

The requirement for deoxynucleoside triphosphates (dNTPs) in mammalian cells is largest during S-phase, when nuclear DNA is replicated. Smaller amounts are required during the cell’s whole life for DNA repair and mitochondrial (mt) DNA replication. DNA repair utilizes a few hundred nucleotides per damaged site, and total mtDNA corresponds to only a few percent of the nuclear DNA. More than 10 DNA polymerases operate in the different forms of DNA synthesis consuming each of the four dNTPs in roughly equimolar amounts. The intracellular concentrations of dNTPs are at least 10 times larger during S-phase than outside S, possibly reflecting the substrate affinities of the individual polymerases. Correct concentrations of dNTPs are essential for normal cellular function, and either deficiency or excess of even a single dNTP increases mutation rates and causes genetic diseases. This rule applies to nuclear DNA and also to mtDNA. The first example of a mtDNA disease caused by a genetically determined dNTP pool imbalance was discovered as late as in 1999.1
Deoxynucleotides are produced by two pathways (Fig. 1). A cytosolic ribonucleoside diphosphate reductase (RNR) synthesizes them de novo from ribonucleotides, and four different cytosolic or mt deoxynucleoside kinases salvage deoxynucleosides by phosphorylation. The two pathways communicate via nucleotide carriers in the inner mt membrane. The anabolic kinases are counteracted in substrate cycles by catabolic 5′-nucleotidases that dephosphorylate deoxynucleoside monophosphates, protecting cells from overproduction of dNTPs.2 A recently discovered dNTP triphosphohydrolase further limits the concentrations of dNTPs producing deoxynucleosides by removal of the triphosphate group.3
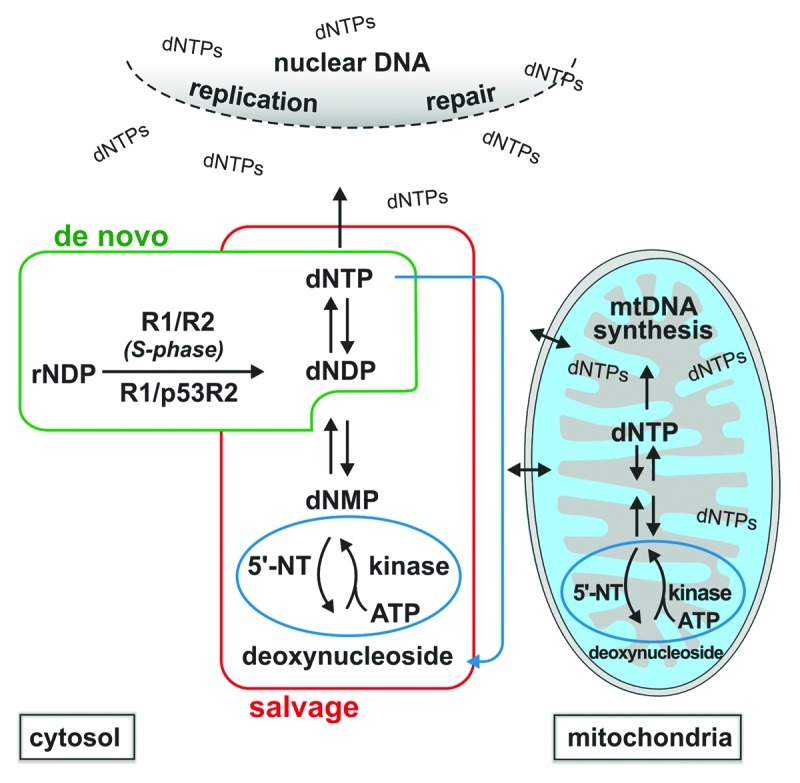
Figure 1. Pathways of dNTP synthesis. Deoxynucleoside triphosphates (dNTPs) are produced both in the cytosol and in mitochondria. They move freely across the nuclear envelope and protein carriers in the inner mt membrane establish a bidirectional communication between cytosol and mitochondria. dNTPs are used for DNA replication and repair in the nucleus and in mitochondria. Synthesis occurs by two pathways, ribonucleotide reductase-dependent de novo synthesis, exclusively cytosolic, and salvage of deoxynucleosides, performed by two parallel sets of kinases in the cytosol and in mitochondria. The subunit composition of ribonucleotide reductase differs during S-phase (R1/R2) and in non-dividing/differentiated cells (R1/p53R2). The first committed step of the salvage pathway, catalyzed by deoxynucleoside kinases, is an irreversible reaction counteracted by the catabolic activity of 5′-nucleotidases (5′-NT). The two classes of enzymes create “substrate cycles” (highlighted in blue) with regulatory functions. The newly discovered catabolic dNTP triphosphohydrolase SAMHD1 (blue arrow) curbs dNTP pool sizes converting dNTPs to deoxynucleosides.
In this picture, RNR occupies center-stage (Fig. 1). The enzyme is intrinsically able to produce each deoxynucleotide.4 Its sophisticated allosteric control not only prevents overproduction of dNTPs, but also directs the enzyme’s substrate specificity. The canonical mammalian enzyme contains two non-identical subunits, named protein R1 and protein R2, both required for catalysis. R1 contains the business end of the enzyme, where the ribonucleotide is reduced under the direction of the allosteric effector; R2 generates and contains a stable free tyrosyl radical required for catalysis.
The transcription of both R1 and R2 is activated at the onset of S-phase, resulting in expansion of the dNTP pools despite their extensive consumption during DNA replication. At this stage, cells contain approximately equal amounts of R1 and R2. On leaving S-phase RNR activity decreases dramatically, with strongly curtailed pool sizes, because both RNR subunits are no longer transcriptionally activated, and, in addition, R2 is specifically degraded during G2 and mitosis. Postmitotic cells lose essentially all R2 activity. Synthesis of dNTPs outside S-phase was earlier ascribed to deoxynucleoside salvage, but in 2000, two independent groups discovered a stable alternative R2 subunit, lacking the degradation signals responsible for R2 disappearance outside S-phase.5,6 The protein, coded by RRM2B and named p53R2 because of its p53-induciblility, was suggested to be imported into the cell nucleus and to replace R2 for DNA repair.5 Both suggestions did not appear fully convincing to us. In cultured cycling fibroblasts we could not confirm an intranuclear localization of p53R2 after DNA damage7 and the cellular concentration of p53R2 was only a small fraction of that of R2, indicating that R1/R2 and not R1/p53R2 is the catalytically active enzyme. A new perspective was opened by a second important discovery: p53R2 mutations cause profound depletion of mtDNA in differentiated cells with lethality in early infancy.8 The new data clearly established a function of p53R2 in mtDNA maintenance. Was this the real function of p53R2 in vivo?
To investigate the role of p53R2 in non-cycling cells where R2 is not expressed, we used normal fibroblasts from a patient with a homozygous lethal missense mutation in RRM2B, a more appropriate tool than the transformed human cell lines used previously in the literature. In cycling cultures, the growth rate and the dNTP pools of the mutated cells were identical to the control. Instead, after prolonged serum starvation, when the mutated fibroblasts had become quiescent, RNR activity was strongly diminished, and the dCTP and dGTP pools were halved, but mtDNA remained unchanged.9 We decided to test the mutant cells’ ability to cope with increased requests for dNTPs during quiescence and either depleted them of mtDNA by incubation with ethidium bromide or induced DNA damage by UV irradiation.10 In both instances, the mutated phenotype became apparent. The p53R2-mutated fibroblasts were unable to fully recover their mtDNA complement after removal of ethidium bromide, and their DNA repair capacity was compromised. These defects were largely overcome by addition of deoxynucleosides, indicating that the observed negative effects of the mutation were indeed caused by a deficiency of dNTPs.10 Our results indicate that p53R2 provides precursors both for mtDNA replication and for nuclear DNA repair, and that its function becomes essential only in the absence of R2. In agreement with these data, in vivo p53R2 deficiency targets terminally differentiated cells that do not express R2.
Among multicellular animals p53R2-like proteins have been found only in vertebrates. The presence of two alternative small subunits of ribonucleotide reductase optimizes dNTP concentrations for nuclear and mtDNA syntheses during the cell cycle and appears to represent a specific evolutionary adaptation of higher metazoans. It also may represent a cell defense mechanism against invading viral DNAs by decreasing in resting cells the concentrations of dNTPs required for viral DNA synthesis, similar to the recently discovered restricitve action of the dNTP triphosphohydrolase SAMHD1 against HIV infection.
Acknowledgments
Supported by Italian Telethon (Grant GGP09019), Italian Association for Cancer Research (Grant 1091), and the University of Padova, Strategic Projects 2008 to V.B.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22324
References
- 1.Nishino I, et al. Science. 1999;283:689–92. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 2.Rampazzo C, et al. Mutat Res. 2010;703:2–10. doi: 10.1016/j.mrgentox.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Goldstone DC, et al. Nature. 2011;480:379–82. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 4.Nordlund P, et al. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka H, et al. Nature. 2000;404:42–9. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 6.Nakano K, et al. Oncogene. 2000;19:4283–9. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 7.Pontarin G, et al. Proc Natl Acad Sci USA. 2008;105:17801–6. doi: 10.1073/pnas.0808198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourdon A, et al. Nat Genet. 2007;39:776–80. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 9.Pontarin G, et al. J Biol Chem. 2011;286:11132–40. doi: 10.1074/jbc.M110.202283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pontarin G, et al. Proc Natl Acad Sci USA. 2012;109:13302–7. doi: 10.1073/pnas.1211289109. [DOI] [PMC free article] [PubMed] [Google Scholar]