

Abstract
The early life environment can be crucial in influencing the development of an animal’s long-term physiology. There is now much evidence to suggest that perinatal challenges to an animal’s immune system will result in changes in adult rat behavior, physiology, and molecular pathways following a single inflammatory event during development caused by the bacterial endotoxin lipopolysaccharide (LPS). In particular, it is now apparent that neonatal LPS administration can influence the adult neuroimmune response to a second LPS challenge through hypothalamic-pituitary-adrenal axis modifications, some of which are caused by alterations in peripheral prostaglandin synthesis. These pronounced changes are accompanied by a variety of alterations in a number of disparate aspects of endocrine physiology, with significant implications for the health and well-being of the adult animal. In this review, we discuss the newly elucidated mechanisms by which neonatal immune challenge can permanently alter an animal’s endocrine and metabolic physiology and the implications this has for various disease states.
Keywords: lipopolysaccharide, fever, hypothalamic-pituitary-adrenal axis
Early Life Events Program Physiology Long Term
Early life events can have significant long-term effects on an animal’s physiology. In humans, an adverse early life experience such as emotional neglect can lead to an increased likelihood of the individual developing anxiety, depression, chronic illness, even drug addiction, in later life (15, 32, 39, 54). Similar findings have been demonstrated in primate models, with parental or social deprivation during the neonatal period producing socially maladjusted, timid, depressed adults that have alterations in neuroimmune and hypothalamic-pituitary-adrenal (HPA) axis function (32, 75). In rodents, too, an experience as subtle as a brief period of handling or withdrawal of maternal attention can have pronounced effects on physiology and behavior throughout the life of the animal. For instance, seminal work by Meaney and colleagues has revealed that parenting style, categorized by high- vs. low-intensity nursing and grooming, can program anxiety levels and responsiveness of the HPA axis in the offspring. Thus, rats nursed by dams that impart low-intensity nursing/grooming have exacerbated HPA axis responses to stress (19, 53). They also have alterations in other functions, ranging from their ability to perform cognitive tasks (52) to sexual behavior (17).
Early life events other than social and maternal contact can also have an impact upon the animal’s physiology long term. For instance, neonatal overfeeding can lead to obesity in adulthood coupled with changes to anxiety and HPA axis responses to stress (84). Also, a painful inflammatory event experienced at critical time points during development alters perception of a painful stimulus in later life (4). Clearly, the early postnatal developmental period is important in programming adult physiology.
One of the more common types of physical stimulus encountered during childhood is an infection, or challenge to the immune system. The question therefore arises: how does a challenge to the immune system, encountered during development, alter immune function in the long term? It is now well accepted that a neuroimmune challenge in the form of a bacterial or viral infection can permanently alter some aspects of the innate immune response (30). In this review, we discuss the mechanisms by which this occurs, how these mechanisms differ from those of other programming events like maternal attention, and some of the long-term consequences of this altered immune function.
The Immune Response
In responding to an immune challenge, the body employs two major strategies, the acquired and the innate immune responses. Whereas the acquired immune response is specifically targeted to particular previously encountered antigens, reviewed in Ref. 28, the innate immune response functions to recognize more generalized and conserved microbial patterns and responds by initiating a rapid nonspecific immune cascade, culminating in fever and sickness behaviors that combat the pathogen. This review focuses on the changes in the innate immune response after neonatal interventions.
Briefly, when an immune stimulus is encountered, it is recognized by pathogen-associated molecular pattern recognition receptors, called Toll-like receptors (TLR), that are found on several cell types, including monocytes, macrophages, and adipocytes. A bacterial infection, such as is mimicked by the application of lipopolysaccharide (LPS), the pyrogenic moiety of the outer membrane of the Gram-negative bacterial cell wall, activates TLR4 (27, 45). This activation of TLR4 initiates a complement cascade contributing to the initiation of the febrile response and local release of prostaglandin (PG)E2 from the liver that acts on the vagus to signal the brain (9, 20), causing an initial elevation in glucocorticoids (35, 68). Subsequently, a number of phosphorylation steps are activated, leading to the phosphorylation of inhibitory (I)κB, which releases nuclear factor (NF)-κB from its complex (24). This NF-κB is then translocated to the nucleus, where it initiates transcription of proinflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)α, as well as anti-inflammatory cytokines such as IL-1 receptor antagonist and IL-10 (18, 20). Cytokines are released into the blood stream and stimulate cyclooxygenase (COX)-2, the rate-limiting enzyme in the conversion of arachidonic acid to PGE2. PGE2 acts in the brain, principally in the vascular organ of the lamina terminalis and in the ventromedial preoptic area of the anterior hypothalamus, to stimulate heat conservation via cutaneous vasoconstriction and reduction of sweating or panting, and heat production via increases in brown adipose tissue metabolism (60), the end result of which is fever (Fig. 1). Additional activation of the HPA axis also helps to control the magnitude of the inflammation.
Fig. 1.
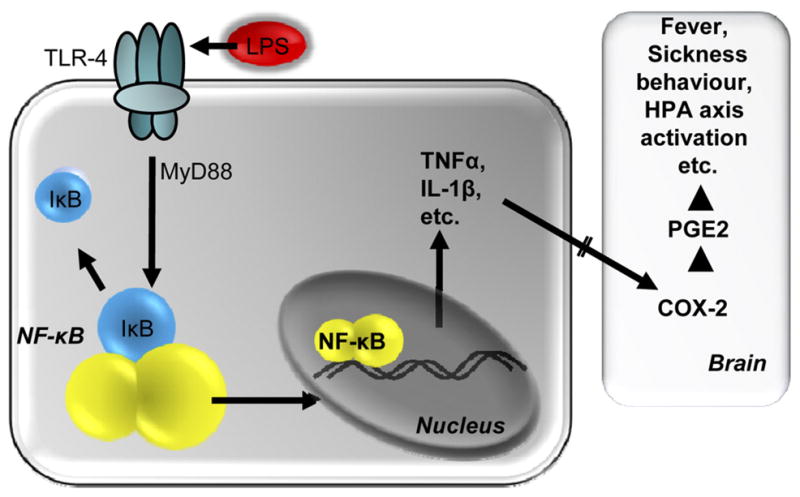
Central responses to an immune challenge. Pathogens [e.g., lipopolysaccharide (LPS)] bind to Toll-like receptors (e.g., TLR4) on monocytes, macrophages, and adipocytes. A complement cascade is initiated, resulting in local release of prostaglandins (PG; e.g., PGE2) that act on the brain. Subsequently, the myeloid differentiation primary response gene (MyD)88-dependent phosphorylation of inhibitory (I)κB is triggered within the immune cell. The transcription factor NF-κB is released, resulting in the synthesis and release of proinflammatory cytokines (e.g., TNFα, IL-1β). Cytokines act directly or indirectly on the brain to activate inducible cyclooxygenase (COX)-2, leading to PGE2 production. This cascade culminates in fever, sickness behavior, and hypothalamic-pituitary-adrenal (HPA) axis activation, combating the invading pathogen.
Concurrent with the development of fever, a variety of sickness behaviors occur that are manifested as withdrawal of social interactions, anhedonia, and anorexia (21). These associated functions are thought to be activated following the synthesis and action of additional cytokines within the brain and action of other mediators such as leptin (21, 69).
The development of fever appears to be important for survival. When animals are prevented from developing a fever, they are more likely to have higher pathogen loads and are more likely to die (38, 49). Fever probably increases our survival ability by altering the body temperature enough that it is suboptimal for bacterial and viral proliferation and survival (43). It also enhances existing host defense mechanisms such as phagocytosis of pathogens by neutrophils (38). Activation of the innate immune response is followed by an acquired immune response wherein antibodies are raised against invading pathogens (28). In the aftermath of an infection, there is often a period when the organism is refractory and therefore less responsive to a subsequent immune challenge, a phenomenon known as “tolerance” (8). However, there are not thought to be any persistent changes to the innate, or more generalized, immune response resulting from an acute infection experienced in adulthood. Infections are also commonly encountered during the perinatal period, however, and it is becoming clear that, depending on the timing and severity of the insult, such infections can impose more permanent alterations in the response to further infections of the same type.
Neonatal Programming of Adult Immune Responses
In the mid to late 1990s, Shanks and colleagues established that an immune challenge experienced at postnatal days (P)3 and 5 of the neonatal period in the rat can lead to permanent alterations in HPA axis function and immune regulation (71, 72). Subsequent studies have addressed the effects of and mechanisms for such alterations.
Results from our laboratory have shown that even a single peripheral injection of LPS (Escherichia coli) given at a critical period in development programs an attenuated neuroimmune response to a similar stimulus in later life (Fig. 2). This phenomenon occurs in both male and female Sprague-Dawley rats (11, 78) as well as in mice (unpublished observations) and is independent of the occurrence of fever in the neonate (11). There appears to be a critical window after P7 and before P28 in development during which this single immune challenge is able to exert these programming effects (82). Similar attenuated neuroimmune responses to LPS in adulthood are seen, however, if consecutive injections of LPS are administered earlier e.g., P3 and P5, implying that a large or chronic challenge may over-ride any protective mechanisms that would otherwise occur at this age (86). Interestingly, the mechanism by which neonatal LPS causes alterations in adult immune function appears to be different depending on age at exposure. This point is discussed further below in Unresolved Issues.
Fig. 2.
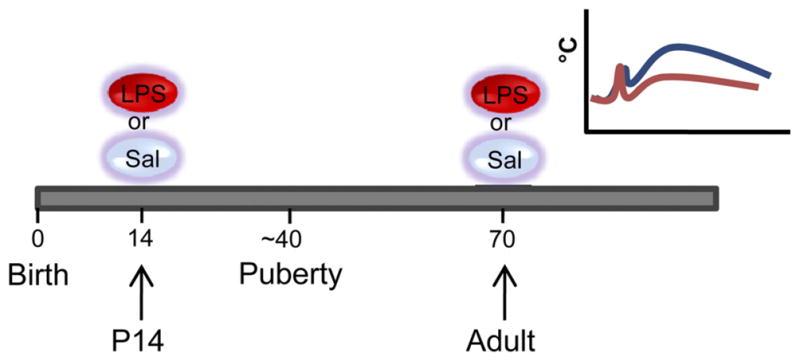
Time line of treatment protocol. On postnatal day (P)14, 50% of the rats from each litter are given an ip injection of 100 μg·kg−1·ml−1 E. coli LPS. The remaining 50% are given a similar volume of pyrogen-free saline. They are left to grow to adulthood, and at approximately P70 are given either 50 μg·kg−1·ml−1 E. coli LPS or saline. Inset: depiction of typical febrile response to LPS in adult animals pretreated at P14 with saline (blue line) or LPS (red line). Neonatally LPS-treated rats have attenuated febrile responses to the same dose of LPS.
The attenuated febrile response to a second challenge with LPS after a single exposure to LPS at P14 is due to permanent changes in the innate and not the acquired immune response. Antibodies to LPS are raised against the O-antigen portion, which is not conserved between species. When P14 rat pups are treated with an injection of Salmonella enteritidis LPS, which has a different O-antigen sequence, attenuated febrile responses to E. coli LPS in adulthood are still seen (11). In addition, this phenomenon also cannot be attributed to merely an extended “tolerance” to the immune challenge. Firstly, immune tolerance typically lasts less than a week (8), whereas the attenuated febrile response is seen with at least a 50-day interval between the first and second LPS challenges. Second, the same attenuated fever is not seen if the initial LPS exposure is given in adulthood (or even as early as P28) (82). These findings therefore describe a long-lasting altered innate immune response following a single low-dose immune challenge that is encountered, in the rat, at a developmental period after P7 and before P28. These alterations in innate immune function with neonatal LPS also appear to be independent of any changes in both the anorectic and anhedonic responses to LPS (47, 79).
Mechanisms of Neonatal Programming of Adult Immune Function
There are a number of components of the immune response that are potentially permanently altered by a neonatal exposure to an immune challenge (Fig. 1). For instance, reduced febrile responses in adulthood could be due to an inability to produce heat or to an alteration in the proinflammatory cytokine profile induced by LPS. The former is unlikely, as rats respond with robust hyperthermia to the stress brought about by an injection. Furthermore, normal fevers are seen with both an intraperitoneal injection of the proinflammatory cytokine IL-1β and with an intracerebrocentricular (brain) injection of PGE2 (11) or LPS (61), indicating that these rats have a normal ability to respond to proinflammatory cytokines and a normal ability of PGE2 and LPS to elaborate the febrile response when the brain is activated centrally. These neonatally LPS-exposed animals are capable of mounting robust fevers but do not do this in response to LPS.
Neonatal LPS does affect circulating proinflammatory cytokine responses to a further LPS challenge, and it is this difference that is likely responsible for the attenuated fever, at least in males (47). Thus, circulating concentrations of the proinflammatory cytokines IL-1β, IL-6, and TNFα are significantly attenuated after an adult LPS challenge in neonatally LPS-treated rats compared with saline-treated controls (25, 47). This reduction in circulating proinflammatory cytokine production is associated with reduced IκB phosphorylation in the liver and spleen (25), which would lead to attenuated NF-κB release, reduced translocation, and less initiation of cytokine transcription. The resultant dampening of circulating cytokine production also results in reduced activation of PGE2-catalyzing COX-2 in the brain (11, 47). It is noteworthy that cytokine levels in adult Fischer 344 rats are not attenuated if the first exposure to LPS occurs earlier in life, i.e., at P3 and P5 (86), indicating that age-, dose-, or strain-specific mechanisms may be involved.
These findings led to the possibility that this dampening of the response to LPS, at least to a single dose between P7 and P28, could be due to altered negative feedback by corticosterone. Glucocorticoids are known to modify cytokine expression, chiefly via their actions on NF-κB and HPA axis activity (35, 68) and have previously been shown to be altered by neonatal immune challenge, albeit under different conditions (40, 71, 72). Glucocorticoid levels or actions therefore represent good candidate mechanisms to explain our altered cytokine expression and febrile responses. In support of this hypothesis, corticosterone is strongly elevated compared with controls in the first hour after LPS in adult rats treated with LPS as neonates, and blocking the ability of corticosterone to respond to LPS with adrenalectomy, or with the glucocorticoid receptor antagonist RU-486, completely restores normal febrile responses and plasma cytokine concentrations (25).
Exploration of the mechanisms by which corticosterone was enhanced after a second LPS challenge revealed that animals that received LPS during development had a greater increase in corticotropin-releasing hormone (CRH) mRNA within the first hour after LPS, although basal levels were similar. Proopio-melanocortin (POMC), the parent peptide for adrenocortico-tropic hormone (ACTH) mRNA, was also increased in the same manner as CRH by the neonatal treatments and following the adult LPS injection. Indeed, increases in CRH and POMC mRNA translated into elevated serum levels of ACTH that were more pronounced after the adult LPS challenge in the rats given LPS neonatally (61). These findings indicate an enhanced HPA axis reactivity to LPS in adulthood in neonatally LPS-treated rats that may account for the amplified corticosterone release.
The HPA axis response to a stressor is under tight negative feedback regulation via glucocorticoids acting at glucocorticoid receptors on neurons in the hypothalamus, hippocampus, and elsewhere that inhibit the paraventricular nucleus (PVN) of the hypothalamus CRH neuron activation. The amplified corticosterone release after a second LPS challenge occurs without changes in this glucocorticoid negative feedback. That is, neonatally LPS-treated rats have amplified corticosterone responses to LPS in adulthood despite no significant changes in glucocorticoid receptor mRNA (37) or protein (61) in the hippocampus of adult animals treated with LPS at P14 compared with controls. In further support of the contention that there are no changes in glucocorticoid feedback, activation of the viral receptor TLR3 in adulthood in LPS-treated neonates resulted in unaltered febrile and corticosteroid responses compared with controls despite the involvement of a similar constellation of immune mediators (26). These findings pointed to a critical role for the LPS receptor TLR4 in the programming response. TLR4 is required for the recognition of the LPS molecule, and animals deficient in this receptor show pronounced febrile hyporesponsiveness when given LPS (41, 66). In contrast to the expectation that TLR4 would be downregulated after neonatal LPS treatment (thus accounting for reduced cytokine levels), adult rats treated with LPS on P14 have significantly higher amounts of TLR4 receptor mRNA in the liver and spleen than controls (61). The question remains as to how changes to TLR4 in the liver and spleen may be responsible for downstream alterations in corticosterone, especially since these occur at an early time point, before they are influenced by cytokine activation of the brain.
Enhanced TLR4 expression in the livers of neonatally LPS-treated rats after a second dose of LPS led to the suggestion that these rats were displaying an early vagally mediated activation of the brain. The vagus nerve plays a large role in the activation of HPA responses following peripheral inflammatory stimuli (31, 44), and acute-phase mediators released from the liver can activate it sufficiently rapidly to precede the synthesis and release of cytokines responsible for fever (10, 51, 65). However, Fos immunoreactivity in the nucleus of the solitary tract (where vagal afferents terminate) was attenuated, not enhanced, in response to LPS in neonatally LPS-treated adults. There was also no change in c-fos mRNA in this region and no reversal of the enhanced corticosterone response to LPS in neonatally LPS-challenged adults after hepatic vagotomy (61), essentially ruling out a role for a vagally mediated component to these effects.
COX-2 plays an important role in mediating the effects of LPS and is a crucial catalyst not only for the production of PG leading to fever (63, 85), but also for the early rise in plasma PGE2. Although neonatally saline-treated rats do not have appreciable levels of liver COX-2, the enzyme can be seen in measurable quantities in those treated neonatally with LPS. Thus, in contrast to control animals, where the enzyme has to be induced, normally resulting in very little early PGE2 production, the neonatally LPS-exposed animals have sufficient enzyme readily available in the liver to quickly produce PGE2. In support of this contention, adult LPS in these animals causes a significantly greater early elevation in plasma PGE2 (61), which can then cause an early activation of the HPA axis.
Collectively, these results point to a collaborative TLR4-and COX-2-mediated mechanism by which early life exposure to LPS causes attenuated febrile responses to the same challenge in adulthood. Early exposure to LPS leads to greater expression of TLR4 in the liver and constitutive COX-2; thus, LPS acting on these more numerous TLR4, coupled with high peripheral levels of available liver COX-2, results in increased early PGE2 production, amplified early HPA axis activity, and subsequent downregulated cytokine production (Fig. 3).
Fig. 3.

Mechanism for attenuated febrile responses in neonatally LPS-treated animals. A: in animals not treated with LPS as neonates, i.e., in the normal innate immune response, LPS acts on TLR4 on immune cells. An immune cascade is initiated whereby COX-2 is activated in liver macrophages (Kupffer cells and possibly other immune cells elsewhere), promoting PG (e.g., PGE2) release into the circulation to signal to the brain, contributing to the onset of fever. At the level of the immune cell, NF-κB also triggers synthesis and release of proinflammatory cytokines (e.g., TNFαand IL-1β or -6), which activate the brain, stimulating COX-2 to catalyze further production of PGE2. In addition to contributing to the onset of fever, PG cause an initial ACTH release from the anterior pituitary and glucocorticoid release from the adrenal cortex. Glucocorticoids have anti-inflammatory actions to limit peripheral cytokine synthesis. B: in animals treated with LPS as neonates, there is a constitutive upregulation of TLR4 on immune cells in the spleen and liver as well as constitutive upregulation of COX-2. In these animals, adult LPS causes a rapid enhanced rise in plasma PGE2 that results in an early glucocorticoid surge that acts on the immune cells to suppress cytokine production, thereby resulting in a reduced febrile response.
It is important to note that this phenomenon is not restricted to LPS. Thus, there is similar programming in response to a viral infection, and this likely occurs via a similar mechanism. We have seen that exposure to the viral mimetic polyinosinic: polycytidylic acid (Poly I:C) at P14 leads to a similar attenuated febrile response to a second, adult, exposure to the same virus (26). This programming of the immune response is specific to the type of pathogen. Neonatal LPS leads to an amplified HPA axis response to further LPS via activation of TLR4, which is more abundantly expressed in these animals. Poly I:C presumably leads to a similar amplification of the HPA axis response to further Poly I:C to suppress fever, but it does so via activation of TLR3, of which there is greater expression (26). How these changes are brought about by a bacterial or viral challenge at specifically this time window and whether they can be reversed remains to be determined.
In support of a critical role for TLRs in the programming response, we found that there was no alteration to the corticosteroid response to restraint stress after neonatal LPS or to the brief stress-induced hyperthermia in response to the needle prick of the injection (61, 79). However, it is interesting to note that, in a recent study by Soriano and Branco in which they examined open-field-induced stress fever in rats treated at P14 with LPS (76), there was a corticosterone-dependent attenuation in stress-induced PGE2 production and fever in these animals similar to what we see with LPS. At this stage, we have no explanation for the differential ability of neonatal LPS to alter responses to open-field relative to restraint stress, but it could be related to the affective magnitude of the stressor to the rat.
Interestingly, the TLR4-PG changes underlying the attenuation of the febrile response are in contrast to those seen with other programming events, such as with changes to the HPA axis caused by altered maternal attention. In this case, a different type of perinatal event (reduced maternal attention) also has permanent programming effects on HPA axis function, but it does so by chronically downregulating hippocampal glucocorticoid receptors, leading to reduced negative feedback (56, 57). Offspring of high-licking and -grooming mothers probably have a serotonin-mediated increase in nerve growth factor-inducible factor A (NGFI-A) expression. NGFI-A binds to its recognition sequence on the glucocorticoid receptor, resulting in increased histone acetylation, facilitating demethylation of the glucocorticoid receptor promoter and thus greater activity of the glucocorticoid receptor. The opposite occurs with reduced maternal attention (58).
Unresolved Issues
After P14 LPS treatment, the febrile, cytokine, and corticosterone responses to adult LPS are all altered. In association with these alterations, the pronounced hyperalgesia usually seen after LPS is also attenuated, most likely via the same mechanisms involving the reduced cytokine levels and reduced brain COX-2 activation (12). However, other responses to LPS, such as sickness behaviors like anorexia (78) and anhedonia (47), are unchanged. As the former most likely involves leptin (70) and insulin (48) action, and the latter the central monoamine systems in addition to the cytokine influence (14, 22), it will be important to determine why they do not respond in the same manner as the febrile, cytokine, and corticosterone responses. Additionally, social withdrawal is an important component of sickness behavior involving central cytokines (46), and we are unaware of any reports concerning how this behavior is altered.
We have discussed here a mechanism by which an LPS challenge at P14 can alter the neuroimmune response to a further challenge long term. However, the mechanism by which an earlier challenge (P3 and P5) affects later immune responses appears to differ. Although an altered adult corticosterone profile to LPS in P3/P5 LPS-treated rats was reported, and this is associated with a decreased glucocorticoid receptor density in the hypothalamus and hippocampus (71), this was not correlated with changes in immunological function (5, 6). That is, these rats had similar increases in plasma corticosterone with adjuvant-induced arthritis as their neonatally saline-treated counterparts but were protected from paw inflammation in this model. Bilbo et al. (5, 6) have conducted a number of elegant studies into the effects of live E. coli exposure at P4 in Sprague-Dawley rats. Unlike the effects of LPS at a similar time point (72), live E. coli during the neonatal period does not alter the adult corticosterone response to LPS (5, 6). The plasma cytokine profile is also not altered. Although it is possible that the effects of a live bacterial infection represent a much more severe inflammation than does a single low dose of LPS, these studies, along with those of Walker et al. (86) at a similar time point, suggest that the differences are due to the neonatal age when exposure takes place.
A possible explanation for these discrepancies may lie in the specific developmental time windows for immune and HPA axis function. As pointed out by Bilbo et al. (7), the very early postnatal period is a time when certain cytokine levels in the brain are naturally elevated. For instance, brain IL-1β is high just after birth in the rat, declining to adult levels by approximately P6 (33). An immune challenge at this time (at least with live E. coli) is able to further elevate brain IL-1β (5), and this potentially exacerbates or prolongs the developmental influence of this cytokine on the brain. Conversely, P14 is a point in development where HPA axis responsiveness to stress begins to acquire an adult-like profile (50), potentially a vulnerable stage in the development of HPA axis function. In this regard, we have also observed, under some housing conditions, changes in adult body weight after an LPS challenge at P7 (82, 83), an observation that has been replicated in rats treated at P10 (42). The hypothalamic pathways regulating body weight and metabolism are known to mature, in the rodent, between P6 and P12 (2, 13), making this a particularly sensitive time for these pathways. It will be essential for future studies to determine how LPS interacts with the central nervous system at particular vulnerable times during development to alter these systems.
Implications
In human newborns, bacterial infections are the primary cause of infection and hospital admissions, particularly in developing countries (64), and pre- and postnatal inflammation can have serious immediate and long-term consequences. For instance, fetal inflammation is a likely cause of preterm birth in many cases (74), and severe perinatal inflammation may also lead to neurodevelopmental impairments, including delays in cognition and psychomotor function (1), if not mortality (64). Viral infections, too, are known to have significant long-term effects on neurodevelopment, with maternal influenza being linked to increased susceptibility to schizophrenia in the offspring (16).
It is less clear what the consequences are of a very mild immune challenge at critical time points of vulnerability in humans, and this remains to be determined. It is difficult and controversial to equate developmental stages between humans and rodents. However, it is generally assumed that late gestation and the early postnatal period in the rat and mouse are roughly equivalent to the third trimester of pregnancy in humans, whereas the later postnatal period in the rodent may be more developmentally similar to a child of six months of age or more (3, 30, 34, 36). While little is known about the impact of inflammation at this time, we do know that the brains of young children (<4 years of age) are more vulnerable to deficits in motor and cognitive function than older children after traumatic brain injury (67). HPA axis function is also vulnerable during infancy to functional alterations. For instance, child abuse can lead to epigenetic regulation of glucocorticoid receptors, causing downstream alterations of CRH and potentially altering the risk for depression (55).
It is highly likely that an inflammatory challenge at this time will also have long-term effects in the human. In terms of whether these effects are likely to be negative or positive for the subject, again, more research is needed. The long-term effects of an inflammatory challenge at or near parturition in the human appear to be overwhelmingly negative (1, 16, 64, 74). However, it is also known that lack of exposure to appropriate immune stimuli in childhood can enhance the risk of developing autoimmune diseases (62, 73). We have seen in the rodent that a P14 challenge with LPS does not lead to obvious deficits in the later life ability to respond to a dose of LPS sufficicent to produce sepsis (79). We have also seen minimal effects on the animals’ behavioral profiles (80). On the other hand, we do see a very severe and clearly detrimental response to experimental colitis in the adult rat after neonatal LPS (81), an effect that appears to be reflected in the human (23, 87). Thus, the survival value or otherwise of an early-life immune challenge appears to depend largely on the physiological system under study and will also depend on genetic and other environmental factors already at play.
It is now abundantly clear that experiencing an immune challenge during the perinatal period can have far-reaching and long-term effects on many aspects of physiology. Current research has produced conflicting results regarding some of these aspects, but it is evident that there are critical windows during development when the animal is vulnerable to such challenges, and the outcomes may depend on the age, dose, type, and duration of the stimulus. It will be essential for future studies to define these critical windows of vulnerability for the system of interest and the different mechanisms by which an immune challenge can cause long-term effects at each period. It will also be important to start to translate these critical windows of vulnerability into similar developmental periods in humans. Given increasing evidence for epigenetic alterations brought about via a wide range of neonatal interventions (57–59), it is imperative that epigenetic analysis be applied to components of the innate immune response after neonatal inflammation. Whatever the mechanism, the immunological experience of a developing animal has profound implications for its long-term physiology and pathophysiology and, therefore, prognosis and treatment in the face of a variety of disorders. For those aspects thought to be detrimental to adult health [such as increased susceptibility to seizures, stroke, pain etc. (12, 29, 77)] it will be particularly interesting, from a medical perspective, to understand how to reverse the effects of a neonatal immune challenge. Given that exposure to pathogens is most likely a normal consequence of infant growth outside the relatively sterile world of an animal research facility, it would also be interesting to learn whether the altered innate immune response that results is associated with better or worse outcomes after disease.
Acknowledgments
GRANTS
Support was provided by the Australian Research Council, Canadian Institutes of Health Research (CIHR), Canadian Heart and Stroke Foundation (HSF), and personnel awards from the National Health and Medical Research Council Australia, Endocrine Society Australia, CIHR, HSF, Astra Zeneca Canada, Alberta Heritage Foundation for Medical Research Canada, the Natural Sciences and Engineering Research Council of Canada, and the Killam Trust.
Footnotes
DISCLOSURES
No conflicts of interest are reported by the authors.
References
- 1.Adams-Chapman I, Stoll BJ. Neonatal infection and long-term neuro-developmental outcome in the preterm infant. Curr Opin Infect Dis. 2006;19:290–297. doi: 10.1097/01.qco.0000224825.57976.87. [DOI] [PubMed] [Google Scholar]
- 2.Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. 1998;101:1020–1027. doi: 10.1172/JCI1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benatti C, Alboni S, Capone G, Corsini D, Caggia F, Brunello N, Tascedda F, Blom JM. Early neonatal inflammation affects adult pain reactivity and anxiety related traits in mice: genetic background counts. Int J Dev Neurosci. 2009;27:661–668. doi: 10.1016/j.ijdevneu.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. 2005;25:8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- 7.Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Blatteis CM. The onset of fever: new insights into its mechanism. Prog Brain Res. 2007;162:3–14. doi: 10.1016/S0079-6123(06)62001-3. [DOI] [PubMed] [Google Scholar]
- 10.Blatteis CM, Li S, Li Z, Perlik V, Feleder C. Signaling the brain in systemic inflammation: the role of complement. Front Biosci. 2004;9:915–931. doi: 10.2741/1297. [DOI] [PubMed] [Google Scholar]
- 11.Boisse L, Mouihate A, Ellis S, Pittman QJ. Long-term alterations in neuroimmune responses after neonatal exposure to lipopolysaccharide. J Neurosci. 2004;24:4928–4934. doi: 10.1523/JNEUROSCI.1077-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boisse L, Spencer SJ, Mouihate A, Vergnolle N, Pittman QJ. Neonatal immune challenge alters nociception in the adult rat. Pain. 2005;119:133–141. doi: 10.1016/j.pain.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 13.Bouret SG, Simerly RB. Developmental programming of hypothalamic feeding circuits. Clin Genet. 2006;70:295–301. doi: 10.1111/j.1399-0004.2006.00684.x. [DOI] [PubMed] [Google Scholar]
- 14.Brebner K, Hayley S, Zacharko R, Merali Z, Anisman H. Synergistic effects of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha: central monoamine, corticosterone, and behavioral variations. Neuropsychopharmacology. 2000;22:566–580. doi: 10.1016/S0893-133X(99)00166-9. [DOI] [PubMed] [Google Scholar]
- 15.Briggs ES, Price IR. The relationship between adverse childhood experience and obsessive-compulsive symptoms and beliefs: the role of anxiety, depression, and experiential avoidance. J Anxiety Disorders. 2009;23:1037–1046. doi: 10.1016/j.janxdis.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- 17.Cameron N, Del Corpo A, Diorio J, McAllister K, Sharma S, Meaney MJ. Maternal programming of sexual behavior and hypothalamic-pituitary-gonadal function in the female rat. PloS one. 2008;3:e2210. doi: 10.1371/journal.pone.0002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cartmell T, Ball C, Bristow AF, Mitchell D, Poole S. Endogenous interleukin-10 is required for the defervescence of fever evoked by local lipopolysaccharide-induced and Staphylococcus aureus-induced inflammation in rats. J Physiol. 2003;549:653–664. doi: 10.1113/jphysiol.2002.037291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Champagne F, Meaney MJ. Like mother, like daughter: evidence for non-genomic transmission of parental behavior and stress responsivity. Prog Brain Res. 2001;133:287–302. doi: 10.1016/s0079-6123(01)33022-4. [DOI] [PubMed] [Google Scholar]
- 20.Conti B, Tabarean I, Andrei C, Bartfai T. Cytokines and fever. Front Biosci. 2004;9:1433–1449. doi: 10.2741/1341. [DOI] [PubMed] [Google Scholar]
- 21.Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De La Garza R., 2nd Endotoxin- or pro-inflammatory cytokine-induced sickness behavior as an animal model of depression: focus on anhedonia. Neurosci Biobehav Rev. 2005;29:761–770. doi: 10.1016/j.neubiorev.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 23.Delco F, Sonnenberg A. Exposure to risk factors for ulcerative colitis occurs during an early period of life. Am J Gastroenterol. 1999;94:679–684. doi: 10.1111/j.1572-0241.1999.00936.x. [DOI] [PubMed] [Google Scholar]
- 24.Doyle SL, O’Neill LA. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol. 2006;72:1102–1113. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Ellis S, Mouihate A, Pittman QJ. Early life immune challenge alters innate immune responses to lipopolysaccharide: implications for host defense as adults. FASEB J. 2005;19:1519–1521. doi: 10.1096/fj.04-3569fje. [DOI] [PubMed] [Google Scholar]
- 26.Ellis S, Mouihate A, Pittman QJ. Neonatal programming of the rat neuroimmune response: stimulus specific changes elicited by bacterial and viral mimetics. J Physiol. 2006;571:695–701. doi: 10.1113/jphysiol.2005.102939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukocyte Biol. 2010;87:989–999. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- 28.Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet. 2010;11:47–59. doi: 10.1038/nrg2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28:6904–6913. doi: 10.1523/JNEUROSCI.1901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galic MA, Spencer SJ, Mouihate A, Pittman QJ. Postnatal programming of the innate immune response. Integr Comp Biol. 2009;49:237–245. doi: 10.1093/icb/icp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaykema RP, Dijkstra I, Tilders FJ. Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology. 1995;136:4717–4720. doi: 10.1210/endo.136.10.7664696. [DOI] [PubMed] [Google Scholar]
- 32.Gilmer WS, McKinney WT. Early experience and depressive disorders: human and non-human primate studies. J Affect Disord. 2003;75:97–113. doi: 10.1016/s0165-0327(03)00046-6. [DOI] [PubMed] [Google Scholar]
- 33.Giulian D, Young DG, Woodward J, Brown DC, Lachman LB. Interleukin-1 is an astroglial growth factor in the developing brain. J Neurosci. 1988;8:709–714. doi: 10.1523/JNEUROSCI.08-02-00709.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gottlieb A, Keydar I, Epstein HT. Rodent brain growth stages: an analytical review. Biol Neonate. 1977;32:166–176. doi: 10.1159/000241012. [DOI] [PubMed] [Google Scholar]
- 35.Goujon E, Laye S, Parnet P, Dantzer R. Regulation of cytokine gene expression in the central nervous system by glucocorticoids: mechanisms and functional consequences. Psychoneuroendocrinology. 1997;22(Suppl 1):S75–S80. doi: 10.1016/s0306-4530(97)00009-7. [DOI] [PubMed] [Google Scholar]
- 36.Grove KL, Grayson BE, Glavas MM, Xiao XQ, Smith MS. Development of metabolic systems. Physiol Behav. 2005;86:646–660. doi: 10.1016/j.physbeh.2005.08.063. [DOI] [PubMed] [Google Scholar]
- 37.Harre EM, Galic MA, Mouihate A, Noorbakhsh F, Pittman QJ. Neonatal inflammation produces selective behavioural deficits and alters N-methyl-D-aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci. 2008;27:644–653. doi: 10.1111/j.1460-9568.2008.06031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 39.Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49:1023–1039. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- 40.Hodgson DM, Knott B, Walker FR. Neonatal endotoxin exposure influences HPA responsivity and impairs tumor immunity in Fischer 344 rats in adulthood. Pediatr Res. 2001;50:750–755. doi: 10.1203/00006450-200112000-00020. [DOI] [PubMed] [Google Scholar]
- 41.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 42.Iwasa T, Matsuzaki T, Kinouchi R, Fujisawa S, Murakami M, Kiyokawa M, Kuwahara A, Yasui T, Irahara M. Neonatal LPS injection alters the body weight regulation systems of rats under non-stress and immune stress conditions. Int J Dev Neurosci. 2010;28:119–124. doi: 10.1016/j.ijdevneu.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 43.Jiang Q, Cross AS, Singh IS, Chen TT, Viscardi RM, Hasday JD. Febrile core temperature is essential for optimal host defense in bacterial peritonitis. Infect Immunol. 2000;68:1265–1270. doi: 10.1128/iai.68.3.1265-1270.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kapcala LP, He JR, Gao Y, Pieper JO, DeTolla LJ. Subdiaphragmatic vagotomy inhibits intra-abdominal interleukin-1 beta stimulation of adrenocorticotropin secretion. Brain Res. 1996;728:247–254. doi: 10.1016/0006-8993(96)00511-2. [DOI] [PubMed] [Google Scholar]
- 45.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 46.Kent S, Bluthe RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, Vannice JL. Different receptor mechanisms mediate the pyrogenic and behavioral effects of interleukin 1. Proc Natl Acad Sci USA. 1992;89:9117–9120. doi: 10.1073/pnas.89.19.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kentner AC, McLeod SA, Field EF, Pittman QJ. Sex-dependent effects of neonatal inflammation on adult inflammatory markers and behavior. Endocrinology. 2010;151:2689–2699. doi: 10.1210/en.2009-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YW, Kim KH, Ahn DK, Kim HS, Kim JY, Lee DC, Park SY. Time-course changes of hormones and cytokines by lipopolysaccharide and its relation with anorexia. J Physiol Sci. 2007;57:159–165. doi: 10.2170/physiolsci.RP003407. [DOI] [PubMed] [Google Scholar]
- 49.Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. Role of fever in disease. Ann NY Acad Sci. 1998;856:224–233. doi: 10.1111/j.1749-6632.1998.tb08329.x. [DOI] [PubMed] [Google Scholar]
- 50.Levine S. Primary social relationships influence the development of the hypothalamic-pituitary-adrenal axis in the rat. Physiol Behav. 2001;73:255–260. doi: 10.1016/s0031-9384(01)00496-6. [DOI] [PubMed] [Google Scholar]
- 51.Li Z, Blatteis CM. Fever onset is linked to the appearance of lipopolysaccharide in the liver. J Endotoxin Res. 2004;10:39–53. doi: 10.1179/096805104225003825. [DOI] [PubMed] [Google Scholar]
- 52.Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci. 2000;3:799–806. doi: 10.1038/77702. [DOI] [PubMed] [Google Scholar]
- 53.Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- 54.McEwen BS. Early life influences on life-long patterns of behavior and health. Ment Retard Dev Disabil Res Rev. 2003;9:149–154. doi: 10.1002/mrdd.10074. [DOI] [PubMed] [Google Scholar]
- 55.McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meaney MJ, Diorio J, Francis D, Widdowson J, LaPlante P, Caldji C, Sharma S, Seckl JR, Plotsky PM. Early environmental regulation of forebrain glucocorticoid receptor gene expression: implications for adrenocortical responses to stress. Dev Neurosci. 1996;18:49–72. doi: 10.1159/000111395. [DOI] [PubMed] [Google Scholar]
- 57.Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005;28:456–463. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 59.Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13:269–277. doi: 10.1016/j.molmed.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 60.Morrison SF, Nakamura K, Madden CJ. Central control of thermogenesis in mammals. Exp Physiol. 2008;93:773–797. doi: 10.1113/expphysiol.2007.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mouihate A, Galic MA, Ellis SL, Spencer SJ, Tsutsui S, Pittman QJ. Early life activation of toll-like receptor 4 reprograms neural anti-inflammatory pathways. J Neurosci. 2010;30:7975–7983. doi: 10.1523/JNEUROSCI.6078-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okada H, Kuhn C, Feillet H, Bach JF. The “hygiene hypothesis” for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ootsuka Y, Blessing WW, Steiner AA, Romanovsky AA. Fever response to intravenous prostaglandin E2 is mediated by the brain but does not require afferent vagal signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1294–R1303. doi: 10.1152/ajpregu.00709.2007. [DOI] [PubMed] [Google Scholar]
- 64.Osrin D, Vergnano S, Costello A. Serious bacterial infections in newborn infants in developing countries. Curr Opin Infect Dis. 2004;17:217–224. doi: 10.1097/00001432-200406000-00008. [DOI] [PubMed] [Google Scholar]
- 65.Perlik V, Li Z, Goorha S, Ballou LR, Blatteis CM. LPS-activated complement, not LPS per se, triggers the early release of PGE2 by Kupffer cells. Am J Physiol Regul Integr Comp Physiol. 2005;289:R332–R339. doi: 10.1152/ajpregu.00567.2004. [DOI] [PubMed] [Google Scholar]
- 66.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 67.Pullela R, Raber J, Pfankuch T, Ferriero DM, Claus CP, Koh SE, Yamauchi T, Rola R, Fike JR, Noble-Haeusslein LJ. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev Neurosci. 2006;28:396–409. doi: 10.1159/000094166. [DOI] [PubMed] [Google Scholar]
- 68.Rivest S. How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology. 2001;26:761–788. doi: 10.1016/s0306-4530(01)00064-6. [DOI] [PubMed] [Google Scholar]
- 69.Rummel C, Inoue W, Sachot C, Poole S, Hubschle T, Luheshi GN. Selective contribution of interleukin-6 and leptin to brain inflammatory signals induced by systemic LPS injection in mice. J Comp Neurol. 2008;511:373–395. doi: 10.1002/cne.21850. [DOI] [PubMed] [Google Scholar]
- 70.Sachot C, Poole S, Luheshi GN. Circulating leptin mediates lipopolysaccharide-induced anorexia and fever in rats. J Physiol. 2004;561:263–272. doi: 10.1113/jphysiol.2004.074351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shanks N, Larocque S, Meaney MJ. Neonatal endotoxin exposure alters the development of the hypothalamic-pituitary-adrenal axis: early illness and later responsivity to stress. J Neurosci. 1995;15:376–384. doi: 10.1523/JNEUROSCI.15-01-00376.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shanks N, Windle RJ, Perks PA, Harbuz MS, Jessop DS, Ingram CD, Lightman SL. Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proc Natl Acad Sci USA. 2000;97:5645–5650. doi: 10.1073/pnas.090571897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sherriff A, Golding J. Hygiene levels in a contemporary population cohort are associated with wheezing and atopic eczema in preschool infants. Arch Dis Child. 2002;87:26–29. doi: 10.1136/adc.87.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skogstrand K, Hougaard DM, Schendel DE, Bent NP, Svaerke C, Thorsen P. Association of preterm birth with sustained postnatal inflammatory response. Obstet Gynecol. 2008;111:1118–1128. doi: 10.1097/AOG.0b013e31817057fb. [DOI] [PubMed] [Google Scholar]
- 75.Smith EL, Batuman OA, Trost RC, Coplan JD, Rosenblum LA. Transforming growth factor-beta 1 and cortisol in differentially reared primates. Brain Behav Immun. 2002;16:140–149. doi: 10.1006/brbi.2001.0629. [DOI] [PubMed] [Google Scholar]
- 76.Soriano RN, Branco LG. Reduced stress fever is accompanied by increased glucocorticoids and reduced PGE(2) in adult rats exposed to endotoxin as neonates. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 77.Spencer SJ, Auer RN, Pittman QJ. Rat neonatal immune challenge alters adult responses to cerebral ischaemia. J Cereb Blood Flow Metab. 2006;26:456–467. doi: 10.1038/sj.jcbfm.9600206. [DOI] [PubMed] [Google Scholar]
- 78.Spencer SJ, Boisse L, Mouihate A, Pittman QJ. Long term alterations in neuroimmune responses of female rats after neonatal exposure to lipopolysaccharide. Brain Behav Immun. 2006;20:325–330. doi: 10.1016/j.bbi.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 79.Spencer SJ, Field E, Pittman QJ. Neonatal programming by neuroimmune challenge: effects on responses and tolerance to septic doses of lipopolysaccharide in adult male and female rats. J Neuroendocrinol. 2010;22:272–281. doi: 10.1111/j.1365-2826.2010.01967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spencer SJ, Heida JG, Pittman QJ. Early life immune challenge-effects on behavioural indices of adult rat fear and anxiety. Behav Brain Res. 2005;7;164:231–238. doi: 10.1016/j.bbr.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 81.Spencer SJ, Hyland NP, Sharkey KA, Pittman QJ. Neonatal immune challenge exacerbates experimental colitis in adult rats: potential role for TNF-α. Am J Physiol Regul Integr Comp Physiol. 2007;292:R308–R315. doi: 10.1152/ajpregu.00398.2006. [DOI] [PubMed] [Google Scholar]
- 82.Spencer SJ, Martin S, Mouihate A, Pittman QJ. Early-life immune challenge: defining a critical window for effects on adult responses to immune challenge. Neuropsychopharmacology. 2006;31:1910–1918. doi: 10.1038/sj.npp.1301004. [DOI] [PubMed] [Google Scholar]
- 83.Spencer SJ, Mouihate A, Galic MA, Ellis SL, Pittman QJ. Neonatal immune challenge does not affect body weight regulation in rats. Am J Physiol Regul Integr Comp Physiol. 2007;293:R581–R589. doi: 10.1152/ajpregu.00262.2007. [DOI] [PubMed] [Google Scholar]
- 84.Spencer SJ, Tilbrook A. Neonatal overfeeding alters adult anxiety and stress responsiveness. Psychoneuroendocrinology. 2009;34:1133–1143. doi: 10.1016/j.psyneuen.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, Roberts JL, Kobayashi S, Matsumura K, Sawchenko PE, Romanovsky AA. Cellular and molecular bases of the initiation of fever. PLoS Biol. 2006;4:e284. doi: 10.1371/journal.pbio.0040284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walker FR, Hodyl NA, Krivanek KM, Hodgson DM. Early life host-bacteria relations and development: long-term individual differences in neuroimmune function following neonatal endotoxin challenge. Physiol Behav. 2006;87:126–134. doi: 10.1016/j.physbeh.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 87.Wurzelmann JI, Lyles CM, Sandler RS. Childhood infections and the risk of inflammatory bowel disease. Dig Dis Sci. 1994;39:555–560. doi: 10.1007/BF02088342. [DOI] [PubMed] [Google Scholar]