides42 and reactive oxygen species.43 Interestingly, our group has observed the suppression of a number of C. elegans antimicrobial peptide genes known to abate bacterial colonization such as lys-7 and spp-1 during Bp R15 infection (Fig. S3). Upon RNAi knock-down of these antimicrobial peptide genes, intestinal lumen colonization by P. aeruginosa and S. Typhimurium is aggravated.31,32 Nevertheless, even in a background of reduced antimicrobial peptide expression, Bp R15 still failed to fully colonize the worm gut. This reinforces our suggestion that B. pseudomallei is inherently less adept at colonizing the worm gastrointestinal tract and instead, relies on other mechanisms to kill the host. Understanding why Bp R15 fails to colonize the host intestinal lumen may help uncover host factors inhibitory to B. pseudomallei growth or survival in vivo which may aid the future development of anti-infective drugs against melioidosis.
C. elegans feeding rates were instantly and dramatically reduced upon B. pseudomallei infection, which implies a low nutritional value of the B. pseudomallei lawn as perceived by the host. Previous studies have demonstrated that the worm has the ability to avoid or move away from a population of pathogenic bacteria.44 However, our study is the first report of a decrease in host feeding as a response to the presence of virulent bacteria. Although the decreased C. elegans feeding rate was not a contributory factor to the limited Bp R15 colonization, it is possible that this unique host response may confer an advantage to host survival as it could provide the host with time to cope with the toxic insults incurred from consuming the pathogen. Song and Avery recently demonstrated that food intake in C. elegans consists of two actions, pharyngeal pumping and isthmus peristalsis, and activation of both are dependent on the interaction between serotonin and its receptor SER-7 present in a small number of motorneurons.45 A search for genes known to govern these pharyngeal motions from our previous microarray data surprisingly revealed that a handful of genes regulating serotonin production and signaling (tph-1, bas-1, ser-7 and cat-4)46 were indeed suppressed during Bp R15 infection (Lee et al., in preparation). Furthermore, we noted the induction of a number of neuropeptide genes (flp-1, flp-3 and flp-13)47 known to potently inhibit pharyngeal pumping. Taken together, these data may explain how the host feeding rate was markedly alleviated upon Bp R15 infection. It is tempting to speculate that B. pseudomallei, as a successful environmental survivor, secretes certain chemorepellents to ward off predation by bacteriophagous nematodes.
Our infection assay data indicated that a low number of intestinal Bp R15 bacterial cells were sufficient to evoke the observed symptoms and importantly to elicit death of C. elegans. From the pathogen’s perspective, Bp R15 may deploy other mechanically distinct yet successful virulence mechanisms that can compensate for the lack of intestinal lumen colonization. In our previous study, we found that diffusible toxins played a minor role in C. elegans killing.19 It is thus plausible to propose that, while remaining in the intestinal lumen, Bp R15 secretes extracellular toxins to damage the host. To support this proposition, our interrogation of the spatio-temporal expression of pgp-5, a known detoxifying ABC transporter, revealed that the infected worms actively engaged in a detoxification process upon Bp R15 infection. In addition, the loci that displayed heightened pgp-5 induction—the pharynx and intestine—coincided with the locality of Bp R15 colonization, suggesting that the low number of Bp R15 cells secreted toxins into the nearby cells and triggered a host detoxification response. We believe that these toxins may translocate to distant cells upon prolonged infection as pgp-5 induction was also observed in other cell types (gonad) during late infection. Interestingly, the pharyngeal pgp-5 induction observed in our study is not seen in P. aeruginosa infection but only in worms exposed to 100 µM cadmium.38 This raises the question of whether Bp R15-secreted toxins are distinct from that produced by other pathogens. Similar to P. aeruginosa and S. Typhimurium infections,38 reducing or abrogating C. elegans pgp-5 expression resulted in higher sensitivity to Bp R15 infection. Hence, we have shown that host detoxification constitutes an important immune defense mechanism against Bp R15 attack, and by extension, this implies that Bp R15 secretes toxins to kill the host.
Our previous study demonstrated that efficient killing of C. elegans necessitates continuous contact with Bp R15 since transient exposure to the pathogen failed to exert complete host killing.19 This strict demand for exposure to live Bp R15 may be a reflection of the inability of Bp R15 to proliferate within the C. elegans digestive tract as well as the strong and lost-lasting induction of the host detoxification machinery. Nevertheless, over the extended period of infection (25–30 h), continuous intoxication by Bp R15 was sufficient to overwhelm the host immune system, causing an abrupt drop in the number of live worms (Fig. S2C).
In conclusion, our study presents further evidence for the requirement of prolonged B. pseudomallei presence and supplementary intoxication in host killing as previously suggested.17-19 Our findings also led us to infer that B. pseudomallei kills C. elegans via continuous and specific secretion of extracellular toxins. Hence, there is an urgent need to identify the toxins responsible for the rapid demise of C. elegans. This in turn will advance our understanding of the interaction between B. pseudomallei and C. elegans, and may push the development of therapeutic countermeasures against acute melioidosis.
Materials and Methods
Bacterial and C. elegans strains and growth conditions
The bacterial strains, worm strains and plasmids used in this study as well as the related information are listed in Table 1. Wild-type Bp R15 was routinely propagated on Ashdown agar whereas R15-GFP was cultured on modified Luria-Bertani (LB) agar (3% tryptone, 1% yeast extract and 1.5% bacteriological agar) supplemented with 100 µg/ml Cm. For cloning experiments, E. coli TOP10 transformants harboring pSMompAgfp were selected on LB agar with 30 µg/ml Cm. E. coli HT115 expressing dsRNA directed against the cdc-25.1 gene (cdc-25.1 RNAi clone) was grown on LB agar containing 100 µg/ml carbenicillin (Cb). All bacterial cultures were aerobically incubated at 37°C unless otherwise stated. All the experiments involving Bp R15 were performed in a BSL2+ level laboratory and approved by the Universiti Kebangsaan Malaysia Animal Ethics Committee (UKMAEC). Wild-type C. elegans Bristol N2 (N2), tnt-3(aj3) and rrf-3(pk1426);glp-4(bn2) strains were obtained from the Tan Laboratory, Stanford University whereas the ppgp-5::gfp transgenic strain was provided by the Shapira Laboratory, University of California Berkeley. All worm strains were handled as previously described.29
Table 1. Bacterial, worm strains and plasmids used in this study.
| Strain/Plasmid | Description | References |
|---|---|---|
|
B. pseudomallei | ||
| R15 |
Wild-type clinical isolate; GmR |
19 and 51 |
| R15-GFP |
Isogenic strain of R15 transformed with pSMompAgfp; GmR CmR |
This study |
|
E. coli | ||
| TOP10 |
F–mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu) 7697 galU galK rpsL (SmR) endA1 nupG |
Invitrogen |
| OP50 |
Derivative of E. coli B strain; uracil auxotroph; lacks O-antigen component of its outer membrane; SmR |
Caenorhabditis Genetics Center (CGC), 52 |
|
cdc-25.1 RNAi clone |
E. coli HT115(DE3) expressing dsRNA complementary to C. elegans cdc-25.1 gene; CbR |
48 |
|
pgp-5 RNAi clone |
E. coli HT115(DE3) expressing dsRNA complementary to C. elegans pgp-5 gene; CbR |
Open Biosystems |
|
C. elegans | ||
| Bristol N2 |
Wild-type nematode isolated from mushroom compost near Bristol, England. |
CGC |
|
tnt-3(aj3) |
WE5006; contains a recessive mutation in the troponin T gene expressed in the pharyngeal muscles |
32 and 33 |
| ppgp-5::gfp |
WE5172; contains an integrated transgene ppgp-5::gfp (transgene constructed by Baillie Laboratory, Simon Fraser University) |
38 |
|
rrf-3(pk1426);glp-4(bn2) |
WE5134; contains loss-of-function mutations in the rrf-3 and glp-4 genes; exhibits germ line proliferation defect at 25°C and enhanced RNAi sensitivity |
53 and 54 |
|
Plasmids | ||
| pSM202 |
Derivative of pBRR1MCS; 3.686 kb; bears rep, mob, cat (CmR) genes and two multiple cloning sites (MCS 1 and 2). |
Mohamad S, unpublished data |
| pSMompAgfp | pSM202 with the PompA::gfp construct inserted into the MCS2; ~4.62 kb | This study |
Abbreviations: Cb, carbenicillin; Cm, chloramphenicol; Gm, gentamicin; Sm, streptomycin.
Fluorescent labeling of Bp R15
For construction of the recombinant plasmid pSMompAgfp, a 234-bp promoter region of the outer membrane protein A precursor gene (PompA) (GenBank: BPSL2522) was amplified from B. pseudomallei D286 genomic DNA with primers OMPAF1 (5′-CACATACTAGTGACCGATGTTAGGGTGGGG-3′) and OMPAR1 (5′-CTCACCATATTTCTCCTCTCGAAATTGAGA-3′). The 742-bp Aequorea coerulescens gfp ORF was amplified from pAcgfp1 (BD Biosciences Clontech, 632468) with primers GFPF1 (5′- CGAGAGGAGAAATATGGTGAGCAAGGGC-3′) and GFPR1 (5′-CGCCTGCAGTCACTTGTACAGCTCATCCAT-3′). SpeI and PstI restriction sites were incorporated into the 5′ end of OMPF1 and GFPR1 primers respectively (underlined sequence) to generate cohesive termini compatible with pSM202 multiple cloning site 2. Using overlap-extension PCR, both amplicons were fused at a 21-bp overlapping region generated at the 5′ end of PompA negative strand and gfp ORF positive strand (italicized sequence), then amplified with OMPAF1 and GFPR1 to form the PompA::gfp construct. Subsequently, the construct was double-digested and ligated with pSM202, forming pSMompAgfp, which was then transformed into E. coli TOP10 using standard CaCl2-mediated transformation. Transformants were screened by PCR and the sequence was verified through dideoxy DNA sequencing. High-fidelity Pfu DNA polymerase (Promega, M7741) was used for all DNA amplifications.
For preparation of electrocompetent cells and electroporation, a B. pseudomallei glycerol stock was diluted 100-fold in 50 ml SOB medium and cultured at ambient temperature (20–25°C) until the cell density reached OD595 nm of ~0.4. The bacterial culture was chilled on ice for 30 min and then harvested by centrifugation at 6,000 g for 5 min at 4°C. Next, the bacterial cells were washed three times and subsequently suspended in an appropriate volume of ice-cold 10% glycerol to shield the cell surface charges. Approximately 300 ng purified pSMompAgfp was added into 200 µl electrocompetent B. pseudomallei suspension and incubated on ice for 30 min. After that, the cell suspension was transferred into a 2 mm gap-width electroporation cuvette and subjected to electrical pulse (2 kV; 186 Ω; 40 µF; BTX Electroporation Apparatus, Harvard). Immediately after electroporation, 1 ml of SOC was added to the cell suspension followed by incubation at 37°C with agitation for 4 h. The electroporated cell suspension was concentrated 10-fold, after which 100 µl cells were spread onto modified LB agar supplemented with 100 µg/ml Cm and grown for 36 h. The presence of green fluorescence within the transformants was assessed using a Leica M205 FA stereomicroscope equipped with a GFP2 filter cube (bandpass 480/40 nm) (Leica Microsystems GmbH).
Preparation of Glp worms
N2 worms used in all assays were sterilized by RNAi knockdown of the cdc-25.1 gene. This gene encodes a CDC25 phosphatase homolog which affects embryonic viability and is necessary for cell proliferation of the germ line.48 The dsRNA directed against cdc-25.1 was introduced into worms by feeding. Briefly, 100 ml of the cdc-25.1 RNAi clone was cultured overnight at 37°C in LB medium supplemented with 100 µg/ml Cb. The culture was concentrated 25-fold, seeded onto NG agar supplemented with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Promega, V3953) and 100 µg/ml Cb and incubated at room temperature for at least 24 h. Wild-type N2 gravids were picked onto cdc-25.1 RNAi plates for two successive rounds of egg laying, each lasting for 4 h at 25°C. The worms were then removed and the eggs were allowed to hatch and develop into sterile worms on the plates.
Infection of N2 and tnt-3(aj3) mutant worms
The conditions used in all assays involving R15-GFP were standardized and are described in this paragraph. To prepare the bacterial lawn, 2–3 freshly streaked R15-GFP colonies were inoculated into BHI medium supplemented with 100 µg/ml Cm and grown with agitation for 22 h at 37°C. By using 60 mm diameter plates, 55 µl of the overnight culture was spread onto NG agar supplemented with 100 µg/ml Cm and incubated for 24 h at 37°C. The assay plates were then equilibrated to room temperature for a further 24 h. For all assays conducted, age-matched Glp adult worms were either hand-picked onto the bacterial lawn or washed twice in sterile distilled water prior to depositing on the bacterial lawn. All infection assays were performed at 25°C unless otherwise stated. Worms whose germ line was not abrogated by cdc-25.1 RNAi knock-down (known as “Emb”) were excluded from the statistical analysis.
For the infection of N2 worms, at 4, 8, 12, 22 and 28 h post-exposure, 75–100 live worms were paralyzed with 100 mM levamisole (Lev) (Sigma-Aldrich, L9756) and mounted on a 2% agarose gel pad for visualization at 400× magnification under a Leica DM5000B upright microscope equipped with an I3 filter cube (bandpass 450–490 nm). For tnt-3(aj3) mutants, the same number of worms was scored at 4, 6, 8 and 11 h post-exposure. When necessary, the N2.1 filter cube (bandpass 515–560 nm) was used to discern the non-specific gut autofluorescence. Fluorescence photomicrographs were collected using a Jenoptik ProgRes Link camera and CapturePro (Jenoptik AG) or Leica Application Suite software (Leica Microsystems GmbH). Each image series was captured with identical settings. To avoid microscopic slide stress and GFP photobleaching, the worms were viewed and scored in batches. At least two independent replicates were performed for the experiments.
Colony forming unit (CFU) assay
At each time point, 10 live worms were randomly picked and briefly anesthetized in 25 mM Lev. The worms were washed twice in 200 µl antibiotic cocktail comprising 25 mM Lev and 500 µg/ml kanamycin followed by incubation for at least 45 min to completely kill bacterial cells associated with the worm cuticle. Next, the worms were washed three times with 200 µl of 25 mM Lev to eliminate the killed bacteria and residual antibiotic. During the last washing step, the Lev was removed, leaving about 5 µl in the tube. Prior to adding 50 µl 1% Triton X (Sigma-Aldrich, X100), the worms were enumerated and then homogenized with a motorized pestle. Serial dilutions were performed on the worm lysates. Briefly, 10 µl of the worm lysate was spotted on Ashdown agar supplemented with 100 µg/ml Cm using the drop plate method with modifications.49 Colonies were counted after incubating the plates at 37°C for 48 h. Average colony numbers obtained from visually separate colonies were used for statistical analysis. Bacterial CFU per worm was calculated using the formula: (average colony number × dilution factor × 55 µl worm lysate)/(10 µl worm lysates plated × number of worms). Three independent replicates were performed for the experiment. The phrase “55 µl worm lysate” (50 µl 1% Triton X + 5 µl Lev containing the worms) refers to the total worm lysate of which 10 µl was used for plating.
For the enumeration of R15-GFP within serotonin-treated N2 worms, the method was identical to that described above, except 5 mM of serotonin hydrochloride (Sigma-Aldrich, H9523) was added into the NG agar one day before spreading the R15-GFP culture. Serotonin was prepared freshly for every experiment. At least two independent replicates were performed for the experiment.
Defecation rate assay
The experiment was performed as previously described with modifications.50 Three live worms were observed for 5 min at 1, 4, 8, 12, 24 and 28 h post-infection to determine the frequency of expulsion (Exp), a step in the defecation motor program that is characterized by enteric muscle contraction and release of gut content. The average Exp frequency was used in the statistical analysis. At least two independent replicates were performed for the experiment. The protocol was also used for determining defecation rates of serotonin-induced worms.
Feeding rate assay
The experiment was conducted as that described by Song and Avery with minor modifications.45 At 1, 4, 8, 12, 24 and 28 h post-infection, 30 live worms were observed in three batches (10 worms/batch, 5 sec/worm) to count the number of pharyngeal pumps. A pharyngeal pump was strictly defined as a backward contraction of the terminal bulb which is distinctly different from the convulsion-like head movement. The average number of pharyngeal pumps in 10 worms (pumps/sec) was calculated and used for statistical analysis. The same protocol was also used in determining the pharyngeal pumping rate of serotonin-induced worms, and all experiments were performed in at least three replicates.
qRT-PCR analysis of pgp-5 induction
The mRNA levels of pgp-5 (GenBank, C05A9.1) were measured using total DNase-treated RNA extracted from worms exposed to Bp R15 for 4 h and 12 h. RNA extraction and qRT-PCR analysis were performed as described by Evans et al.31 using the forward primer (5′-GGAAAATCAGAATGGGACGA-3′) and reverse primer (5′-TGTGGTAAGTACCGTTAGTT-3′). qRT-PCR was performed using the iScript One-Step RT-PCR kit with SYBR green (Bio-Rad, 170-8892) according to the manufacturer's instructions. The Bio-Rad iCycler (Bio-Rad) was used for amplification and quantification of the products. Specificity of amplification was confirmed by melt curve analysis after amplification. Normalized threshold cycle (Ct) values were used to calculate fold change of mRNA levels in Bp R15-infected worms as compared with worms fed on E. coli OP50. The Ct values were normalized to changes in three genes [ama-1, F44B9.5 and pan-actin (act-1, 3, 4)] that were found to be unchanged with infection. The experiment was performed in triplicate.
Scoring of Bp R15-infected ppgp-5::gfp transgenic worms
Bp R15 was used to infect the ppgp-5::gfp transgenic worms. Twenty live transgenic worms were picked for observation and scoring of green fluorescence at 100× magnification 4, 12 and 28 h post-infection. The average of the sum of scores was calculated and used for statistical analysis. Three independent replicates were performed.
RNAi knock-down of pgp-5 and survival assay
The RNAi knock-down was performed by placing the eggs of rrf-3(pk1426);glp-4(bn2) double mutant worms on NGM/IPTG/Cb plates pre-seeded with E. coli bearing the pgp-5 RNAi clone, which was acquired from the C. elegans ORF-RNAi library (Open Biosystems, RCE1181). The eggs were allowed to hatch and develop into Glp adult worms on the RNAi plates. For the survival assay, 120 RNAi-treated Glp worms were transferred onto the Bp R15 lawn (40 worms/plate) and incubated at 16°C. Worms were considered dead when they were no longer responsive to probing with a platinum wire picker. Worms that crawled up to the plate wall and died as a result of desiccation were censored from the analysis. The experiment was performed in duplicate.
Statistical analysis and image processing
Differences in nematode killing between Bp R15 and R15-GFP as well as the survival between pgp-5 RNAi-treated and untreated worms were assessed by the Log-rank test using StatView version 5.0.1 (SAS Institute, Inc.). For the remainder of assays, the data sets were first assessed using the Kolmogorov-Smirnov normality test. Only data sets that passed the test were analyzed with two-tailed Student’s t-test, the rest of the data sets were analyzed using non-parametric Mann-Whitney U-test. All the analyses were performed using GraphPad Prism version 5.04 (GraphPad Software). p-values of < 0.05 and < 0.01 were considered as statistically significant. All fluorescence photomicrographs were cropped, overlaid or compressed using Adobe Photoshop version 7.0 (Adobe) and assembled into figures using GraphPad Prism.
Supplementary Material
Acknowledgments
We thank Dr Suriani Mohamad (Universiti Sains Malaysia) for kindly providing the pSM202. We gratefully acknowledge Dr Man-Wah Tan (Genentech) for his constructive advice on this study as well as Dr Michael Shapira (University of California at Berkeley) for the transgenic worm strain and image analysis. We also thank Rui-Rui Wong and Nur Afifah Ijap for their technical assistance. This study was supported by the Malaysia Genome Institute-Stanford University International Research Grant awarded to S.N. by the Ministry of Science, Technology and Innovation, Malaysia.
Glossary
Abbreviations:
- Bp R15
B. pseudomallei R15
- ompA
outer membrane protein A gene
- GFP
green fluorescence protein
- R15-GFP
GFP-tagged Bp R15
- Glp
germ line proliferation-deficient
- CFU
colony forming unit
- ABC
ATP-binding cassette
- Cm
chloramphenicol
- Cb
carbenicillin
- TDmean
mean time to death
- Exp
expulsion
- Lev
levamisole
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/virulence/article/21808
Footnotes
Previously published online: www.landesbioscience.com/journals/virulence/article/21808
References
- 1.Galyov EE, Brett PJ, DeShazer D. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol. 2010;64:495–517. doi: 10.1146/annurev.micro.112408.134030. [DOI] [PubMed] [Google Scholar]
- 2.Cheng AC, Currie BJ. Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev. 2005;18:383–416. doi: 10.1128/CMR.18.2.383-416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sprague LD, Neubauer H. Melioidosis in animals: a review on epizootiology, diagnosis and clinical presentation. J Vet Med B Infect Dis Vet Public Health. 2004;51:305–20. doi: 10.1111/j.1439-0450.2004.00797.x. [DOI] [PubMed] [Google Scholar]
- 4.Gan YH. Interaction between Burkholderia pseudomallei and the host immune response: sleeping with the enemy? J Infect Dis. 2005;192:1845–50. doi: 10.1086/497382. [DOI] [PubMed] [Google Scholar]
- 5.Limmathurotsakul D, Peacock SJ. Melioidosis: a clinical overview. Br Med Bull. 2011;99:125–39. doi: 10.1093/bmb/ldr007. [DOI] [PubMed] [Google Scholar]
- 6.Schweizer HP, Peacock SJ. Antimicrobial drug-selection markers for Burkholderia pseudomallei and B. mallei. Emerg Infect Dis. 2008;14:1689–92. doi: 10.3201/eid1411.080431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inglis TJJ, Sagripanti JL. Environmental factors that affect the survival and persistence of Burkholderia pseudomallei. Appl Environ Microbiol. 2006;72:6865–75. doi: 10.1128/AEM.01036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leelarasamee A. Recent development in melioidosis. Curr Opin Infect Dis. 2004;17:131–6. doi: 10.1097/00001432-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Currie BJ. Burkholderia pseudomallei and Burkholderia mallei: melioidosis and glanders. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practices of Infectious Disease. 7th ed. Philadelphia: Elsevier Inc, 2010: 2869-77. [Google Scholar]
- 10.Hodgson KA, Morris JL, Feterl ML, Govan BL, Ketheesan N. Altered macrophage function is associated with severe Burkholderia pseudomallei infection in a murine model of type 2 diabetes. Microbes Infect. 2011;13:1177–84. doi: 10.1016/j.micinf.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Williams NL, Morris JL, Rush C, Govan BL, Ketheesan N. Impact of streptozotocin-induced diabetes on functional responses of dendritic cells and macrophages towards Burkholderia pseudomallei. FEMS Immunol Med Microbiol. 2011;61:218–27. doi: 10.1111/j.1574-695X.2010.00767.x. [DOI] [PubMed] [Google Scholar]
- 12.Chin CY, Monack DM, Nathan S. Delayed activation of host innate immune pathways in streptozotocin-induced diabetic hosts leads to more severe disease during infection with Burkholderia pseudomallei. Immunology. 2012;135:312–32. doi: 10.1111/j.1365-2567.2011.03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gravato-Nobre MJ, Hodgkin J. Caenorhabditis elegans as a model for innate immunity to pathogens. Cell Microbiol. 2005;7:741–51. doi: 10.1111/j.1462-5822.2005.00523.x. [DOI] [PubMed] [Google Scholar]
- 14.Chanchamroen S, Kewcharoenwong C, Susaengrat W, Ato M, Lertmemongkolchai G. Human polymorphonuclear neutrophil responses to Burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun. 2009;77:456–63. doi: 10.1128/IAI.00503-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58. doi: 10.1038/nri2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiersinga WJ, Wieland CW, Roelofs JJTH, van der Poll T. MyD88 dependent signaling contributes to protective host defense against Burkholderia pseudomallei. PLoS One. 2008;3:e3494. doi: 10.1371/journal.pone.0003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Quinn AL, Wiegand EM, Jeddeloh JA. Burkholderia pseudomallei kills the nematode Caenorhabditis elegans using an endotoxin-mediated paralysis. Cell Microbiol. 2001;3:381–93. doi: 10.1046/j.1462-5822.2001.00118.x. [DOI] [PubMed] [Google Scholar]
- 18.Gan YH, Chua KL, Chua HH, Liu B, Hii CS, Chong HL, et al. Characterization of Burkholderia pseudomallei infection and identification of novel virulence factors using a Caenorhabditis elegans host system. Mol Microbiol. 2002;44:1185–97. doi: 10.1046/j.1365-2958.2002.02957.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee SH, Ooi SK, Mahadi NM, Tan MW, Nathan S. Complete killing of Caenorhabditis elegans by Burkholderia pseudomallei is dependent on prolonged direct association with the viable pathogen. PLoS One. 2011;6:e16707. doi: 10.1371/journal.pone.0016707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Schaik EJ, Tom M, Woods DE. Burkholderia pseudomallei isocitrate lyase is a persistence factor in pulmonary melioidosis: implications for the development of isocitrate lyase inhibitors as novel antimicrobials. Infect Immun. 2009;77:4275–83. doi: 10.1128/IAI.00609-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alegado RA, Campbell MC, Chen WC, Slutz SS, Tan MW. Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell Microbiol. 2003;5:435–44. doi: 10.1046/j.1462-5822.2003.00287.x. [DOI] [PubMed] [Google Scholar]
- 22.Sifri CD, Begun J, Ausubel FM. The worm has turned--microbial virulence modeled in Caenorhabditis elegans. Trends Microbiol. 2005;13:119–27. doi: 10.1016/j.tim.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Pizarro-Cerdá J, Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124:715–27. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 24.Mellies JL, Barron AMS, Haack KR, Korson AS, Oldridge DA. The global regulator Ler is necessary for enteropathogenic Escherichia coli colonization of Caenorhabditis elegans. Infect Immun. 2006;74:64–72. doi: 10.1128/IAI.74.1.64-72.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spanier B, Starke M, Higel F, Scherer S, Fuchs TM. Yersinia enterocolitica infection and tcaA-dependent killing of Caenorhabditis elegans. Appl Environ Microbiol. 2010;76:6277–85. doi: 10.1128/AEM.01274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen SJ, Batzloff M, Chehrehasa F, Meedeniya A, Casart Y, Logue CA, et al. Nasal-associated lymphoid tissue and olfactory epithelium as portals of entry for Burkholderia pseudomallei in murine melioidosis. J Infect Dis. 2009;199:1761–70. doi: 10.1086/599210. [DOI] [PubMed] [Google Scholar]
- 27.Lefebre MD, Valvano MA. Construction and evaluation of plasmid vectors optimized for constitutive and regulated gene expression in Burkholderia cepacia complex isolates. Appl Environ Microbiol. 2002;68:5956–64. doi: 10.1128/AEM.68.12.5956-5964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 2010;6:e1000982. doi: 10.1371/journal.ppat.1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shapira M, Tan MW. Genetic analysis of Caenorhabditis elegans innate immunity. In: Ewbank J, Vivier E, eds. Methods in Molecular Biology. Totowa: Humana Press Inc, 2008; 429-42. [DOI] [PubMed] [Google Scholar]
- 30.Avery L, Shtonda BB. Food transport in the C. elegans pharynx. J Exp Biol. 2003;206:2441–57. doi: 10.1242/jeb.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans EA, Kawli T, Tan MW. Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 2008;4:e1000175. doi: 10.1371/journal.ppat.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alegado RA, Tan MW. Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell Microbiol. 2008;10:1259–73. doi: 10.1111/j.1462-5822.2008.01124.x. [DOI] [PubMed] [Google Scholar]
- 33.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–6. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 34.Branicky R, Hekimi S. What keeps C. elegans regular: the genetics of defecation. Trends Genet. 2006;22:571–9. doi: 10.1016/j.tig.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Hart AC, ed. Behavior (July 3, 2006), WormBook. The C. elegans Research Community, Wormbook, http://www.wormbook.org
- 36.Williams SN, Locke CJ, Braden AL, Caldwell KA, Caldwell GA. Epileptic-like convulsions associated with LIS-1 in the cytoskeletal control of neurotransmitter signaling in Caenorhabditis elegans. Hum Mol Genet. 2004;13:2043–59. doi: 10.1093/hmg/ddh209. [DOI] [PubMed] [Google Scholar]
- 37.Srinivasan S, Sadegh L, Elle IC, Christensen AGL, Faergeman NJ, Ashrafi K. Serotonin regulates C. elegans fat and feeding through independent molecular mechanisms. Cell Metab. 2008;7:533–44. doi: 10.1016/j.cmet.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurz CL, Shapira M, Chen K, Baillie DL, Tan MW. Caenorhabditis elegans pgp-5 is involved in resistance to bacterial infection and heavy metal and its regulation requires TIR-1 and a p38 map kinase cascade. Biochem Biophys Res Commun. 2007;363:438–43. doi: 10.1016/j.bbrc.2007.08.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramli NSK, Nathan S, Vadivelu J. Biofilm formation and Acyl-homoserine lactone production is varied amongst large and small colony variants of Burkholderia pseudomallei. PLoS ONE. 2012 In press. [Google Scholar]
- 40.Allman E, Johnson D, Nehrke K. Loss of the apical V-ATPase a-subunit VHA-6 prevents acidification of the intestinal lumen during a rhythmic behavior in C. elegans. Am J Physiol Cell Physiol. 2009;297:C1071–81. doi: 10.1152/ajpcell.00284.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avery L, Thomas JH. Introduction to C. elegans In Riddle DL, Blumenthal T, Meyer BJ, Priess JR eds. C. elegans II. 2nd ed. New York: Cold Spring Harbor Laboratory Press, 1997: 679-81. [PubMed] [Google Scholar]
- 42.Millet ACM, Ewbank JJ. Immunity in Caenorhabditis elegans. Curr Opin Immunol. 2004;16:4–9. doi: 10.1016/j.coi.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Chávez V, Mohri-Shiomi A, Maadani A, Vega LA, Garsin DA. Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics. 2007;176:1567–77. doi: 10.1534/genetics.107.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Zhang Y. Neural-immune communication in Caenorhabditis elegans. Cell Host Microbe. 2009;5:425–9. doi: 10.1016/j.chom.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Song BM, Avery L. Serotonin activates overall feeding by activating two separate neural pathways in Caenorhabditis elegans. J Neurosci. 2012;32:1920–31. doi: 10.1523/JNEUROSCI.2064-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chase DL, Koelle MR. Biogenic amine neurotransmitters in C. elegans (February 20, 2007), WormBook, ed. The C. elegans Research Community, Wormbook, http://www.wormbook.org [DOI] [PMC free article] [PubMed]
- 47.Rogers CM, Franks CJ, Walker RJ, Burke JF, Holden-Dye L. Regulation of the pharynx of Caenorhabditis elegans by 5-HT, octopamine, and FMRFamide-like neuropeptides. J Neurobiol. 2001;49:235–44. doi: 10.1002/neu.1078. [DOI] [PubMed] [Google Scholar]
- 48.Ashcroft N, Golden A. CDC-25.1 regulates germline proliferation in Caenorhabditis elegans. Genesis. 2002;33:1–7. doi: 10.1002/gene.10083. [DOI] [PubMed] [Google Scholar]
- 49.Chen CY, Nace GW, Irwin PL. A 6 x 6 drop plate method for simultaneous colony counting and MPN enumeration of Campylobacter jejuni, Listeria monocytogenes, and Escherichia coli. J Microbiol Methods. 2003;55:475–9. doi: 10.1016/S0167-7012(03)00194-5. [DOI] [PubMed] [Google Scholar]
- 50.Weinshenker D, Garriga G, Thomas JH. Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans. J Neurosci. 1995;75:6975–85. doi: 10.1523/JNEUROSCI.15-10-06975.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SH, Chong CE, Lim BS, Chai SJ, Sam KK, Mohamed R, et al. Burkholderia pseudomallei animal and human isolates from Malaysia exhibit different phenotypic characteristics. Diagn Microbiol Infect Dis. 2007;58:263–70. doi: 10.1016/j.diagmicrobio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 52.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan MW. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci U S A. 2006;103:14086–91. doi: 10.1073/pnas.0603424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, et al. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12:1317–9. doi: 10.1016/S0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.