

Abstract
Cell cycle regulation and DNA repair following damage are essential for maintaining genome integrity. DNA damage activates checkpoints in order to repair damaged DNA prior to exit to the next phase of cell cycle. Recently, we have shown the role of Ada3, a component of various histone acetyltransferase complexes, in cell cycle regulation, and loss of Ada3 results in mouse embryonic lethality. Here, we used adenovirus-Cre-mediated Ada3 deletion in Ada3fl/fl mouse embryonic fibroblasts (MEFs) to assess the role of Ada3 in DNA damage response following exposure to ionizing radiation (IR). We report that Ada3 depletion was associated with increased levels of phospho-ATM (pATM), γH2AX, phospho-53BP1 (p53BP1) and phospho-RAD51 (pRAD51) in untreated cells; however, radiation response was intact in Ada3−/− cells. Notably, Ada3−/− cells exhibited a significant delay in disappearance of DNA damage foci for several critical proteins involved in the DNA repair process. Significantly, loss of Ada3 led to enhanced chromosomal aberrations, such as chromosome breaks, fragments, deletions and translocations, which further increased upon DNA damage. Notably, the total numbers of aberrations were more clearly observed in S-phase, as compared with G₁ or G₂ phases of cell cycle with IR. Lastly, comparison of DNA damage in Ada3fl/fl and Ada3−/− cells confirmed higher residual DNA damage in Ada3−/− cells, underscoring a critical role of Ada3 in the DNA repair process. Taken together, these findings provide evidence for a novel role for Ada3 in maintenance of the DNA repair process and genomic stability.
Keywords: Ada3, ATAC, SAGA, nuclear foci, DNA repair
Introduction
DNA damage response (DDR) is essential for the maintenance of genomic stability,1,2 and an unstable genome leads to accumulations of mutations and cancer development. Inefficient DNA double-strand break (DSBs) repair can result in chromosomal translocations, deletions and chromosome fusions or loss. Cells respond to DSBs by activating the DDR, which blocks cell-cycle progression until the damage is repaired or initiates cell death if the damage is irreparable2-4. There are two main pathways for repair of DNA DSBs: non-homologous end-joining (NHEJ), an error-prone repair pathway predominant during G1 and early S phases of the cell cycle, and homologous recombination (HR), an error-free repair pathway active primarily in late S and G2 phases of the cycle.5 DNA damage signaling involves a large number of proteins that act as sensors, mediators, transducers and effector proteins.6,7 Recruitment of the mediator/adaptor protein H2AX, BRCA1, 53BP1 and CtIP to DNA DSBs is a key event in the DDR.8-10 Defective recruitment of repairosome factors at DNA DSBs, such as delay in appearance or disappearance of these proteins, is associated with defective DDR.
Cell cycle checkpoint regulatory proteins have a critical role in the DNA repair process. The eukaryotic cell cycle progression depends on proper coordination of DNA replication and duplication of chromosomes to daughter cells,11 a process precisely regulated by modification of the components of chromatin that allow the accessibility of factors to target sites involved in transcription.12 Thus, proteins involved in modulating the chromatin structure play an important role in cell cycle progression. In this context, recent studies have shown a role for p53, a well-known tumor suppressor protein, in maintaining chromosomal stability during tetraploidization.13 Additionally, the posttranslational modification of core histones (H2A, H2B, H3 and H4) is an essential process for altering chromatin structure.14,15 Coordination of chromosomal regulation and histone synthesis with DNA replication is required for proper cell division.16,17 Histoneacetyltransferases (HATs) and histone deacetylases (HDACs) are required to maintain steady-state levels of acetylation.18 Several HAT enzymes, such as GCN5 (general control nonderepressible 5), PCAF, p300, CBP and MOF, have been identified over the years as participants in histone acetylations.19-22 Most of the HATs are part of large complexes, such as the human TBP-free TAF complex (TFTC), SPT3/TAF9/GCN5 acetyltransferase complex (STAGA) (human homolog of yeast SAGA complex) and the Ada2a-containing (ATAC) complex, that play a role in several important processes, such as cell cycle and DDR.23-27 The combined presence of Ada3 with GCN5 in a number of distinct HAT complexes and the recent evidence for a role of GCN5 and various HAT complexes in regulating DNA replication as well as repair26-29 suggested that Ada3 may also play a role in DDR.
We have previously identified human Ada3 as a novel HPV 16 E6-binding protein.30 We showed that Ada3 binds and stabilizes the tumor suppressor p53 protein and is required for p53 acetylation by p300.31 Significantly, our recent studies have shown that germline deletion of Ada3 in mouse is embryonic lethal, and adenovirus-Cre mediated conditional deletion of Ada3 in Ada3fl/fl MEFs leads to delay in G1 to S phase of cell cycle and mitotic defects by controlling histone acetylation and several mitotic genes.32
Recently, it has been shown that cyclin-dependent kinase activity and cell cycle phase determine whether DSBs are repaired by NHEJ or HR.33 Central to this regulation are the proteins that initiate the processing of DNA repair by HR, such as the Mre11-Rad50-Nbs1 protein complex and CtIP.33,34 Because Ada3 is a regulator of cell cycle as part of HAT complexes, we determined the role of Ada3 in DDR. Here, we report that loss of Ada3 results in severe chromosome aberrations, which increases post-irradiation and correlates with significant delay in disappearance of repairosomes, thus suggesting the role of Ada3 in DNA replication stress and maintenance of genomic stability.
Results
Increased levels of DNA damage-related proteins in Ada3-null cells
Given the connection of DNA damage and the cell cycle,27,28 we assessed if Ada3 plays a role in the DNA damage response. Cells with and without Ada3 were analyzed for pATM, γH2AX, p53BP1 and pRAD51 as such or after IR exposure. Significantly, Ada3−/− cells exhibited higher levels of phosphorylated forms of these proteins as compared with Ada3fl/fl cells (Fig. 1), indicating that Ada3 deficiency itself led to DNA replication stress-induced DNA damage. However, IR response was intact upon Ada3 deletion, indicating that Ada3 loss has minimum influence on DNA damage sensing.

Figure 1. Ada3 deletion affects ATM activation and other downstream targets in DNA damage response. Total proteins were prepared from Ada3fl/fl and Ada3−/− immortalized MEFs at the indicated times after exposure to 10 Gy IR. Immunoblotting was performed using indicated antibodies. The normalization of each phospho protein with respect to total proteins was calculated by using ImageJ software and shown at top of the corresponding gel.
Ada3 deletion delays disappearance of DNA damage foci
DSBs are critical cellular lesions that can result from ionizing radiation exposure. A well-known marker for DSB is the phosphorylated (Ser139) form of the histone H2 variant H2AX (γH2AX) and recruitment of the damage sensor p53-binding protein 1 (53BP1) to the DSB-containing chromatin, so we next investigated the appearance of IR induced γH2AX and 53BP1 foci. These experiments clearly showed that upon radiation treatment formation of foci of γH2AX, and 53BP1 was not compromised in Ada3−/−cells.
Given the critical role of Ada3 in cell cycle checkpoints and histone acetylation, and emerging evidence that resumption of the cell cycle following DNA damage requires disassembly of DNA damage response foci, we next examined disappearance of foci in Ada3fl/fl and Ada3−/− cells upon IR treatment. These experiments showed that both cells showed maximal numbers of γH2AX foci at 30 min after IR (Fig. 2A); however, at 2 h post-irradiation, only ~65% of Ada3fl/fl cells contained γH2AX foci, whereas almost 80% of Ada3−/− cells retained γH2AX foci. Similarly, 50% of Ada3−/− cells retained 53BP1 foci at 2 h, persisting up to 4 h, as compared with 30% in 2 h and only 15% at 4 h in control Ada3fl/fl cells (Fig. 2B). The persistence of γH2AX and 53BP1 foci in Ada3-deleted cells is indication of a delay in DNA repair process, suggesting a role of Ada3 in the DNA repair process.
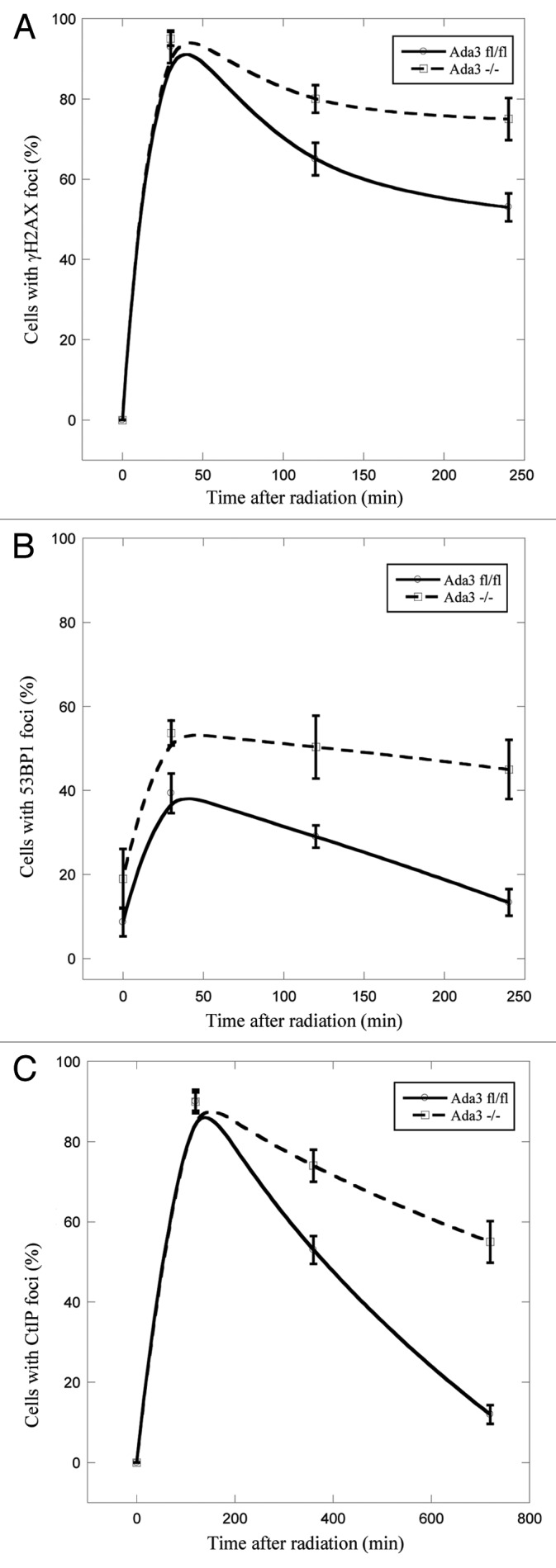
Figure 2. Ada3 regulates disappearence of DNA repair foci after IR treatment. Ada3fl/fl and Ada3−/− immortalized MEFs were immunostained with antibodies against γH2AX, 53BP1 or CtIP after irradiation with 2 Gy, and foci at different time points post-irradiation were quantitated for γH2AX (A), 53BP1 (B) and CtIP (C). Cells with more than five foci were scored positive for each antibody. The data represents mean ± SE from three independent experiments performed in triplicates.
Given the recent findings from our laboratory and that of others’ that Ada3 plays an important role in S and G2/M cell cycle check point and recent evidence of the indispensible role of CtIP in intra-S phase and G2/M checkpoints and DNA repair pathway,25,32 we assessed disappearance of CtIP foci in Ada3fl/fl and Ada3−/− cells upon 2 Gy of IR treatment. Notably, 6 h post-radiation ~50% of Ada3fl/fl cells exhibited CtIP foci, whereas 70% of Ada3−/− cells retained CtIP foci; however, as the time progressed, ~60% of Ada3−/− cells exhibited foci by 12 h, whereas the majority of Ada3fl/fl cells showed no foci (Fig. 2C). Taken together, these results clearly demonstrate that Ada3 plays a role in the DNA repair response.
Ada3-null cells exhibit spontaneous chromosomal aberrations and higher residual aberrations
Based on a vast body of literature in which it is known that proteins that regulate cell cycle, and DNA replication processes are major checkpoints for maintaining genomic stability, we performed karyotypic analysis of Ada3fl/fl MEFs upon Ada3 deletion. For this purpose, we used primary MEFs isolated at passage 1 rather than immortal cells, to avoid in vitro culture artifacts, and infected with adenovirus expressing the Cre recombinase to delete Ada3. Metaphase chromosomes from two independent Ada3fl/fl or Ada3−/− cell lines were prepared and analyzed for chromosome anomalies. Although mitotic chromosome condensation was normal in Ada3-deleted cells, higher frequencies for chromosome aberrations were observed (Fig. 3A, indicated by arrow). As summarized in Figure 3C, 87 aberrations were observed in 50 metaphases obtained from Ada3-deleted cells, as compared with 35 aberrations in control Ada3fl/fl cells. The average number of aberrations per metaphase among Ada3-deleted cells was 1.74 compared with 0.7 in control Ada3fl/fl cells. Furthermore, overall Ada3-deleted cells showed increased chromatid and chromosome abnormalities, including chromosome breaks, fragments, deletions and translocations. Thus, accumulation of spontaneous genome instability in Ada3-deleted mouse cells suggests that Ada3 plays important role in maintaining genome integrity.

Figure 3. Ada3 maintains genome integrity. (A) Representative metaphase images for spontaneous chromosome abnormalities in Ada3fl/fl and Ada3−/− MEFs (arrow indicates one of the aberration seen upon Ada3 deletion). (B) Western blotting for Ada3 protein in Ada3fl/fl and Ada3−/− primary MEFs shows the adeno-cre deletion of Ada3; β-actin was used as a loading control. (C) Summary for spontaneous genomic instability in Ada3fl/fl and Ada3−/− primary MEFs. Metaphase spreads were Giemsa banded and analyzed for chromosomal abnormalities.
Ada3-null cells exhibit more chromosomal aberrations upon DNA damage
Next, to assess the effect of clastogen-induced DNA damage, primary MEFs isolated at passage 1 with adenovirus expressing the Cre recombinase to delete Ada3 were treated with mitomycin (MMC) or Diepoxybutane (DEB) and then examined for various chromosomal aberrations. As summarized in Figure 4C, with the treatment of these clastogens, there is an increase in DNA damage, as measured by the number of chromosomal aberrations. Both MMC and DEB treatments revealed increased chromosome aberrations in Ada3−/− cells as compared with Ada3fl/fl cells. When treated with MMC, the average numbers of aberrations per metaphase of Ada3−/− cells was 10.2 as compared with 7.6 in Ada3fl/fl cells. Treatment with DEB resulted in an average of 8.4 aberrations per metaphase in Ada3−/− cells as compared with 5.9 in Ada3fl/fl cells. Chromosomal abnormalities like dicentrics, rings, tri-radials and quadri-radials were frequently observed in the clastogen-induced aberration analyses (Fig. 4A). Taken together, our chromosomal analyses showed a clear role for Ada3 in maintaining genome integrity.

Figure 4. Ada3-null cells are more prone to DNA damage. (A) Representative images depicting chromosome aberrations in Ada3fl/fl and Ada3−/− primary MEFs upon treatment with Mitomycin C (MMC) and Diepoxybutane (DEB). Chromosome aberrations are indicated by arrows. (B) western blotting for Ada3 expression in Ada3fl/fl and Ada3−/− primary MEFs; β-actin was used as the loading control. (C) Summary for MMC- and DEB-induced chromosome aberrations in Ada3fl/fl and Ada3−/− primary MEFs. Giemsa-banded metaphase spreads were analyzed for chromosome abnormalities.
Ada3-deleted cells exhibit higher frequencies of chromosomal aberrations in S phase after IR treatment
To determine cell cycle stage-specific frequencies of chromosomal aberrations post-irradiation: for G1 specific aberrations, cells were treated with 3 Gy, incubated for 18 h post-irradiation and then subcultured, and metaphases were collected; for G2-specific aberrations analyses, metaphases were harvested following 90 min irradiation. Cell cycle phase-speci□c chromosome aberrations were ascertained based on the frequency of chromosomal and chromatid-type aberrations observed at metaphase. Categories of asymmetric chromosome aberrations were scored: dicentrics, centic rings, interstitial deletions/acentric rings and terminal deletions. These analyses showed that although the frequency of chromosomal aberrations was modestly higher in Ada3−/− cells in G1 as well as in G2, the differences were not statistically significant (Fig. 5A and C). However, a significant increase was obvious in S phase-specific chromosome aberrations (p 0.05) (Fig. 5B). These results indicate that Ada3 plays a role in S phase checkpoint, which is affecting the repair of chromosome aberrations in S phase cells (Fig. 5B). Since HR is predominant in S phase cells, it suggests that Ada3 may be modulating the checkpoint required for the recruitment of repairosome formation during HR.
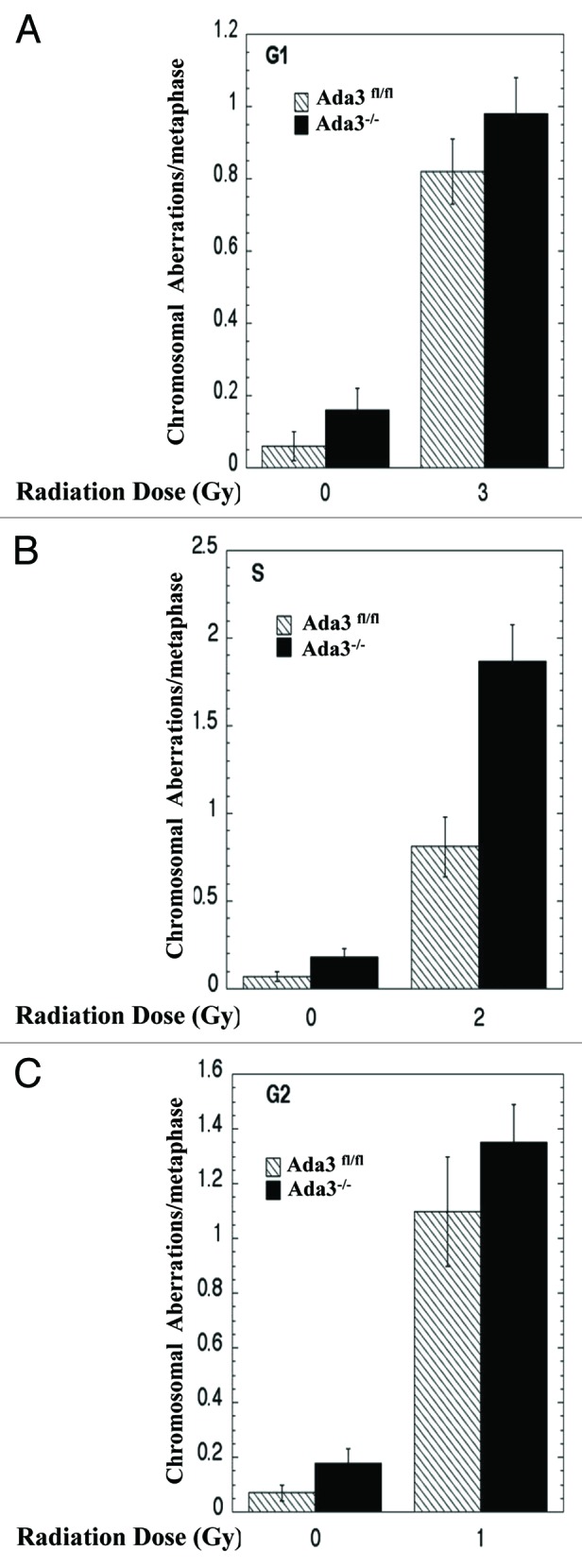
Figure 5. Ada3 deletion followed by IR induces more chromosomal aberrations in S phase of the cell cycle. (A) For G1-specific aberrations, cells in plateau phase of proliferation were irradiated with 3 Gy, incubated for 24 h post-irradiation and then subcultured, and metaphases were examined. (B) For S-phase-specific aberrations cells in exponential phase were irradiated with 2 Gy and analyzed after 4 h post IR, as above, (C) For G2 phase-type aberrations, cells in exponential phase were irradiated with 1 Gy and analyzed as above. For each experiment, 50 metaphases were scored. Data represents mean ± SE of three independent experiments, each done in triplicates.
Ada3-deleted cells exhibit more DNA damage following irradiation
To determine whether Ada3−/− cells exhibit more DNA damage as compared with control Ada3fl/fl cells, we assessed DNA damage post-10 Gy IR exposure using the comet assay. Ada3−/− cells did show more DNA damage as compared with control cells. Notably, consistent with data presented above, Ada3−/− cells showed a significant delay in repair efficiency, having a higher percentage of unrepaired DNA in the comet tail by 8–16 h in comparison to control Ada3fl/fl cells (Fig. 6A and B). These results are consistent with delayed disappearance of nuclear foci, suggesting Ada3 deletion significantly influences the DNA repair process.

Figure 6. Ada3 deletion delays DNA repair process. Ada3fl/fl and Ada3−/− immortalized MEFs were irradiated with 10 Gy of γ irradiation and analyzed for DNA damage-induced comet tail over various time periods (1–16 h) after irradiation. (A) Representative images of neutral comet assays in both cells. (B) Unrepaired DNA was quantified as comet tail length (in pixels) using the CometScore software. At least 50 cells per sample were analyzed, and presented as mean ± SE from three independent experiments.
Discussion
In recent years, emerging evidence of intra-S-phase DNA damage checkpoint and its impact on the cell cycle machinery has provided important insights into the DNA repair process.35,36 Recent research has also provided important insights into chromatin modifications in response to DNA damage. Here we demonstrate that Ada3, a known component of ADA complex that connects HATs to TFs, influences the DNA repair process.
We have recently shown that conditional deletion of Ada3 from Ada3fl/fl mouse embryonic fibroblasts (MEFs) using an adenovirus cre system demonstrated a critical role of Ada3 in cell cycle progression.32 Given the known connection between cell cycle regulators and DNA repair pathways, in this study we first examined the levels of several proteins known to play a role in the DNA repair process. Significantly, several proteins, such as pATM, γH2AX, p53BP1 and pRAD51, were elevated in Ada3−/− cells as compared with control Ada3fl/fl cells without inducing any external damage. These results indicated that loss of Ada3 itself induced DNA damage, probably as a result of DNA replication stress. Next, we assessed levels of well-known proteins in the DNA repair pathway upon radiation treatment. Although, there was no significant difference in the kinetics of various proteins in control and Ada3−/− cells, a significant delay was observed in disappearance of nuclear foci in Ada3 −/− cells in response to radiation induced DNA damage. It has been demonstrated that DNA damage-sensing proteins localize to the sites of DSBs within seconds to minutes following irradiation, resulting in the formation of repairosomes. The observation that Ada3−/− cells showed delayed disappearance of DNA damage foci is consistent with a significant role of Ada3 in the DNA repair process. These results together with our recent observations that Ada3 regulates cell cycle checkpoints underscore a role for ADA complex in the DNA repair pathway. Finally, using comet assays, we demonstrate the persistence of DNA damage in Ada3-deleted cells as compared with the Ada3fl/fl cells upon IR treatment, further confirming the importance of Ada3 in regulating the DNA repair process.
Similar to Ada3, hMOF, the human ortholog of the Drosophila MOF (male absent on the first) is a histone acetyl transferase whose depletion results in early lethality in mice, and hMOF has also been associated with DDR.21,22,37 Another HAT TIP60 has been shown to acetylate core histones H2A, H3 and H438,39 and cells expressing catalytically inactive TIP60 accumulate DSBs.40 Further, TIP60 and its cofactor Trapp directly bind to chromatin near DSBs, and depletion of Trapp impairs DNA damage-induced H4 acetylation and impairs repair of DSBs by HR.41 Similar results were obtained with NuA4, a yeast homolog of TIP60, following HO endonuclease-induced DSBs.42 The NuA4 HAT complex binds directly to the sites of DSBs, which occurs concomitantly with the appearance of γH2AX.42 These studies clearly link histone modification to DSB repair. Our recent observations that Ada3 is in complex with p300/CBP, PCAF and GCN5, and that deletion of Ada3 dramatically downregulates p300 and PCAF is consistent with a role of Ada3 in histone modification.32 Furthermore, we recently directly demonstrated that knockdown of Ada3 significantly decreased the levels of acetylation of several histones, such as H2A-K5, H2B-K5, H3-K9, H3-K14, H3-K56 and H4-K8,32 further underscoring the importance of histone acetylation in DNA repair pathway.
Given the important role of Ada3 in the cell cycle and the observation that DNA damage was induced upon deletion of Ada3 in cells without exogenous treatments, we were prompted to analyze effect of Ada3 on genomic stability. Supporting our hypothesis, karyotypic analysis of Ada3-deleted cells revealed a dramatic increase in aneuploidy and polyploidy as well as higher frequencies for chromosome breaks or fragmentation, suggesting a critical role of Ada3 in maintaining the genomic integrity of cells. Notably, cells lacking Ada3 when exposed to DNA damage further accumulated more genomic instability, as shown by treatment of cells with MMC or DEB and enhanced chromosomal aberrations as compared with untreated cells. Consistent with our recent findings that Ada3 regulates transition of cells from G1 to S and S to G2/M, we observed higher frequencies of chromosomal aberrations in S-phase in Ada3-deleted cells upon exposure to DNA damage as compared with G1 or G2 phase of cell cycle. As maintenance of genomic integrity is coordinated by DNA replication and histone biosynthesis,16,17 our previous results32 together with data presented in this study suggest a critical role of Ada3 in this pathway.
In conclusion, we have demonstrated that Ada3, as part of histone coactivator complexes and a regulator of cell cycle, plays a critical role in maintaining genomic stability. Considering that Ada3 interacts with human papilloma virus 16 E6 oncoprotein and is required for E6 function,30 and our recent observations that Ada3 levels/localization are correlated with bad prognostic markers and predict poor survival in breast cancer patients (Mirza et al., manuscript submitted), we provide evidence of an important link with genomic integrity and cancer.
Materials and Methods
Establishment of MEFs
Embryonic day 13.5 embryos were dissected from Ada3+/fl intercrossed females, and MEFs were isolated and immortalized following the 3T3 protocol.43 MEFs were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum.
Adenovirus infection
Adenoviruses expressing EGFP-Cre or EGFP alone were purchased from University of Iowa (Gene transfer vector core). The protocol followed was as previously described.32
Western blotting and antibodies
Protein was quantitated from cell extracts using the BCA protein assay reagent (Pierce). Immunoblotting was performed with primary antibodies against Ada3;23 other primary antibodies used were anti-phosphoATM (#05-740, Millipore), anti-ATM total (#07-1286, Millipore), anti-Phosho-H2AX (#MABE205, Millipore), anti-H2AX total (DR 1016, Calbiochem), anti- phospho-53BP1 (NB100-1803, Novus Biologicals), anti-53BP1(NB100-304, Novus Biologicals), anti-phospho-RAD51 (SAB4504264, Sigma), anti-RAD51 (#07-1782, Millipore) and β-actin (A5441, Sigma).
Analysis of γH2AX, 53BP1 and CtIP foci by immuno〉uorescence after γ irradiation
Ada3fl/fl and Ada3−/− cells were cultured on coverslips and then irradiated with 2 Gy using a Mark I Cs-137 irradiator. Cells were fixed in 4% paraformaldehyde in PBS for 5 min and analyzed at various time periods after irradiation. Cells were permeabilized and incubated with antibodies against γH2AX, 53BP1 or CtIP (kind gift from Richard Baer) primary antibodies followed by incubation with goat anti-mouse or goat anti-rabbit Alexa Flour 594-conjugated secondary antibodies. Mounted slides were analyzed under fluorescence microscope briefly; three independent observers randomly counted cells displaying greater than 5 foci per nucleus; 100 cells per experiment were counted, and three independent experiments were performed. Representative images were captured using a Zeiss Axiovert epifluorescence microscope equipped with a Hamamatsu ORCA-ER high-resolution digital B/W camera driven by Slidebook acquisition software (Intelligent Imaging).
Cytogenetic studies of spontaneous and clastogen treated MEFs
Primary MEFs (passage 1) were initially plated in DMEM containing 10% FBS until cells reached 60–70% confluence. For clastogen-induced aberration studies, two separate cell cultures were treated with mitomycin-C (MMC) at a final concentration of 0.045 µg/ml and diepoxybutane (DEB) at a final concentration of 0.1µg/ml for 48 h. All cultures were harvested following conventional cytogenetic protocol.44 Briefly, the cell cultures were treated with 0.1µg/ml Colcemid (Irvine Scientific) approximately 40 min before the initiation of harvest. For chromosome preparations, the cells were harvested following conventional cytogenetic protocol of hypotonic treatment (0.7% sodium citrate for 25 min at 37°C) and freshly prepared chilled 3:1 acetone:methanol fixation. This was followed by three additional fixation cycles and air-dried slide preparation. The slides were “aged” in a hot oven (100°C) for 30 min or in a 60°C oven overnight, followed by Giemsa banding with trypsin pretreatment. A total of 50 metaphases were scored for each culture. To avoid selection bias, consecutive metaphases with sufficient, well-defined chromosome morphology were included in the analyses. Structural aberrations, including chromatid/chromosome breaks, fragments, deletions, translocations, dicentrics, rings, triradials and quadriradials were recorded as per the protocol.44 Fifty metaphases (n = 50 cells) from each genotype were analyzed for chromosome aberrations. Chromosome images were acquired using CytoVision® (Leica microsystems).
Analysis of chromosomal aberrations during cell cycle
Ada3fl/fl and Ada3−/− cells were exposed to various doses of γ irradiation, and chromosomal aberrations were assessed in cells at G1, S and G2 phases of the cell cycle using the procedure described previously.45
Analysis of DNA damage using neutral comet assay
Ada3fl/fl and Ada3−/− MEFs were harvested at various time points after γ irradiation (10 Gy) using a Mark I Cs-137 irradiator (J.L. Sheperd and Associates), re-suspended in cold PBS at 5 x 105 cells/ml, mixed with low-melt agarose (1:10), and 50 μl of the cell/agarose mixture was spread on manufacturer’s slide (Trevigen’s Comet Assay kit). Slides were subjected to electrophoresis, immersed 30 min each in DNA precipitate solution (1M NH4Ac in 95% ethanol) and then in 70% ethanol, and dried at 45°C. The dried slides were mounted in mounting media with DAPI (Vector Labs) and analyzed using Zeiss Axioskop epifluorescent microscope, and images were captured with a Zeiss AxioCam HRm camera driven by Axiovision 4.6. TIFF images were converted to bitmap format, and 50 cells per experimental condition were analyzed for tail length and tail DNA percentage using the Comet Score software (TriTek). Three independent experiments done in triplicate were performed.
Acknowledgments
We would like to thank to Dr. Richard Baer, Columbia University Health Science Division for providing us CtIP antibody. This research is supported by the NIH grant CA96844 and CA144027 and Department of Defense grants W81XWH-07-1-0351 and W81XWH-11-1-0171 to V.B.; the NIH grants CA87986, CA105489, CA99163, CA116552 and NCI 5U01CA151806-02 to H.B. and the NCI Core Support Grant to UNMC-Eppley Cancer Center; RO1CA129537, RO1CA154320 to T.K.P. S.M. is supported by Susan G. Komen Postdoctoral Fellowship (KG111248).
Glossary
Abbreviations:
- Ada3
activation/deficiency in alteration-3
- ATAC
Ada2a containing complex
- SAGA
human homologs of yeast SAGA complex
- CtIP
CtBP-interacting protein
- IR
irradiation
- NHEJ
non-homologous end-joining
- HR
homologous recombination
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22613
References
- 1.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–51. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 2.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 3.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–54. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 4.Pierce AJ, Jasin M. NHEJ deficiency and disease. Mol Cell. 2001;8:1160–1. doi: 10.1016/S1097-2765(01)00424-5. [DOI] [PubMed] [Google Scholar]
- 5.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–68. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 7.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guirouilh-Barbat JK, Wilhelm T, Lopez BS. AKT1/BRCA1 in the control of homologous recombination and genetic stability: the missing link between hereditary and sporadic breast cancers. Oncotarget. 2010;1:691–9. doi: 10.18632/oncotarget.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz LB, Chehab NH, Malikzay A, DiTullio RA, Jr., Stavridi ES, Halazonetis TD. The DNA damage checkpoint and human cancer. Cold Spring Harb Symp Quant Biol. 2000;65:489–98. doi: 10.1101/sqb.2000.65.489. [DOI] [PubMed] [Google Scholar]
- 10.Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010;9:3602–10. doi: 10.4161/cc.9.17.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schafer KA. The cell cycle: a review. Vet Pathol. 1998;35:461–78. doi: 10.1177/030098589803500601. [DOI] [PubMed] [Google Scholar]
- 12.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Ho CC, Hau PM, Marxer M, Poon RY. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget. 2010;1:583–95. doi: 10.18632/oncotarget.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 15.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710–8. doi: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell. 2005;17:301–11. doi: 10.1016/j.molcel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 19.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 20.Carrozza MJ, Utley RT, Workman JL, Côté J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19:321–9. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- 21.Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol. 2010;30:3582–95. doi: 10.1128/MCB.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta A, Sharma GG, Young CS, Agarwal M, Smith ER, Paull TT, et al. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25:5292–305. doi: 10.1128/MCB.25.12.5292-5305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guelman S, Kozuka K, Mao Y, Pham V, Solloway MJ, Wang J, et al. The double-histone-acetyltransferase complex ATAC is essential for mammalian development. Mol Cell Biol. 2009;29:1176–88. doi: 10.1128/MCB.01599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–57. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 25.Orpinell M, Fournier M, Riss A, Nagy Z, Krebs AR, Frontini M, et al. The ATAC acetyl transferase complex controls mitotic progression by targeting non-histone substrates. EMBO J. 2010;29:2381–94. doi: 10.1038/emboj.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh S, Pugh BF. Sequential recruitment of SAGA and TFIID in a genomic response to DNA damage in Saccharomyces cerevisiae. Mol Cell Biol. 2011;31:190–202. doi: 10.1128/MCB.00317-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–33. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 28.Burgess RJ, Zhou H, Han J, Zhang Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell. 2010;37:469–80. doi: 10.1016/j.molcel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paolinelli R, Mendoza-Maldonado R, Cereseto A, Giacca M. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat Struct Mol Biol. 2009;16:412–20. doi: 10.1038/nsmb.1583. [DOI] [PubMed] [Google Scholar]
- 30.Kumar A, Zhao Y, Meng G, Zeng M, Srinivasan S, Delmolino LM, et al. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol Cell Biol. 2002;22:5801–12. doi: 10.1128/MCB.22.16.5801-5812.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nag A, Germaniuk-Kurowska A, Dimri M, Sassack MA, Gurumurthy CB, Gao Q, et al. An essential role of human Ada3 in p53 acetylation. J Biol Chem. 2007;282:8812–20. doi: 10.1074/jbc.M610443200. [DOI] [PubMed] [Google Scholar]
- 32.Mohibi S, Gurumurthy CB, Nag A, Wang J, Mirza S, Mian Y, et al. Mammalian alteration/deficiency in activation 3 (Ada3) is essential for embryonic development and cell cycle progression. J Biol Chem. 2012;287:29442–56. doi: 10.1074/jbc.M112.378901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan J, Chen J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J Biol Chem. 2010;285:1097–104. doi: 10.1074/jbc.M109.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond B Biol Sci. 2011;366:3562–71. doi: 10.1098/rstb.2011.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huertas P, Cortés-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–92. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–65. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar R, Hunt CR, Gupta A, Nannepaga S, Pandita RK, Shay JW, et al. Purkinje cell-specific males absent on the first (mMof) gene deletion results in an ataxia-telangiectasia-like neurological phenotype and backward walking in mice. Proc Natl Acad Sci U S A. 2011;108:3636–41. doi: 10.1073/pnas.1016524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto T, Horikoshi M. Novel substrate specificity of the histone acetyltransferase activity of HIV-1-Tat interactive protein Tip60. J Biol Chem. 1997;272:30595–8. doi: 10.1074/jbc.272.49.30595. [DOI] [PubMed] [Google Scholar]
- 39.Kimura A, Horikoshi M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells. 1998;3:789–800. doi: 10.1046/j.1365-2443.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- 40.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–73. doi: 10.1016/S0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 41.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 42.Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16:979–90. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown MG, Lawce HJ. Peripheral Blood Cytogenetic Methods. In: Margaret J. Barch (Editor) TKE, Jack Spurbeck (Editor), ed. The AGT laboratory manual: Lippincott Williams & Wilkins, 1997:77-163. [Google Scholar]
- 45.Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, et al. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006;26:1850–64. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]