

Abstract
Repeat Expansion Diseases result from expansion of a specific tandem repeat. The three Fragile X-related disorders (FXDs) arise from germline expansions of a CGG•CCG repeat tract in the 5′ UTR of the FMR1 gene. We show here that in addition to germline expansion, expansion also occurs in the somatic cells of both mice and humans carriers of premutation alleles. Expansion in mice primarily affects brain, testis and liver with very little expansion in heart or blood. Our data would be consistent with a simple two-factor model for the organ specificity. Somatic expansion in humans may contribute to the mosaicism often seen in individuals with one of the FXDs. Since expansion risk and disease severity are related to repeat number, somatic expansion may exacerbate disease severity and contribute to the age-related increased risk of expansion seen on paternal transmission in humans. Since little somatic expansion occurs in murine lymphocytes, our data also raise the possibility that there may be discordance in humans between repeat numbers measured in blood and that present in brain. This could explain, at least in part, the variable penetrance seen in some of these disorders.
Keywords: Fragile X-related disorders, FMR1, somatic expansion, MutSβ
Introduction
The Repeat Expansion Diseases (REDs) are a group of human genetic conditions that result from expansion of a particular tandem repeat tract in a specific gene. The Fragile X-related disorders (FXDs) result from expansions of a CGG•CCG-repeat tract in the 5′ untranslated region (UTR) of the Fragile X mental retardation 1 gene (FMR1; MIM# 309550). Fragile X (FX)-associated tremor and ataxia syndrome (FXTAS) (Hagerman, et al., 2001) and FX-associated primary ovarian insufficiency (FXPOI) (Allingham-Hawkins, et al., 1999; Conway, et al., 1998; Murray, et al., 1998; Vianna-Morgante, et al., 1996) result from inheritance of FMR1 alleles with 55–200 repeats. Such alleles are referred to as FX premutation alleles (PM). FXTAS is a neurodegenerative disorder that results in gait and balance abnormalities, cognitive decline, dementia and executive function deficits. FXPOI symptoms include menstrual irregularities, fertility problems and an early menopause. FXPOI is thought to account for ~14% of familial cases of infertility and ~3.5% of sporadic cases (Conway, et al., 1998; Marozzi, et al., 2000; Murray, et al., 1998; Uzielli, et al., 1999). Evidence suggests that the pathology in FXTAS, and probably FXPOI, arises because of some deleterious consequence of the elevated expression of RNA containing long CGG-repeat tracts (Clark, et al., 2006; Hashem, et al., 2009; Jin, et al., 2003).
In humans, PM alleles are thought to arise at some low frequency from normal alleles and are prone to expand further on intergenerational transfer. Paternal transmissions of such alleles generate small increases in repeat number with an effect of paternal age being evident (Ashley-Koch, et al., 1998). Inheritance of such expanded alleles can have two consequences. Since PM pathology is related to repeat number (Tassone, et al., 2007), this could result in a gradual increase in disease severity in families with the PM. Furthermore, since repeat number is related to the risk of further expansion, it could also affect expansion risk on subsequent intergenerational transmissions. Maternal transmissions of PM alleles generate large increases in repeat number. When the repeat number exceeds 200, a third disorder, FX syndrome (FXS), the most common heritable cause of intellectual disability, is seen in those who inherit such alleles.
The REDs-associated repeats form a variety of secondary structures including hairpins, tetraplexes and triplexes (reviewed in (Mirkin, 2007)). It is generally thought that expansions arise due to the aberrant processing of these structures. Work done with different REDs-associated repeats in human cell extracts, human cell lines, bacteria and yeast suggest that any one of a variety of biological processes including replication, transcription, DNA repair and recombination could be responsible (reviewed in (Mirkin, 2007)). We have previously shown that the ATM (ataxia-telangiectasia, mutated) and the ATR (ATM and Rad3-related) DNA damage response pathways are involved in reducing the risk of expansion during intergenerational transfer (Entezam and Usdin, 2008; Entezam and Usdin, 2009) and that factors like oxidative stress can exacerbate expansion risk (Entezam, et al., 2010). While the molecular details of the mechanism responsible for repeat expansions remain unclear, these studies suggest that most, if not all, of these intergenerational expansions occur in the gamete by a process that likely involves aberrant DNA repair. However, whether all REDs share the same mechanism for intergenerational expansions is unclear.
Somatic expansions are seen in many of the other REDs (Chong, et al., 1995; De Biase, et al., 2007; Manley, et al., 1999a; Tanaka, et al., 1999; Telenius, et al., 1994; Ueno, et al., 1995). Evidence from mouse models of these diseases suggests that such expansions can exacerbate disease severity and reduce the age at which disease symptoms become apparent (Wheeler, et al., 2003). Whether somatic expansion involves the same mechanism as intergenerational expansion is unknown. Somatic expansion of FX PM alleles has not yet been reported, although mosaicism with respect to CGG•CCG-repeat numbers has been observed in many carriers of FM alleles. While many of the heterogeneous products seen on Southern Blot analysis of FM alleles represent an artifact of electrophoresis in the presence of ethidium bromide, more than one allele size is frequently seen even when analysis is done in the absence of this intercalator (Nolin, et al., 2008). A striking example has been reported of an individual who has unmethylated repeats ranging in size from the PM to the FM range in most parts of the brain but a single methylated FM allele in the parietal lobe and most non-brain tissues studied (Taylor, et. al., 1999). However, without knowing the size of the original allele, it is difficult to know whether the smaller alleles arise from somatic contractions of a larger original allele or the larger alleles arise by somatic expansion of smaller ones. Since the consequences of repeat expansion are thought to result from the combination of FMR1 mRNA levels and the number of repeats (Jin, et al., 2003), true somatic expansion, should it occur in FX PM carriers, would have the potential to produce a transcript that would be more deleterious than the original allele. It could also lead to an increased risk of larger expansions with increasing parental age.
We have previously described the generation of a knock-in mouse model for the FX PM which recapitulates features of both the neurodegeneration (Entezam, et al., 2007) and ovarian dysfunction (Hoffman, et al., 2012) seen in human PM carriers as well as the intergenerational repeat expansion seen in this population (Entezam, et al., 2007; Entezam, et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009). We show here that somatic expansion is also seen in these mice. Some organs are more susceptible to expansion than others, with the brain, liver and gonads being particularly expansion-prone. Expansions are also seen in a lymphoblastoid cell line from a human PM carrier and in the brains of FXTAS patients. Our data may have implications not only for the understanding of the expansion mechanism but also for the assessment of expansion risk in families with PM alleles.
Materials and Methods
Mouse and human samples
The generation of the FX PM mice was described previously (Entezam, et al., 2007). Mice were maintained in accordance with the guidelines of the NIDDK Animal Care and Use Committee and with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1996). Sperm was isolated from mouse epididymis using standard procedures (Nagy, et al., 2003). Somatic and germ line cells were isolated from testes as previously described (Wu, et al., 2009). Cells from different regions of the mouse brain were isolated as follows. Mice were anesthetized with sodium pentobarbital (100 mg/kg, i.p.), decapitated and the brains frozen in dry ice. The cerebellum was dissected and different brain regions were punched by means of Harris Uni-Core (1.25 mm) (Electron Microscopy Sciences, Hatfield, PA) in a Leica cryostat set at −22 °C.
Human tissue samples from individuals with FXTAS were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders (Baltimore, MD). DNA isolated from the blood of one of these individuals was a kind gift from Sally Nolin (Institute for Basic Research in Developmental Disabilities, NJ). The human lymphoblastoid cell line, GM06891, was obtained from the Coriell Institute for Medical Research (Camden, NJ) and maintained in culture using RPMI 1640 with GlutaMax, 10 % heat inactivated fetal bovine serum and 1X antibiotics (Life Technologies, Grand Island, NY) at 37°C in a 5% CO2 atmosphere. The identity of the original cell line and subsequent passages showing expansion was confirmed by SNP analysis of 26 loci using the Sequenom MassARRAY iPLEX™ platform (Bioserve Biotechnologies, Beltsville, MD).
Quantitation of mRNA
Total RNA from different mouse organs was isolated using Trizol™ (Life Technologies) as per the manufacturer’s instructions. The RNA was then treated with DNase I and reverse transcribed using a SuperScript® VILO™ cDNA synthesis kit (Life Technologies). Real time PCR was done in triplicate using TaqMan® Fast universal PCR master mix and the appropriate Taqman probe-primer pairs (Applied Biosystems). Since the expression levels of the endogenous controls tested (β-actin, Gapdh and Ubc9) differed in various organs, they could not be used for normalization. However, equal amounts of RNA were used in these experiments and so normalization was carried out by comparing the Ct value for the mRNA in different organs to the Ct value obtained for heart.
Analysis of repeat length
Genomic DNA was prepared from mouse and human samples as previously described (Entezam, et al., 2007). The size of the mouse CGG•CCG-repeat tract was determined as described previously (Lavedan, et al., 1998) except that primer frax-m4 was either labeled with 6-carboxyfluorescein (FAM) or 4,7,2′,4′,5′,7′-hexachloro-6-carboxyfluorescein (HEX). The 5′ and 3′ primers are 25 and 24 bases long respectively. The primer binding sites are located immediately adjacent to the repeat and so the PCR product corresponds to the length of the repeat with 25 additional base pairs on the 5′ side of the repeat, and 24 additional base pairs on the 3′ side. The human repeats were analyzed in the same way except that HEX-labeled frax-F (5′-AGCCCCGCACTTCCACCACCAGTCTCCTCCA-3′) and frax-AF (5′-GCTGCCAGGGGGCGTGCGGCA-3′) primers were used. This primer pair produces a PCR product that contains the repeat and 77 bp of DNA from the upstream flank and 41 bp from the downstream flank for a total of 124 bp of flanking DNA. The somatic instability index was calculated for each organ using 3–5 mice from each age group (Lee, et al., 2010).
Determination of Msh2, Msh3 and Msh6 protein levels in different mouse organs
Organs were collected from 2 months old PM mice, the proteins isolated, resolved by SDS-PAGE and Western Blotted using standard procedures. Msh2, Msh3 and Msh6 were detected using the antibodies AB70270 (Abcam, Cambridge, MA), AB74607 (Abcam) and 610918 (BD Biosciences, Sparks, MD) respectively, and an ECL prime kit (GE Healthcare) used according to the suppliers’ instructions. Purified human MSH2/MSH3 used as a positive control was a kind gift of Shikha Gupta (NIH).
Results
A unique pattern of organ-specific somatic expansion is seen in FX PM mice
To assess whether somatic instability occurs in FX PM mice we isolated DNA from different organs of young and old male mice with ~130 CGG•CCG repeats. The DNA was amplified by PCR, resolved on a 3130XL Genetic Analyzer and analyzed using GeneMapper 4.0 software. Typically the resultant GeneMapper profiles have a Gaussian distribution of products that differ from each other by one repeat. Products larger and smaller than the mean allele size are sometimes referred to as “stutter-products”. They thought to be due, in part, to strand-slippage during PCR. However, work with mutants that abolish expansion suggests this explanation accounts for the smaller products but not all of the larger products (Lokanga and Usdin, manuscript in preparation). In samples containing a mixture of alleles with different repeat numbers, as would be seen if expansions or deletions had occurred in the tissue from which the sample was derived, the product distribution is broader, right-shifted or left-shifted in the case of expansions and deletions respectively, and may contain multiple peaks as new derivative alleles arise in the cell population.
The number of CGG•CCG-repeats in the DNA from different organs was determined based on the mobility of the most abundant PCR product, and compared to the repeat number found in the tail DNA of the same animals at three weeks of age. When DNA from the organs of a number of PM mice was tested at two months of age, not only was the repeat number no different from the repeat number in the original tail DNA sample in all the mice tested, but the GeneMapper profiles had the same relatively narrow distribution of PCR products (see Fig. 1 for a representative example). This indicates that little or no somatic instability occurs during early post-natal life in FX PM mice. There was one notable exception. Testes DNA showed a small right-shift of the PCR products, with the most abundant product now having a mobility consistent with the addition of one repeat unit. A low level of somatic expansion was seen in animals at 4 months of age as evidenced by a broadening and/or skewing of the GeneMapper peak profile in the case of brain and liver or by a rightwards shift in the peak distribution towards alleles with ~2 additional repeats in testes (Fig. 1, middle of panel A). By 12 months of age expansions were much more extensive in all animals tested, with clear evidence of expansion seen in a variety of organs particularly brain, liver and testes (Fig. 1, right hand side of panel A). Unlike what has been reported for mouse models of other REDs, kidney showed relatively little expansion. Heart and blood showed even less expansion, even in mice that were more than 24 months of age (Fig. 1, panels A and B and data not shown). No deletions were observed. The extent of somatic expansion at any given age varied with repeat number but was comparable in animals with similar numbers of repeats. The change in the average somatic instability index with age for mice with 141–145 repeats is shown in Fig. 1C.
Fig. 1. Age and organ dependent somatic expansions in FX PM mice.

A) GeneMapper profiles were obtained for the repeat tract in different organs of male mice of different ages. Representative results for mice at 3 different ages are shown. The grey dotted vertical line in each panel indicates the position of the original sized allele based on the repeat number in the tail DNA taken at weaning (3 weeks of age; tail 1). The second tail DNA sample in each case (tail 2) was taken after euthanasia. The repeat DNA profiles of the 12 month old animal was generated using a HEX-labeled primer. The repeat profiles for the 2 and 4 month old animals were generated using a FAM-labeled primer. The choice of primer label does not affect the repeat profile. A LIZ600 molecular weight marker was used for the analysis of the 2 and 4 month old mice, and a ROX500 molecular weight marker was used for the 12 month old animal. B) GeneMapper profiles from blood and heart of a 12 month old animal. C) Graph showing the average somatic instability index for mice with 141–145 repeats at 2, 4 and 12 months.
Different expansion profiles are apparent in different organs
In liver and testes the allele profile frequently showed two major allele sizes, one corresponding to the original allele and one that was 7–14 repeats larger. In total brain a pattern was seen that was consistent with the addition of a smaller number of repeats. Different regions within the brain also showed different expansion profiles with thalamus showing relatively little expansion, while the striatum and basolateral amygdala showed more expansions with a “jump size” that was 6 and 8 repeats respectively (Fig. 2).
Fig. 2. Regional variation in the repeat profiles in the brain of a FX PM mouse.

A ROX500 molecular weight marker was used for all samples. The dotted line indicates the mobility of the original allele as assessed from the tail DNA taken at weaning. GeneMapper profiles for the FX repeat were obtained from different regions of the brain of a 12 month old PM male mouse with ~131 repeats. Similar profiles were seen in two other mice that were tested.
High levels of expansion are seen in the non-somatic cells of the testis
The repeat profile in sperm isolated from 12 months old animals differs from that seen in total testes DNA from the same animal (Fig. 3A). In sperm a single peak was seen that was 5 repeats larger than the original allele, while in testes two peaks are seen, one corresponding to the original sized allele and one corresponding to an allele that was 13 repeats larger. Small pool (SP) PCR shows that expansion occurs in ~75% of sperm with deletions being seen at a frequency of ~10% (Fig. 3B and data not shown). The expansion frequency and average expansion size in sperm is very similar to that seen in the offspring of males with the same repeat number (Entezam, et al., 2007; Entezam, et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009). Thus expansions occurring on paternal transmission of the PM allele probably occur predominantly prezygotically consistent with our previous observations (Entezam, et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009). Within the testis of a mouse that was 11 months old expansions are apparent in both the fraction of cells enriched for primary spermatocytes and the fraction enriched for spermatogonial stem cells (Fig. 3C). However, no expansions were seen in the somatic cell fraction. No evidence of expansions was seen in either somatic or germ cells of prepubertal (18 days old) mice (Fig. 3D), suggesting that the expansions do not arise in the primordial germ cells or prospermatogonia, but presumably begin to accumulate in germ cells during the peri-pubertal period as spermatogenesis progresses.
Fig. 3. Cell-specific and age related expansion in the testes.

The dotted line in each panel represents the original allele size (~140 repeats) based on the tail DNA at 3 weeks of age in the case of the data shown in Panels A, B and C and 18 days in the case of the data shown in Panel D. The repeat profile in Panel A was obtained with a HEX-labeled primer and the repeat profiles shown in B, C and D were obtained using a FAM-labeled primer. The choice of primer label does not affect the repeat profile. The numbers in panel A, B and C refer to the number of repeats added to the major allele in the population relative to the repeats present in the original allele. A) Comparison of the repeat number in tail DNA taken at weaning and in total testes DNA and sperm DNA from a 12 month old animal. B) Comparison of the repeat number in tail taken at weaning with the repeat number in the products of 3 independent small pool PCRs (labeled a, b and c) carried out on the sperm of the same animal at 12 months of age. C) and D) Comparison of the repeat profile in the tail DNA taken at weaning and in the fractions enriched for the indicated cell type in the testes of mice at 11 months (C) and 18 days (D).
Somatic expansion is not related to proliferative capacity
It has been suggested that expansion is the result of a problem arising during DNA replication. Liver is an organ that has one of the highest levels of expansion, with this first becoming apparent in adult mice. Yet very little cell division or DNA synthesis occurs in the livers of adult rodents (Bohman, et al., 1985). Similarly brain, which contains a high proportion of slow growing glia and non-dividing neurons, shows extensive expansion. The amount of expansion differed in different regions of the brain with the striatum, basolateral amygdala, and cerebellum showing larger expansions than the frontal cortex or thalamus (Fig. 2). In some regions of the brains of some mice, expansion was so extensive that most alleles present in the population were larger than the original allele (for example in the cerebellum shown in Fig. 2).
FMR1 transcription levels do not explain the tissue specificity of expansion
Transcription of the gene in which the repeat is located has also been suggested to be a major factor in the expansion process. We thus measured Fmr1 mRNA levels in the organs of the FX PM mice. While Fmr1 mRNA levels are low in heart (Fig. 4A), which could explain the low rate of expansion in this organ, Fmr1 mRNA levels in kidney, which shows very little expansion, are much higher than they are in liver, which shows high levels of expansion. Hence, there is no simple relationship between the levels of Fmr1 transcription and somatic expansion frequency.
Fig. 4. Fmr1 mRNA and Msh2, Msh3 and Msh6 protein levels in different organs of FX PM mice.

A) Fmr1 mRNA levels in different organs were determined as described in the Materials and Methods. The average mRNA levels from 3 animals was calculated and the Fmr1 transcript levels in brain, kidney, liver and testis normalized to the levels in heart. B) Levels of Msh2, Msh3 and Msh6 proteins in different organs. Representative data from one mouse is shown. B: brain, H: heart; L: liver; K: kidney; and T: testes. Similar levels of proteins were seen in two other animals tested.
Levels of the mismatch repair proteins Msh2, Msh3 and Msh6 also do not explain the tissue specificity
The mismatch repair proteins Msh2 and Msh3 form a complex, MutSβ, which is required for expansion in mouse models of some of the other REDs (Foiry, et al., 2006; Manley, et al., 1999b; Tome, et al., 2009; Wheeler, et al., 2003) and which our data suggests is also required for germline and somatic expansion in the FX PM mice (Lokanga and Usdin, manuscript in preparation). Since the mRNA levels for Msh2, Msh3, and the alternate binding partner of Msh2, Msh6, do not correlate well with the levels of their respective proteins (compare Fig. 4A with Supp. Figure S1), we compared the levels of these proteins with the tendency of the heart, brain, liver, kidney and testes to show expansion. Since Msh3 levels are thought to be rate limiting for the formation of the MutSβ complex (Chang, et al., 2000; Genschel, et al., 1998; Marra, et al., 1998), and free Msh3 is rapidly degraded, the level of Msh3 in any organ is thought to be a good indicator of the amount of MutSβ present. Since brain and testes, organs that have high levels of expansion also have high levels of Msh3, this would be consistent with the idea that the levels of Msh3/MutSβ determine the propensity of a given organ to undergo expansion. However, the fact that heart, kidney and liver all have similar levels of Msh3, would not be consistent with that hypothesis since all of these organs show very different levels of somatic expansion.
Expansion from a PM allele to an FM allele can be seen in a human lymphoblastoid cell line
The somatic expansion seen in mice led us to examine human DNA samples in our collection that had been isolated from a lymphoblastoid cell line, GM06891, over a period of 42 months. This cell line originally had a single predominant allele of ~118 repeats (t0). We found that in samples isolated after two years of intermittent propagation, very little of the original allele was detectable and alleles that were ~16, ~35 and 51 repeats larger were apparent with the largest allele predominating (t1 in Fig. 5). A later sample (t2) showed that the most prominent allele in the sample had ~195 repeats, i.e., it was 77 repeats larger than the original allele. In addition there was a small amount of alleles that were ~45, 62 and 93–96 repeats larger than the original allele. Since the PCR yield decreases with increasing repeat number, the low yield of these fragments is not surprising. Nonetheless, these results were verified by Southern Blotting and a different PCR technique by the Institute for Basic Research on Developmental Disabilities (Staten Island, NY). The results, which confirm our findings, are shown in Supp. Figure S2.
Fig. 5. Somatic expansions in a human PM lymphoblastoid cell line.

GeneMapper profiles for the PM repeat obtained for the GM06891 lymphoblastoid cell line after intermittent growth in tissue culture. Three different samples are shown, t0, corresponding to the DNA isolated from cells when they were first obtained from the cell repository, and samples t1 and t2 that were isolated at 2 later time points. In all 3 samples the same amount of LIZ1200 standard was used. However because of the poor PCR yield of the sample at t2, a phenomenon typical of amplification through long repeat tracts, this data is shown on an expanded scale. The last panel shows the repeat profiles from all three time points superimposed on one another with the red profile representing the original allele and the green and blue profiles corresponding to samples t1 and t2. The data in this panel are all shown on the same scale. The dotted line indicates the size of the original allele. The numbers associated with each peak indicates the number of repeats added from the original allele at t0.
The identity of the original cell line and subsequent passages showing expansion was confirmed by SNP analysis using 26 loci. The chance that the cell populations containing expansions were not derived from the original cell line is 1.042×10−11. Since selection is thought to operate in favor of shorter alleles that are likely to make more FMRP (Salat, et al., 2000), clonal selection of pre-existing alleles in the population seems unlikely an explanation for the emergence of these larger alleles. Thus the changes in repeat number that we observed likely represent bona fide somatic expansions. As with some mouse tissues, expansion in this cell line also seemed to occur in a saltatory fashion with a jump size in this instance of 15–19 repeats. In this case, the cumulative expansions resulted in the conversion of the original PM allele to a small FM-sized allele in a subset of cells. Work is currently in progress to determine the rate at which these expansions arise and to identify the factors that led to their generation.
Somatic expansions are also seen in human brain
The in vitro expansion of the FX repeats in a human cell line raised the possibility that somatic expansions may also occur in humans in vivo. To test this idea we examined autopsy samples from three individuals with FXTAS. These individuals had repeat numbers in blood of 67, 82 and 91 at diagnosis. The 91 repeat allele had one AGG interruption at the 5′ end of the allele (S. Nolin, personal communication). The interspersion pattern of the other alleles is unknown. Only two regions of the brain were available from the patient with 67 repeats, the prefrontal cortex and the cerebellum. For the patient with the 67 repeat allele, very similar GeneMapper profiles were obtained for both regions with a narrow distribution of PCR products consistent with little or no instability (data not shown). In the case of the individual with 82 repeats, tissue from heart was available. Since mouse heart shows little, if any, instability we rationalized that the human heart sample may have an allele size very similar to the original. Very subtle differences in the repeat profile were seen in this individual (Supp. Figure S3). No heart DNA was available from the individual with 91 repeats. However, a blood sample was available that had been taken from that individual 6 years prior to death. While no evidence of a change in the size of the allele relative to blood was found in the striatum or cerebellum, the amygdala showed evidence of the presence of a small amount of an allele that was ~13 repeats larger than the allele seen in blood (Fig. 6). The frontal cortex showed a slight shift in the major peak corresponding to an increase of one repeat. There was also a small peak corresponding to an allele that was ~8 repeats larger, and a smaller peak just discernable above background that was ~19 repeats larger than the allele seen in blood. Whether the expansions occur uniformly in these brain regions or occur preferentially in specific cell populations or subregions of the cortex and amygdala remains to be seen.
Fig. 6. Expansions differ in different regions of the brain of a FXTAS patient with 91 repeats.
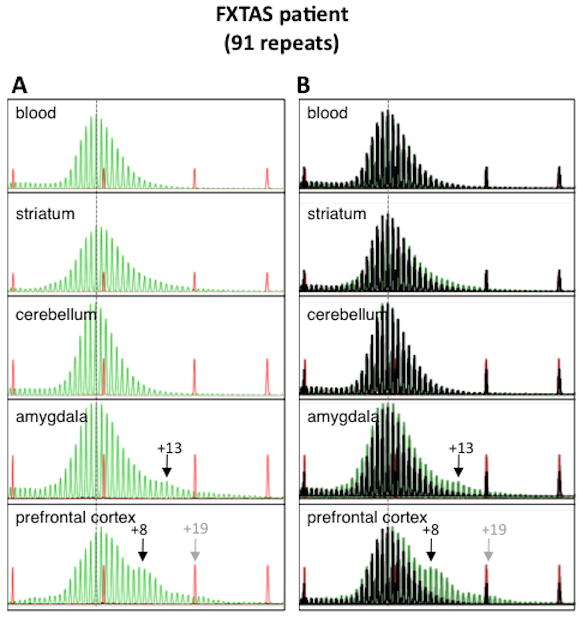
A) GeneMapper profiles from different brain regions of this patient compared to the profile seen in DNA isolated from blood isolated at the time of diagnosis 6 years before death. B) Repeat profiles for different brain regions superimposed with the repeat profile from heart shown in black. The numbers refer to the number of repeats added relative to the major allele in blood. This profile was reproducible and all peaks were seen with a range of different DNA concentrations.
Discussion
We have shown here that FX PM mice show age-dependent somatic expansion of the CGG•CCG-repeat that occurs primarily in the brain, liver and the non-somatic cells of the testes (Fig. 1, 2 and 3). Areas of the mouse brain that show particularly high levels of expansion include the amygdala (Fig. 2). This is particularly interesting given the fact that dysfunction of this brain region has been reported in men with FXTAS (Hessl, et al., 2007). Expansion also occurs in human PM cells in tissue culture converting the PM allele stepwise into a FM allele (Fig. 5). Expansion was also detected in the brains of individuals with FXTAS (Fig. 6 and Supp. Figure S3). While the expansions seen in human brain were small, even when the presence of interruptions in the largest allele is taken into consideration, it should be noted that the largest human allele examined was ~50 repeats shorter than the mouse alleles studied here. Since the repeat number in the starting allele has a significant effect on both the size and frequency of expansion in the germline (Entezam, et al., 2007), it may well be that it also affects the frequency and number of repeats added during somatic expansion. Furthermore, we know that in mice both genetic and environmental factors can exacerbate expansion risk (Entezam, et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009). Thus, depending on repeat size, genetic background and environmental exposure, somatic expansion may be more extensive in some human PM carriers than others. Our demonstration that human lymphoblastoid cells with an initial starting allele with 118 repeats show significant expansion suggests that somatic expansion may be particularly significant in that subset of PM carriers that have >100 repeats. The fact that little, if any, expansion was seen in the blood of PM mice, also raises the possibility that in humans there may also be discordance between the repeat number seen in blood in these individuals and that seen in more expansion-prone regions like the brain, liver and gonads.
The high level of expansion in the brain and liver of the FX PM mice, organs in which most cells are post-mitotic, suggests that cell proliferation is not required for expansion. Thus, somatic expansion in FX PM mice may be more likely to result from aberrant DNA damage repair than replicative DNA synthesis. In this respect, somatic expansion in the PM mice resembles what is seen on germline transmission of PM alleles where expansion is seen in oocytes and is exacerbated by oxidative damage (Entezam, et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009).
Somatic expansion is also seen in mouse models of other REDs. However, the tissue specificity of this expansion differs. For example, in mouse models for CAG•CTG-repeat expansions, the highest levels of somatic expansion are seen in the kidney (Fortune, et al., 2000; Gomes-Pereira, et al., 2001; Lia, et al., 1998; Mangiarini, et al., 1997; van den Broek, et al., 2002), while in FX PM mice, this organ shows relatively little propensity to expand (Fig. 1). In mouse models of Friedreich ataxia, FRDA, a GAA•TTC-RED, very little expansion is seen in kidney either (Clark, et al., 2007). However, in contrast to the FX PM mice that show high levels of expansion in sperm, the FRDA mice do not. Understanding the reasons for the differences in tissue-specificity may help us identify some of the factors important for driving expansions in different disease models and thus ultimately shed light on the expansion mechanism.
A variety of different explanations have been advanced to explain the tissue specificity of expansion in other REDs. Transcription of the affected gene has been suggested to be important for expansion (Lin, et al., 2009; Lin, et al., 2010; Lin and Wilson, 2007; McIvor, et al., 2010; Mochmann and Wells, 2004; Nakamori, et al., 2011; Schumacher, et al., 2001). However, the fact that heart, which shows almost no expansion, and liver and testes, which show extensive expansion have similar levels of Fmr1 mRNA (Fig. 4A), suggests that Fmr1 mRNA transcription alone does not explain the organ specificity of expansion in the FX PM mice.
Variations in the levels of different proteins involved in DNA replication and/or repair have also been suggested to account for the tissue-specificity of repeat expansion. For example, differences in the levels of the Flap endonuclease, FEN1, and Polβ, enzymes involved base excision repair, have been suggested to account for the difference in expansions in cerebellum and striatum in a mouse model of HD (Goula, et al., 2009). However, direct evidence for a role of these proteins in the REDs has not yet been demonstrated and the level of these proteins was not examined in other tissues (Goula, et al., 2009). High levels of MutSβ have been suggested to account for the fact that expansions seen in induced pluripotent stem cells (iPSCs) derived from patients with FRDA are much more extensive than those seen in the fibroblasts from which they were derived (Ku, et al., 2010; Seriola, et al., 2011). Similarly expansion is higher in embryonic stem cells (ESCs) derived from embryos with Myotonic dystrophy type 1 (DM1), a CTG•CAG-RED, than it is in differentiated cells produced from these ESCs (Seriola, et al., 2011). However, a larger study comparing multiple tissues concluded that the levels of these proteins did not account for the tissue specificity of expansion in mouse model for another CTG•CAG-RED, Huntington Disease (HD) (Lee, et al., 2010). One caveat with this data is that its conclusion was based on the Msh2/3 mRNA levels, which we have shown to not correlate well with the Msh2/3 protein levels in the FX PM mice (Fig. 4B and Supp. Figure S1). The fact that in the FX PM mice, the levels of these proteins are very high in both brain and testis would be consistent with a role for MutSβ levels in determining organ specificity. However, very similar levels of Msh3 are seen in heart, kidney and liver despite the fact that these organs have very different propensities to expand. Thus when protein levels are considered and multiple tissues are compared there is not a good correlation between the levels of MutSβ and the expansion frequency.
However, it is unnecessary to invoke complex models to explain our data as has been done for other REDs (Lee, et al., 2010). Our observation that expansion in the FX PM mice likely occurs via a process involving aberrant DNA repair raises the possibility that some combination of the levels of MutSβ and the amount of DNA damage may account for the organ specificity. The amount of DNA damage as assessed by the number of γ-H2AX foci, does not alone account for the organ specificity since brain and heart have very similar numbers of foci (Hudson, et al., 2011; Wang, et al., 2009), despite their very different levels of expansion. However, when considered in conjunction with MutSβ levels, the data for all five mouse organs can be reconciled. For example, expansions would be low in tissue like heart where MutSβ and DNA damage levels are low. Expansions would be high in brain and testes because the levels of MutSβ are relatively high. Finally, while MutSβ is low in liver, the 3–10-fold higher level of DNA damage seen in liver relative to kidney (Hudson, et al., 2011; Wang, et al., 2009) increases the likelihood that expansion will occur in that organ. The prediction from this model would be that agents that increase the amount of DNA damage in organs that normally show little somatic instability would produce increased levels of expansion in those organs. Work is in progress to test this hypothesis.
While the details of the mechanism responsible for somatic expansion of FX PM alleles remains unknown, our demonstration that expansions can also occur in human carriers of such alleles is of potential clinical relevance. Somatic expansion in humans may have health consequences since an increase in repeat number would be expected to increase the deleterious effect of transcripts produced from such alleles. The inter-tissue variability in expansion risk also raises the possibility that there might be discordance between the repeat number in blood and in affected tissue, particularly when there are genetic or environmental factors that affect somatic expansion risk. In addition, expansion in testes has the potential to affect the size of the repeat transmitted by a PM father to his daughter. This could account for the observed effect of paternal age on the size of the transmitted PM allele in humans (Ashley-Koch, et al., 1998). We have previously shown that environmental factors like oxidative stress can increase intergenerational expansion risk (Entezam, et al., 2010). Identifying factors that reduce somatic expansion may lead to the development of strategies to reduce disease severity in PM carriers and the risk of parental transmissions of expanded alleles.
Supplementary Material
Acknowledgments
We would like to thank Huiyan Liu (NIDDK) for invaluable assistance and Xiao-Nan Zhao (NIDDK) for very helpful discussions. We would also like to thank Sally Nolin and Carl Dobkin (Institute for Basic Research on Developmental Disabilities), for their testing of some of our human DNA samples. We are also grateful for the hard-working technicians that take care of our mouse colony without whose efforts this work would not be possible. This work was supported by funding from the Intramural Program of NIDDK to KU (DK057808-05).
Grant Sponsor: Intramural program of the NIDDK, NIH (DK057808)
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Holden JJ, Yang KT, Lee C, Hudson R, Gorwill H, Nolin SL, Glicksman A, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study--preliminary data. Am J Med Genet. 1999;83:322–5. [PMC free article] [PubMed] [Google Scholar]
- Ashley-Koch AE, Robinson H, Glicksman AE, Nolin SL, Schwartz CE, Brown WT, Turner G, Sherman SL. Examination of factors associated with instability of the FMR1 CGG repeat. Am J Hum Genet. 1998;63:776–85. doi: 10.1086/302018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohman R, Tamura CT, Doolittle MH, Cascarano J. Growth and aging in the rat: changes in total protein, cellularity, and polyploidy in various organs. J Exp Zool. 1985;233:385–96. doi: 10.1002/jez.1402330307. [DOI] [PubMed] [Google Scholar]
- Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275:18424–31. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR, Zoghbi HY. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1995;10:344–50. doi: 10.1038/ng0795-344. [DOI] [PubMed] [Google Scholar]
- Clark RM, Bhaskar SS, Miyahara M, Dalgliesh GL, Bidichandani SI. Expansion of GAA trinucleotide repeats in mammals. Genomics. 2006;87:57–67. doi: 10.1016/j.ygeno.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Clark RM, De Biase I, Malykhina AP, Al-Mahdawi S, Pook M, Bidichandani SI. The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum Genet. 2007;120:633–40. doi: 10.1007/s00439-006-0249-3. [DOI] [PubMed] [Google Scholar]
- Conway GS, Payne NN, Webb J, Murray A, Jacobs PA. Fragile X premutation screening in women with premature ovarian failure. Hum Reprod. 1998;13:1184–7. doi: 10.1093/humrep/13.5.1184. [DOI] [PubMed] [Google Scholar]
- De Biase I, Rasmussen A, Endres D, Al-Mahdawi S, Monticelli A, Cocozza S, Pook M, Bidichandani SI. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007;61:55–60. doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–34. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Lokanga AR, Le W, Hoffman G, Usdin K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. 2010;31:611–6. doi: 10.1002/humu.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Usdin K. ATR protects the genome against CGG. CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2008;36:1050–6. doi: 10.1093/nar/gkm1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Usdin K. ATM and ATR protect the genome against two different types of tandem repeat instability in Fragile X premutation mice. Nucleic Acids Res. 2009;37:6371–7. doi: 10.1093/nar/gkp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiry L, Dong L, Savouret C, Hubert L, te Riele H, Junien C, Gourdon G. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet. 2006;119:520–6. doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- Fortune MT, Vassilopoulos C, Coolbaugh MI, Siciliano MJ, Monckton DG. Dramatic, expansion-biased, age-dependent, tissue-specific somatic mosaicism in a transgenic mouse model of triplet repeat instability. Hum Mol Genet. 2000;9:439–45. doi: 10.1093/hmg/9.3.439. [DOI] [PubMed] [Google Scholar]
- Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273:19895–901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- Gomes-Pereira M, Fortune MT, Monckton DG. Mouse tissue culture models of unstable triplet repeats: in vitro selection for larger alleles, mutational expansion bias and tissue specificity, but no association with cell division rates. Hum Mol Genet. 2001;10:845–54. doi: 10.1093/hmg/10.8.845. [DOI] [PubMed] [Google Scholar]
- Goula AV, Berquist BR, Wilson DM, 3rd, Wheeler VC, Trottier Y, Merienne K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 2009;5:e1000749. doi: 10.1371/journal.pgen.1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–30. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Hashem V, Galloway JN, Mori M, Willemsen R, Oostra BA, Paylor R, Nelson DL. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009;18:2443–51. doi: 10.1093/hmg/ddp182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessl D, Rivera S, Koldewyn K, Cordeiro L, Adams J, Tassone F, Hagerman PJ, Hagerman RJ. Amygdala dysfunction in men with the fragile X premutation. Brain. 2007;130:404–16. doi: 10.1093/brain/awl338. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Le W, Entezam A, Otsuka N, Tong Z-B, Nelson L, Flaws JA, McDonald JH, Jafar S, Usdin K. Ovarian abnormalities in a mouse model of Fragile X-associated primary ovarian insufficiency. J Histochem and Cytochem. 2012;60:439–456. doi: 10.1369/0022155412441002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson D, Kovalchuk I, Koturbash I, Kolb B, Martin OA, Kovalchuk O. Induction and persistence of radiation-induced DNA damage is more pronounced in young animals than in old animals. Aging (Albany NY) 2011;3:609–20. doi: 10.18632/aging.100340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–47. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- Ku S, Soragni E, Campau E, Thomas EA, Altun G, Laurent LC, Loring JF, Napierala M, Gottesfeld JM. Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell. 2010;7:631–7. doi: 10.1016/j.stem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavedan C, Grabczyk E, Usdin K, Nussbaum RL. Long uninterrupted CGG repeats within the first exon of the human FMR1 gene are not intrinsically unstable in transgenic mice. Genomics. 1998;50:229–40. doi: 10.1006/geno.1998.5299. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zhang J, Su AI, Walker JR, Wiltshire T, Kang K, Dragileva E, Gillis T, Lopez ET, Boily MJ, et al. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol. 2010;4:29. doi: 10.1186/1752-0509-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lia AS, Seznec H, Hofmann-Radvanyi H, Radvanyi F, Duros C, Saquet C, Blanche M, Junien C, Gourdon G. Somatic instability of the CTG repeat in mice transgenic for the myotonic dystrophy region is age dependent but not correlated to the relative intertissue transcription levels and proliferative capacities. Hum Mol Genet. 1998;7:1285–91. doi: 10.1093/hmg/7.8.1285. [DOI] [PubMed] [Google Scholar]
- Lin Y, Hubert L, Jr, Wilson JH. Transcription destabilizes triplet repeats. Mol Carcinog. 2009;48:350–61. doi: 10.1002/mc.20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Leng M, Wan M, Wilson JH. Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol. 2010;30:4435–51. doi: 10.1128/MCB.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–17. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- Manley K, Pugh J, Messer A. Instability of the CAG repeat in immortalized fibroblast cell cultures from Huntington’s disease transgenic mice. Brain Res. 1999a;835:74–9. doi: 10.1016/s0006-8993(99)01451-1. [DOI] [PubMed] [Google Scholar]
- Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. 1999b;23:471–3. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- Marozzi A, Vegetti W, Manfredini E, Tibiletti MG, Testa G, Crosignani PG, Ginelli E, Meneveri R, Dalpra L. Association between idiopathic premature ovarian failure and fragile X premutation. Hum Reprod. 2000;15:197–202. doi: 10.1093/humrep/15.1.197. [DOI] [PubMed] [Google Scholar]
- Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci U S A. 1998;95:8568–73. doi: 10.1073/pnas.95.15.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIvor EI, Polak U, Napierala M. New insights into repeat instability: role of RNA*DNA hybrids. RNA Biol. 2010;7:551–8. doi: 10.4161/rna.7.5.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–40. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Mochmann LH, Wells RD. Transcription influences the types of deletion and expansion products in an orientation-dependent manner from GAC*GTC repeats. Nucleic Acids Res. 2004;32:4469–79. doi: 10.1093/nar/gkh787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A, Webb J, Grimley S, Conway G, Jacobs P. Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet. 1998;35:637–40. doi: 10.1136/jmg.35.8.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A laboratory manual: CSH Press 2003 [Google Scholar]
- Nakamori M, Pearson CE, Thornton CA. Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum Mol Genet. 2011;20:580–8. doi: 10.1093/hmg/ddq501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolin SL, Ding XH, Houck GE, Brown WT, Dobkin C. Fragile X full mutation alleles composed of few alleles: implications for CGG repeat expansion. Am J Med Genet A. 2008;146A:60–5. doi: 10.1002/ajmg.a.32087. [DOI] [PubMed] [Google Scholar]
- Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas M, Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Salat U, Bardoni B, Wohrle D, Steinbach P. Increase of FMRP expression, raised levels of FMR1 mRNA, and clonal selection in proliferating cells with unmethylated fragile X repeat expansions: a clue to the sex bias in the transmission of full mutations? J Med Genet. 2000;37:842–50. doi: 10.1136/jmg.37.11.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher S, Pinet I, Bichara M. Modulation of transcription reveals a new mechanism of triplet repeat instability in Escherichia coli. J Mol Biol. 2001;307:39–49. doi: 10.1006/jmbi.2000.4489. [DOI] [PubMed] [Google Scholar]
- Seriola A, Spits C, Simard JP, Hilven P, Haentjens P, Pearson CE, Sermon K. Huntington’s and myotonic dystrophy hESCs: down-regulated trinucleotide repeat instability and mismatch repair machinery expression upon differentiation. Hum Mol Genet. 2011;20:176–85. doi: 10.1093/hmg/ddq456. [DOI] [PubMed] [Google Scholar]
- Tanaka F, Reeves MF, Ito Y, Matsumoto M, Li M, Miwa S, Inukai A, Yamamoto M, Doyu M, Yoshida M, et al. Tissue-specific somatic mosaicism in spinal and bulbar muscular atrophy is dependent on CAG-repeat length and androgen receptor--gene expression level. Am J Hum Genet. 1999;65:966–73. doi: 10.1086/302578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Adams J, Berry-Kravis EM, Cohen SS, Brusco A, Leehey MA, Li L, Hagerman RJ, Hagerman PJ. CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS) Am J Med Genet B Neuropsychiatr Genet. 2007;144B:566–9. doi: 10.1002/ajmg.b.30482. [DOI] [PubMed] [Google Scholar]
- Taylor AK, Tassone F, Dyer PN, Hersch SM, Harris JB, Greenough WT, Hagerman RJ. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am J Med Genet. 1999;84:233–9. [PubMed] [Google Scholar]
- Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, Adam S, Greenberg C, Ives EJ, Clarke LA, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. 1994;6:409–14. doi: 10.1038/ng0494-409. [DOI] [PubMed] [Google Scholar]
- Tome S, Holt I, Edelmann W, Morris GE, Munnich A, Pearson CE, Gourdon G. MSH2 ATPase domain mutation affects CTG*CAG repeat instability in transgenic mice. PLoS Genet. 2009;5:e1000482. doi: 10.1371/journal.pgen.1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Kondoh K, Kotani Y, Komure O, Kuno S, Kawai J, Hazama F, Sano A. Somatic mosaicism of CAG repeat in dentatorubral-pallidoluysian atrophy (DRPLA) Hum Mol Genet. 1995;4:663–6. doi: 10.1093/hmg/4.4.663. [DOI] [PubMed] [Google Scholar]
- Uzielli ML, Guarducci S, Lapi E, Cecconi A, Ricci U, Ricotti G, Biondi C, Scarselli B, Vieri F, Scarnato P, et al. Premature ovarian failure (POF) and fragile X premutation females: from POF to to fragile X carrier identification, from fragile X carrier diagnosis to POF association data. Am J Med Genet. 1999;84:300–3. [PubMed] [Google Scholar]
- van den Broek WJ, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJ, Wieringa B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. 2002;11:191–8. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- Vianna-Morgante AM, Costa SS, Pares AS, Verreschi IT. FRAXA premutation associated with premature ovarian failure. Am J Med Genet. 1996;64:373–5. doi: 10.1002/(SICI)1096-8628(19960809)64:2<373::AID-AJMG28>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–23. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12:273–81. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- Wu Z, Falciatori I, Molyneux LA, Richardson TE, Chapman KM, Hamra FK. Spermatogonial culture medium: an effective and efficient nutrient mixture for culturing rat spermatogonial stem cells. Biol Reprod. 2009;81:77–86. doi: 10.1095/biolreprod.108.072645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.