

Abstract
Tumors are composed of highly proliferate, migratory, invasive, and therapy-evading cells. These characteristics are conferred by an enormously complex landscape of genomic, (epi-)genetic, and proteomic aberrations. Recent efforts to comprehensively catalogue these reversible and irreversible modifications have began to identify molecular mechanisms that contribute to cancer pathophysiology, serve as novel therapeutic targets, and may constitute biomarkers for early diagnosis and prediction of therapy responses. With constantly evolving technologies that will ultimately enable a complete survey of cancer genomes, the challenges for discovery cancer science and drug development are daunting. Bioinformatic and functional studies must differentiate cancer-driving and - contributing mutations from mere bystanders or ‘noise’, and have to delineate their molecular mechanisms of action as a function of collaborating oncogenic and tumor suppressive signatures. In addition, the translation of these genomic discoveries into meaningful clinical endpoints requires the development of co-extinction strategies to therapeutically target multiple cancer genes, to robustly deliver therapeutics to tumor sites, and to enable widespread dissemination of therapies within tumor tissue. In this perspective, I will describe the most current paradigms to study and validate cancer gene function. I will highlight advances in the area of nanotechnology, in particular, the development of RNA interference (RNAi)-based platforms to more effectively deliver therapeutic agents to tumor sites, and to modulate critical cancer genes that are difficult to target using conventional small-molecule- or antibody-based approaches. I will conclude with an outlook on the deluge of challenges that genomic and bioengineering sciences must overcome to make the long-awaited era of personalized nano-medicine a clinical reality for cancer patients.
1. Introduction
Personalized cancer medicine, i.e., the design of therapeutic regimens informed by tumor genotyping, has recently entered oncological practice. FDA-approved ALK kinase inhibitor crizotinib and the BRAF inhibitor vemurafenib are the most recent examples of tailored cancer therapy, which have been successfully advanced for the treatment of ALK-translocated lung cancer, and BRAF-mutated melanoma, respectively.1, 2 These successes demonstrate how the study of DNA-associated abnormalities can guide drug development and clinical trials to pharmacologically target these tumorigenic perturbations, and to stratify patients for treatment. The vast majority of the dauntingly complex genomic datasets, however, have yet to be translated into meaningful therapeutic strategies. Exigent barriers for the rapid and cost-effective translation of the genome into clinical practice have become obvious, and are beginning to galvanize multidisciplinary teams of geneticist, computational scientists, cancer biologists, and bioengineers to develop the next generations of computational algorithms, preclinical cell and animal models, and refined therapeutic conjugates. In this article, I will highlight the most recent successes in translating genomic information into clinical practice; I will describe advances in the preclinical interrogation of gene function in silico, and in cell and animal models in vivo, and will summarize efforts in the bioengineering sciences to develop nanotechnological platforms that enable more effective, and less toxic targeting of multiple oncogenic lesions. I will conclude with an outlook on future challenges to further advance and integrate functional cancer genomics and material sciences.
2. Personalized cancer medicine – where we stand
Comprehensive surveys of somatic mutations in cancer genomes have revealed a compendium of aberrations that confer growth and/or survival advantage to a tumor, and have enabled the development of therapies that specifically target these oncogenic signatures. The FDA approval of small molecule inhibitors (SMIs) and biotherapeutic antibodies targeting amplified, over-expressed, and/or mutated receptor tyrosine kinases (RTKs) embodied the early promises of tailored therapies aimed at neutralizing the mitogenic, pro-invasive, pro-migratory, and anti-apoptotic activities of the cancer cell receptor kinome. These drugs include the anti-ErbB2 (HER2) monoclonal antibody trastuzumab and the SMI lapatinib for the treatment of HER2-amplified breast cancers3, the Abl kinase SMI imatinib, which blocks the activity of a Bcr-Abl fusion protein in chronic myeloid leukemia (CML)4, the SMI gefitinib, which targets mutated, hyperactivated epidermal growth factor receptor (EGFR) in advanced non-small cell lung cancers (NSCLCs) as first-in-line treatment5–7, and the VEGF-neutralizing, anti-angiogenic monoclonal antibody bevacizumab (Avastin) for the treatment of Glioblastoma (GBM), NSCLCs, and metastatic colorectal and kidney cancer.8–12 Building on these early clinical successes, the past few years have witnessed an exceedingly rapid translation of genomic discoveries into clinical endpoints (see Figure 1 detailing the timeline for the translation of genomic findings into drug development). Activating mutations in the serine/threonine kinase BRAF acting downstream of membrane-proximal RTK activation were identified in >50% of melanoma patients.13 Functional studies revealed that mutated BRAF (BRAFMUT) could directly activate mitogen-activated kinases, thereby uncoupling MEK-ERK signaling from growth factor-driven RTK activation. Mirroring remarkable activities in preclinical cancer models14, 15, clinical trials with the BRAFMUT inhibitor vemurafenib revealed extraordinarily high response rates in therapy-refractory patients with metastatic, BRAF-mutated melanoma.2 Equally dramatic clinical responses were observed with SMI crizotinib, which targets an aberrant and constitutively active ALK fusion protein in NSCLC patients1. The rapid preclinical and clinical validation of these targeted therapeutics, together with the development of companion diagnostic tests to assess BRAF mutation and ALK translocations in lung and melanoma patients, represent the most important achievements in personalized cancer medicine to date (see Figure 1).
Fig. 1. Discovery of cancer mutations and its translation into drug development.
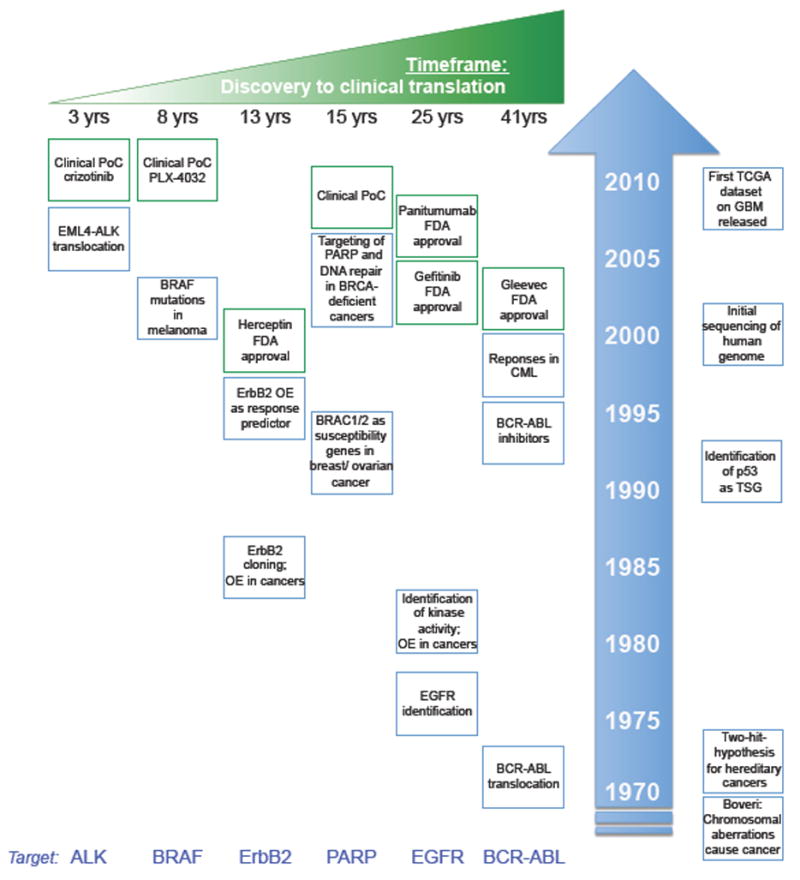
Cancer genetics accelerated the clinical development of mutation-specific targeted drugs. Highlighted examples include targeted therapies that inactivate BCR-ABL, EGFR, PARP, ERBB2, BRAF and ALK (left panels). On the right, seminal discoveries in cancer biology and cancer genetics are highlighted, e.g., Boveri’s discovery of the genetic basis of cancer in 1902, and the characterization of human glioblastoma and ovarian cancer genome in 2008. Gleevec received FDA-approval more than 40 years after the initial discovery of BCR-ABL translocation in chronic myelogenous leukemia (CML), and the EGFR inhibitor gefitinib was finally approved in 2003, almost 25 years after the cloning of the EGFR gene, and more than 20 years after the identification of EGFR overexpression as a cancer-driving event. Clinical translation of more recent discoveries of ALK translocation and BRAF mutations in non-small cell lung carcinoma (NSCLC) and melanoma patients, respectively, has been translated into clinical endpoints much faster. Here, crizotinib, originally discovered as a cMet inhibitor, has entered clinical phase I/II trials 3 years after the discovery of ALK translocations, and the BRAF inhibitor PLX4032 has been enrolled into clinical proof of concept (PoC) studies in melanoma patients 8 years after the initial discovery of BRAF mutations. In addition, the more rigorous mapping of cancer-associated driving and collaborating mutations enabled prognostication. Specifically, Her2 overexpression (OE) has been correlated with favorable responses toward Her2-targeting herceptin, and lead to the development of the diagnostic HercepTest. Similarly, the presence of BRAC1/2 mutations dictates responses toward PARP inhibitors. TSG, tumor suppressor gene.
Additional kinase inhibitors currently in clinical trials target activated JAK2 V617F in myelofibrosis16, mutated RET in medullary thyroid carcinoma17, and PI3K, Akt, and FGFR in various malignancies (see review by Courtney et al18). Finally, non-kinase SMI currently being evaluated in the clinic include the Smoothened (SMO) SMI GDC-0449 (vismodegib)19, and SMIs targeting the DNA repair enzyme poly (ADP) ribose polymerase I (PARP1).20 SMO becomes hyperactivated and triggers constitutive activation of the Hedgehog pathway in basal cell carcinomas and a subset of medulloblastomas due to loss-of-function mutations of the tumor suppressor and SMO inhibitor PTCH1 (see review by Rubin and Sauvage21). PARP SMIs are effective in breast and ovarian cancers with incapacitated homologous recombination (HR) due to loss-of-function of two critical DNA repair enzymes, BRCA1 or BRCA2. HR-deficient cancers are dependent on PARP-driven alternative mechanisms of DNA repair, and consequently, PARP inhibitors show synthetic lethality in the setting of BRCA1/2 mutation.22, 23
Recent genome sequencing efforts identified additional and ‘druggable’ mutations, such as recurrent activating mutations in the heterotrimeric G-protein α-subunit GNAQ, which triggers MAPK activation in uveal melanoma24, activating NOTCH1 mutations in chronic lymphocytic leukemia (CLL)25, and various mutations within several genes of the NF-κB pathway critical for the development of multiple myeloma.26 Currently available MEK, NOTCH, and NF-κB signaling inhibitors can readily be enrolled into (pre-)clinical testing for the treatment of these malignancies. In addition, gain-and loss-of-function mutations of enzymes implicated in chromatin modification, e.g., histone (de)methyltransferases and components of the SWI-SNF complex,27, 28 (see review by Albert and Helin29), DNA methylation (e.g., DNMT3A),30 and pathways generating important metabolites critical for the function of these enzymes (e.g., isocitrate dehydrogenase 1 (IDH1)31, 32, or ten-eleven-translocation gene 2 (TET2)),33 have emerged as additional drug targets in lymphoid, myeloid and solid tumors. While a more detailed understanding of their roles in tumorigenesis is still pending, these epigenetic regulators define a novel class of cancer-associated aberrations, and may drive the development of pathway-specific drugs for the treatment of genomically defined cancers.
The rapidly growing field of cancer genomics has, thus, identified myriad genetic and epigenetic perturbations within cancer genomes. Drugs targeting some of these mutations have already been translated into oncological practice with clear benefits for genomically defined patient populations. Where do we go from here? The confluence of several areas of cancer discovery science, i.e., genome surveys, medicinal chemistry processes, computational science approaches, and high-throughput genome-scale interrogation of cancer gene function will be critical for prognostication and advancing personalized drug design in the near future. These efforts will address important questions in basic and clinical cancer sciences: Which genes with aberrant copy number and/or expression are critical for tumorigenesis? How do cancer-associated mutations dictate phenotypic hallmarks of proliferation, angiogenesis, migration, invasion, and therapy resistance? What are the most important molecular markers to match individual patients with the most appropriate therapy to achieve the best possible outcome?
3. Making sense of complex genomes and transcriptomes
Surveys of oncogenomes have delivered extensive lists of candidate cancer genes (CCGs) with increasing accuracy and resolution. The Cancer Genome Atlas Project (TCGA) is the most recent and most impressive example describing the landscape of genetic and epigenetic changes in GBM and ovarian cancers.34, 35 Some CCGs have well-annotated functions in disease pathogenesis and are characterized by high-level recurrent copy number alterations and expression; consequently, these are classified as important ‘driving’ mutations. Examples include amplification and mutation of EGFR, deletions of the tumor suppressors CDKN2a or PTEN, and mutations in TP53, RAS, BRAF, or PI3K, which are found across many different solid tumors (see review by Chin and Gray36). Other CCGs with low-amplitude genomic and genetic perturbations, including non-coding RNAs, have yet to be mechanistically characterized. Some of these less prevalent lesions may represent drivers or contributors, while the vast majority constitute ‘passenger’ mutations randomly acquired during the lifespan of the tumor. Differentiating between drivers/contributors and genomic ‘noise’ is a dauntingly difficult task that continues to challenge multi-disciplinary research teams of computational scientists and cancer biologists. The integration of several multi-dimensional cancer datasets in concert with comparative interspecies cancer genomics provide the substrate for knowledge-based pathway and epistasis analyses, and for subsequent functional cell culture-based and animal studies to document the roles of CCGs in cancer pathogenesis. Figure 2 illustrates the workflow of functional genomics to identify, characterize, and therapeutically target cancer-associated mutations.
Fig. 2. Form cancer gene discovery to clinical evaluation of mutation-specific therapeutics.

(A) Multi-dimensional analysis of human and murine cancers include identification of copy number variations (assessed by Fluorescence In Situ Hybridization and arrayed Comparative Genomic Hybridization, aCGH), DNA mutation (evaluated by DNA sequencing), promoter methylation (determined by chromatin immunoprecipitation and microarray analysis of immunoprecipitated DNA), and aberrant mRNA and protein expression (assessed by microarray analysis and e.g., by antibody arrays). Each of these genomic, genetic, epigenetic, and proteomic changes can alter the function, the splicing patterns, or the expression levels of candidate cancer genes (CCGs) that potentially can drive cancer initiation, progression, and dissemination. Additional model systems not depicted here include zebrafish, nematodes, fruitflies and yeast. (B) Integrative, cross-species bioinformatics, and genome-scale interrogation of gene function inform mutation-specific drug design and evaluation. Here, the assessment of genomic, genetic and proteomic aberrations, cross-species, comparative analyses, and the identification of similarities between different tumor types enable the identification of critical CCGs. Subsequently, CCGs will be functionally interrogated in massively parallel high-through put (HT) gain- and loss-of-function (GOF/LOF) studies to assess the modus operandi and impact on tumorigenic signaling cascades. Subsequently, validated CCGs are enrolled into detailed clinicopathological analyses (i.e., CGC expression analyses in primary tumors), and cell culture- and animal model-based experiments. CCGs with strong cancer-promoting activities can be used as therapeutic targets and/or biomarkers, prompting further clinical evaluation. Importantly, this cascade has to be an iterative process, where the result from each step can inform and refine preceding analyses to help improve entire drug development process. TICs, tumor initiating cells; GEMMs, genetically engineered mouse models.
3.1 Integrative and interspecies comparative cancer genomics
Analysis of the cancer genome using multiple ‘omic’ dimensions is critical to understanding how CCG function is compromised (Figure 2A). Is a CCG crippled by mutations of its coding or non-coding sequences, by DNA or histone modifications, by changes to DNA structure, or by expression of antagonizing non-coding RNAs? Because of their importance in malignant transformation, drivers are often inactivated via several mechanisms. The tumor suppressor INK4a (encoded by CDKN2a) is inactivated by homozygous deletion, epigenetic silencing (via promoter methylation), and by point mutation (see review by Sharpless37); hyperactivation of oncogenic PI3CA is driven by amplification, overexpression and mutation;38, 39 and Tie-2/Tek, a critical effector of endothelial cell survival and vascularization, is regulated at the levels of copy number, promoter methylation, mutation, and mRNA expression (Stegh and Kesari, unpublished observation). Consequently, integrative analyses assessing genomic, genetic, and epigenetic regulation across multiple tumor types represent a critical first step in assessing the pathobiological importance of a given CCG, and in evaluating its potential as a therapeutic target or prognostic biomarker (Figure 2B).
Murine and human tumors harbor orthologous genomic lesions, and many human oncogenes are transforming in murine cells and vice versa despite low-level evolutionary conservation. Consequently, more advanced computational approaches use murine cancer models in cross-species, comparative analyses. As critical cancer-driving genes and pathways are likely conserved across different species, evolutionary conservation is used in these approaches as yet another criterion to assess disease relevance of a given CCG. Conserved genomic signatures were found upon K-RAS activation in lung cancers,40 and NOTCH1 mutations were identified in T cell acute lymphoblastic leukemia of both human and murine origin.41 In addition, comparison of human and murine gene signatures also uncovered novel oncogenes. Chin and colleagues analyzed genomic profiles of metastatic human and murine K-Ras-driven melanoma, and identified NEDD9 as a novel pro-migratory and pro-invasive gene that exhibited amplification-driven overexpression in these tumors.42 Similar analyses in human and murine hepatocellular and mammary carcinomas identified recurrent co-amplification of oncogenic Yap and cIAP-1 in liver,43 and GRB7 and 14-3-3-σ as contributors to ERBB2-driven carcinogenesis in breast cancer.44
3.2 High-throughput interrogation of genomic datasets
Although integrative inter-species analyses of oncogenomes have identified critical CCGs, and have illuminated a path toward diagnostics and therapy development, the prioritization of the vast majority of CCGs buried in highly rearranged and mutated human oncogenomes remains a major challenge for cancer discovery sciences, and fervidly demands a systematic approach to interrogate gene function on a genome-scale. Such mechanistic studies not only provide information on the modus operandi of a given CCG in a particular cancer cell lineage, they also aid in identifying collaborating non-mutated genes with essential functions within CCG-driven pathways that may serve as important drug targets. Stunning advances in the development of cDNA, RNAi, and chemical libraries over the past few years have made genome-scale functional studies an attainable reality. When combined with the structural characterization of oncogenomes, and further improved in terms of sensitivity, specificity, cost, and throughput, these emerging technologies will continue to identify and validate novel CCGs as putative drug targets.
Here, I will focus on the most recent examples of near genome-scale functional studies that resulted in the identification of critical CCGs across many different malignancies; retrovirus-/transposon-based mutagenesis studies, the development, technical specifications and applications of ORF, cDNA, miRNA, and chemical SMI-based libraries are described and reviewed in detail elsewhere.45, 46
Using a lentiviral shRNA library with more than 54,000 shRNAs targeting ~11,000 genes, Hahn and colleagues assessed functional consequences of RNAi-mediated gene knockdown on cellular growth and survival. Screening more than 100 human cell lines including 25 ovarian cancer lineages, and integrating these functional data with genomic surveys of oncogenomes revealed strikingly different oncogene dependencies across several tumor types. In-depth analyses of genetic vulnerabilities of ovarian cancers identified 54 genes critical for ovarian cancer cell survival. These genes also showed amplification and overexpression in primary ovarian tumors and cells. One of these genes, Paired box gene (Pax)8, is focally amplified in 16% of high-grade serous ovarian cancers, and its RNAi-mediated knockdown induces apoptosis in ovarian cancer cells, suggesting that Pax8 represents a lineage-specific survival factor in ovarian tumorigenesis.47 Similarly, an RNAi screen in 34 breast cancer cell lines using kinome-targeting pooled siRNA oligonucleotides revealed selective RNAi-induced lethality in genomically defined cell lineages. For example, viability of PTEN-deficient breast cancer cells was dependent on genes controlling the mitotic spindle assembly checkpoint, e.g., the checkpoint kinase TTK/Msp1, and consequently, RNAi-mediated knockdown or SMI-mediated inhibition of TTK showed synthetic lethality with PTEN loss-of-function.48 Using a comparable screening strategy, Barbie et al. and Elledge and colleagues identified the non-canonical IκB kinase TBK1, and regulators of mitosis (e.g., the mitotic kinase PLK1, the anaphase-promoting complex/cyclosome, and the proteasome) as genes and gene networks that when inhibited pharmacologically or via RNAi resulted in death of KRas mutant cells and derived xenografts.49, 50 Additional studies pointed to SGK2 and PAK3 kinases as essential genes in p53-deficient cells,51 and identified MET, CDK6, and MEK1 as critical survival factors in cells lacking VHL.52
Rather than focusing on the identification of individual CCGs displaying synthetic lethal interactions with oncogenic drivers, several groups began to construct comprehensive gene interaction maps using high-throughput (HT), combinatorial RNAi approaches to generate double-deletion mutants in yeast. Comparing such interaction maps under normal and DNA-damaging conditions using differential epistasis mapping lead to the identification of dynamic genetic interactions and ascribed novel roles of Slt2 kinase, Pph3 phosphatase, and histone variant Htz1 in DNA repair.53 Feasibility studies in Drosophila targeting 96 genes with two independent siRNA oligonucleotides per gene generated approximately 18,000 possible double-deletion mutants. This co-extinction approach revealed that combinatorial knockdown of >600 genes triggered phenotypic changes that could not be predicted from perturbations of individual genes.54 While the generation of such interaction maps for mammalian genomes represents an enormous challenge, the systematic probing of genetic interactions will be critical to understanding cancer cell circuitry, and to unraveling genetic co-dependencies. These approaches promise to answer the question of which combination of genes has to be neutralized in order to impact cell growth and survival, and what is the most effective combinatorial treatment regimen for a specific, genomically, genetically and epi-genetically characterized cancer.
3.3 Cell and animal models
To further test and validate CCG function, over the past few years a wide spectrum of different model systems has been developed, which have added levels of sophistication to the standard arrays of transformed human cancer cell lines. These include primary, patient-derived cells, most importantly cancer stem cells (CSCs), CSC-derived orthotopic xenograft models, and genetically engineered mouse models (GEMMs), along with their primary and transformed cell derivatives, and secondary syngeneic explants (see review by Chin et al45). If enrolled into the most adequate applications/assays, each of these cell and in vivo tumor models represents a powerful experimental system to study CCG activity and/or anti-CCG drugs.
Transformed and primary patient-derived cells are invaluable tools for studying gene function in low- and high-throughput applications. These cell culture models are relatively inexpensive, easy to maintain, and easy to manipulate. In addition, efforts to verify cellular effects of RNAi-mediated loss-of-function or cDNA complementation across a spectrum of genetically diverse cells also minimize the risk that an observed phenotype is cell-line specific. On the other hand, cell lines grown on plastic do not capture the intricacies of human cancers, such as tumor-microenvironment interactions, and are often only partially characterized on genomic levels. In addition, the oncogenome of transformed, long-term cultured cells does not necessarily capture the genomic aberrations seen in primary tumors, and the genotype of patient-derived lineages represents only a limited and often less prevalent combination of genetic and epigenetic aberrations resident in clinical samples. Given the importance of genetic context to understand CCG function, re-enforcing complementary data from other cancer models are always required to validate initial cell culture-based mechanistic studies. Primary, non-transformed, genetically engineered cells have emerged as a powerful cell system that in the absence of full transformation could be transfected with CCGs to survey their cancer-relevant functions in the context of a defined mutational spectrum. These minimally transformed cells also permit the generation of isogenic cell lines that mirror mutational profiles in specific tumor subtypes. Finally, GEMMs represent the most elaborate model system to assess the role of CCGs in tumorigenesis. Inducible knockout and transgenic alleles crossed onto tumor-prone strains can determine the role of a CCG in tumor maintenance, and consequently, these models represent decisive tools to identify driver mutations (reviewed by Sharpless and Depinho55). In addition, refined GEMM provide faithful pheno- and genocopies of human malignancies, and consequently, have emerged as invaluable tools for interspecies oncogenomics. It is important to consider, however, that murine tumors are characterized by few, if any, chromosomal gains or losses, and exhibit less complex profiles of point mutations.56 Consequently, the low-level genomic instability of murine tumors limits the application of large-scale comparative approaches to prioritize extensive atlases of human mutations. In contrast, murine tumor models associated with telomerase dysfunction more faithfully replicates the complex genomic profiles seen in human cancers, as telomere-based crises cause high-level non-reciprocal translocations and regional copy number variations. Importantly, these perturbations non-randomly overlap with chromosomal aberrations observed in several human oncogenomes. These findings point to a similar malignant evolution of human and murine cancers, and underline the usefulness of telomerase-deficient murine cancer models as critical filters for the prioritization of human oncogenomic datasets.57
The extensive time frame and high costs associated with the generation and characterization of GEMMs, however, has limited their application for high throughput assays. Non-germline GEMMs, i.e., genetically engineered, tissue-restricted stem and progenitor cells orthotopically implanted into a syngeneic host, represent a valid alternative (reviewed by Heyer et al58). Here, primary, minimally transformed cells can be transfected with ORF or RNAi libraries and implanted into recipients. The subsequent development of syngeneic tumors can then be analyzed for the selection of cooperating events that in concert with signature mutations drive explant growth. Examples include the stem cell transgenesis system developed by Bachoo et al. using minimally transformed murine cortical astrocytes and neural stem cells,59 and a hematopoietic progenitor cell system derived from Eμ-myc transgenic mice, which has been used successfully in HT RNAi applications to identify haploinsufficient tumor suppressors driving lymphomagenesis.60
4. Realizing the limitation of conventional drug development – what about the undruggable oncogenome?
HT surveys of cancer genomes, e.g., the TCGA, have provided a periodic table of key genetic elements that drive and contribute to cancer development. So far, 474 genes have been described as professional cancer genes (Cancer Gene Census) encoding 2% of the human proteome. With advances in genomic technologies, this number will increase by several fold, and will likely trigger an equally impressive evolution of technologies to mechanistically understand and ultimately prosecute these genes as putative targets. As pointed out above, these functional studies have to provide a deep mechanistic understanding of selected CCGs as a function of cooperating oncogenic and tumor suppressive signatures. Such studies have to explain CCG relevance for cancer development in the context of a complex, intertwined tumor circuitry, rather than as linear, isolated pathways. They also have to consider that cancers represent highly heterogeneous neo-organs, which are composed of cells with different genomic profiles, a multitude of histopathological phenotypes, and staggering heterotypic interactions with the host microenvironment.
Currently identified, clinically validated, and most importantly, ‘druggable’ targets, such as BRAF or ALK, are ‘low-hanging’ fruits for drug development. The vast majority of CCGs, however, will likely represent unprecedented, non-enzymatic targets with unknown modi operandi. How can multiple cooperating, undruggable, and uncharacterized genes be functionally compromised? While it is conceivable that the rapid evolution of genomics can address the challenges posed by high-level plasticity of cancers, the most critical question becomes whether ‘conventional’ drug development pipelines focusing on SMI and biotherapeutic antibodies are equipped to tackle the challenge of drugging the undruggable. In addition, many targeted therapies have failed in the clinic because they cannot be effectively delivered to tumors sites, and exhibited suboptimal intratumoral dissemination. Effective drug delivery is a critical challenges for solid organ tumors, in particular cancers of the brain and pancreas. Here, tailored therapies bound for the central nervous system have to negotiate passage through the blood-brain barrier (BBB), the blood-cerebrospinal fluid barrier (BCSF), and the blood-tumor barrier (BTB), and must withstand the substantial dynamic force in the brain interstitium caused by CSF flow, edema-associated intratumoral, and tumor mass-related pressure (reviewed by Abbott61). Similarly, pancreatic cancer drugs have to extravasate from the tumor vasculature and permeate thick fibrotic tissue to target tumor cells.62, 63
Finally, cancers can compensate for functional ablation of one genetic element by activating other circuitry components, suggesting that co-extinction strategies are required to halt tumor progression. Combinatorial therapies utilizing multiple SMI- or antibody-based agents have to consider significant drug-drug interactions, systemic toxicity due to pronounced off-target effects, and the emergence of target gene mutations leading to drug resistance.64
5. Preclinical validation of nucleic acid-based nano-conjugates to generate radically novel options for cancer therapy
Nanotechnology, i.e., the science of engineering materials on a molecular, nanometer scale, provides fundamentally different approaches to cancer therapy. Nano-drug delivery systems with a size of <100 nm can accumulate at higher concentrations within tumors compared to unconjugated therapeutics through the enhanced permeability and retention (EPR) effect, and consequently have lower dose-limiting adverse side effects, and higher therapeutic efficacies65 (reviewed by Maeda et al and Petros and Desimone66, 67). Several formulations have been described, including liposomes, polymeric nanoparticles, dendrimers, metal nanoparticles, and molecular targeted nanoparticles.
Importantly, several nano-conjugates have successfully been used to deliver small interfering RNAs (siRNAs) to tumor sites, and have been found to harness the great promise of RNAi-mediated biotherapeutic gene silencing to neutralize virtually any gene, including undruggable oncogenes, and to overcome the major challenges of RNA interference (RNAi)-based therapy, i.e., poor cellular uptake, lack of intracellular stability, and rapid renal clearance following systemic administration (reviewed by Dillon et al and Reischl and Zimmer68, 69). I will describe the most prominent nano-RNAi approaches, i.e., chemically modified siRNAs, lipid-, polymer-based delivery strategies, and RNAi-functionalized metal nanoparticles, and their potential to drive the concept of personalized cancer medicine toward clinical opportunity.
5.1 The RNAi approach
RNAi is a potent mechanism of gene silencing capable of blocking the translation of mRNA, and thus reducing the expression of pathological proteins, in particular those that are difficult to target by traditional pharmacological approaches. siRNAs are generated by cleavage of long double-stranded (ds)RNAs into ~20 nucleotide-containing siRNAs by the enzyme Dicer. Unwinding of siRNAs into two single-stranded (ss)RNAs, incorporation of the guide strand into the RNA-induced silencing complex (RISC), and binding of siRNAs to complementary mRNA triggers the degradation of endogenous mRNA by Argonaute, the catalytic component of the RISC complex (Figure 3; reviewed by Hannon and Rossi70). siRNA oligonucleotides can silence expression of various cancer genes implicated in growth, apoptosis, migration, and invasion, and consequently, have motivated myriad preclinical studies to assess the potential of RNAi as anti-cancer therapeutics. Due to the negative charge of the RNA backbone, siRNA oligonucleotides require delivery systems to overcome negatively charged membranes, and to counteract electrostatic repulsion. In addition, systemic delivery strategies have to prevent rapid renal and hepatic clearance and degradation of siRNAs by nucleases, and most importantly, have to display favorable safety profiles. While local delivery may overcome some of these difficulties, the cancerous tissue to be targeted, however, may not be easily accessible or may be too extensive for local treatment, e.g., in the case of metastatic disease. The first RNAi-based viral vectors faced challenges relating to significant adverse side effects, high costs associated with the production of sufficient viral stocks, and suboptimal dissemination. While late-generation viral platforms for nucleic acid delivery may overcome some of these shortcomings, the development of non-viral, less toxic alternatives for RNAi delivery is highly desirable (reviewed by Akhtar and Benter71).
Fig. 3. Mechanism of RNAi.

Long double-stranded (ds)RNA is processed by the RNase III enzyme Dicer to 21–23 nucleotide siRNA-duplex-like intermediates. The duplex is unwound (mediated by the RNA helicases Armitage and R2D2), and the RISC complex with single-stranded siRNAs is formed to mediate cleavage of target mRNAs.
With physico-biological properties significantly dependent on particle size, charge, and hydrophobicity (reviewed by Petros and DeSimone67), nanomaterials at the submicron scale have features that make them ideal carrier systems for RNAi delivery.
In addition to intra- and extracellular stability and biocompatibility, the design of these nanoconstructs has to consider several critical aspects of conjugate chemistry, i.e., particle aggregation, endosomal escape of particles into the cytosol to gain access to the RISC signaling machinery, and off-target effects of siRNAs. Aggregation of siRNA particles can be counterbalanced by reducing surface charge, e.g., by co-functionalization with poly(ethylene glycol) (PEG), sugar molecules (e.g., cyclodextrin), or hyaluronic acid (HA).67, 72–75 pH- or reduction-sensitive polymers can enhance endosomal escape, as sharp pH differences and a redox potential exist between the intra-endosomal and the cytosolic compartment. Consequently, cationic polymers with a pKA slightly below physiological pH, e.g., poly(ethylene imine) (PEI), the most widely used pH-responsive polymer (reviewed by Kim and Kim, Schaffert and Wagner, and Midoux et al76–78) can absorb protons, increase the intra-endosomal osmotic pressure, and ultimately rupture endosomal membranes— a phenomenon referred to as the “proton sponge effect” 69, 79. Other strategies to enhance endosomal escape have utilized (a) polymers undergoing hydrophilic-to-hydrophobic transitions,80 (b) fusogenic peptides aimed at disrupting endosomal membranes,81, 82 and (c) protein transduction peptides that facilitate membrane penetration of siRNAs through an as yet only partially characterized mechanism.75 Lastly, the most critical aspect of RNAi biology and therapeutic application is the reduction of off-target effects, i.e., the unspecific binding of siRNAs to non-target mRNAs, which may result in increased cellular toxicity and immunogenicity (reviewed by Aigner83). Chemical modification of the siRNA guide strand represents one strategy to limit off-target effects. Here, the modified guide strand anneals to a passenger strand and abrogates off-target effects mediated by passenger strand complementarity (see reviews by Rao and colleagues84, 85).
5.2 Chemically modified siRNAs
While the delivery of naked siRNA oligonucleotides via hydrodynamic injection and electroporation is not suited for in vivo (systemic) delivery due to RNA instability in serum, conjugation of the guide strand with small molecules, peptides, or polymers can increase RNA stability. Standard modifications of siRNAs include 2′-O-methyl, 2′-fluoro, 2′-O-methoxyethyl and phosphorothioate (reviewed by Wilson and Keefe86). Additional approaches involving conjugation of siRNAs with cell-penetrating peptides (CPPs), PEG, cholesterol and its derivatives, long-chain fatty acids (>C18), bile-salt derivatives, and acid-responsive polymers containing PEG and an N-acetylgalactosamine (NAG) have been described (reviewed by De Paula et al87). These constructs resulted in potent gene silencing in cell culture in vitro and in organ systems in vivo but may be associated with increased off-target effects, attenuated therapeutic efficacy, and production of toxic metabolites due to degradation of RNA-modifying compounds.
5.3 Cationic polymers and lipids
Cationic polymers, e.g., the PEI-derivatives PEI–PEG36,88, 89 cyclodextrin-containing polycations, polylysine, chitosan, and cationic peptides and proteins such as CADY,90 MPG-8,91 and CPP-modified proteins92 have been used to neutralize the negatively charged phosphate backbone to stabilize siRNAs and enhance cellular delivery via binding to the negatively charged plasma membranes (reviewed by Kim and Kim76). Co-functionalization of cationic polymers, e.g., via PEGylation, is critical for reducing material toxicity and unspecific binding caused by the positive charge.93 Additional strategies include cross-linking of PEI via reversible disulfide bonds, amine modification, e.g., acetylation and introduction of negatively charged propionic acid or succinic acid groups, or ketalizing branched PEI.94, 95 Importantly, such nano-constructed PEI derivatives demonstrated high transfection efficiencies and substantial target knockdown in vivo coupled with robust endosomal escape, but displayed marked, albeit lower, toxicity compared to the unmodified polymeric cation. Chitosan particles, in contrast, effectively and safely delivered siRNAs in vivo upon intranasal and intravenous administration (reviewed by Andersen et al96). Additional rationally designed cationic polymers with significant knockdown efficacies and biocompatibility include mPEG45-b-PCL100-b-PPEEA12, an amphiphilic and cationic tri-block copolymer consisting of monomethoxy PEG, poly(3-caprolactone), and poly(2-aminoethyl ethylene phosphate),97 a transferrin-conjugated β-propionamide-cross-linked oligoethylenimine,98 a polyplex composed of poly(amido ethylenimine) and linear PEI,99 a poly (β-amino ester) (PbAE)/PEI/PEG hybrid conjugate,100 and a poly(amino ester glycol urethane) (PaEGU)/siRNA polyplex.101
Lipid nanoparticles (LNP) are one of the most widely used platforms to deliver siRNAs to cells, tissue and tumors. LNP platforms vary significantly with regard to lipid composition, lipid ratios, particle size, and overall structure. 1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) represents one of the first cationic lipids engineered for in vivo delivery of siRNAs, and unlike polymers, results in faster siRNA decomplexation into the cytosol, and consequently, in more effective gene knockdown (reviewed by Kim and Kim76). The stable nucleic acid lipid particle (SNALP) represent a prominent class of LNPs. This platform has a diameter of approximately 80 nm, and contains four different lipids, i.e., a PEG moiety (mPEG2000-C-DMA) to stabilize and to prevent aggregation of the construct, cholesterol, a neutral helper lipid (DPPC), and the ionizable lipid DLinDMA, which triggers fusion with endosomal membranes to release the siRNAs into the cytosol, and mediates condensation of lipid and anionic RNAi components during particle formation.102 Importantly, modification of the cationic lipid moiety of SNALP had dramatic impact on the in vivo efficacy of the conjugate: SNALP based on the ester-containing lipid DLinDAP exhibited reduced knockdown efficacy in vivo when compared to particles containing the alkoxy-containing lipid DLinDMA, and insertion of one additional methylene group into the headgroup (DLin-KC2-DMA) resulted in a 4-fold increase in potency compared to DLin-K-DMA.102 In addition, Akinc et al.56 developed a high-through put synthesis scheme to rapidly generate a library of amino-alkyl-acrylate and -acrylamide materials termed ‘lipidoids’, which were tested in cell and animal models for RNAi delivery and efficacy. Similar to SNALP formulations, lipoids required siRNA dosages greater than 1 mg/kg. Using epoxide chemistry, Love et al. generated a lipid library that enabled systemic delivery of siRNAs at doses below 0.01 mg/kg, and is capable of silencing multiple hepatic genes simultaneously after only one i.v. injection.103 Specifically, epoxide-derived lipidoids formulated with five siRNAs targeting genes implicated in cholesterol metabolism, i.e., ApoB, PCSK9, Xbp1, and SORT1, triggered gene knockdown greater than 65% in murine liver tissue in vivo upon i.v. injection,103 demonstrating the high-level efficacy and the potential to target entire oncogenic signatures in cancer in the near future. Clinical proof-of-concept studies of the SNALP platform include a phase I single-dose study of SNALP-ApoB (Tekmira Pharmaceuticals) in patients with elevated LDL cholesterol (reviewed by Barros and Gollob104), and of ALN-TTR01 (Alnylam Pharmaceuticals) for the treatment of ATTR (i.e., amyloidosis triggered by mutations in the transthyretin (TTR) gene). ATTR is characterized by the accumulation of pathogenic deposits of mutant and wild-type TTR protein in liver and in multiple extra-hepatic tissues, including the peripheral nervous system, heart, and the gastrointestinal tract. ALN-TTR01 was well-tolerated, and lead to substantial and persistent knockdown of serum TTR protein (reviewed by Barros and Gollob104). ALN-VSP (Alnylam) is a second SNALP-based conjugate, which carries two types of siRNA oligonucleotides targeting VEGF and kinesin family member 11 (KIF11, or KSP), genes with critical function in tumor angiogenesis and mitotic spindle formation. Multi-dose phase I clinical trials in patients with advanced solid tumors revealed that ALN-VSP was well tolerated, without signs of liver toxicity, only modest adverse side effects including fatigue, nausea and fever, and transient immunogenicity. Dose-limiting side effects in a small number of patients included transient grade 3 thrombocytopenia, and grade 3 hypokalemia. Importantly, RNAi-mediated cleavage of VEGF and KSP mRNAs within tumor biopsies confirmed activity of delivered siRNAs, and translated into partial responses, including stable disease greater than 2 months, and reduction in tumoral angiogenesis (see alnylam.com for more information). Additional SNALP conjugates include SNALP-PLK1, which utilize siRNAs targeting polo-like kinase 1 (PLK1), a cell cycle kinase with critical functions in mitosis and cytokinesis.105 Results from multi-dose phase-I clinical trials in patients with advanced solid tumors and primary liver cancers or liver metastases are expected in late 2012. Finally, the cationic liposomal conjugate ATU-027 (Silence Therapeutics), comprised of the cationic lipid AtuFect01, a neutral fusogenic DPhyPE helper lipid, PEG-, and siRNA targeting protein kinase N3 (PKN3), is another example of successful implementation of the lipid nanoparticle platform into early clinical trials. ATU-027 was well tolerated in the absence of dose-limiting toxicities, with few patients exhibiting disease stabilization and partial regression.106 (For a detailed review of these and additional cationic lipids, see Huang and Liu107 and Schroeder et al. 108).
5.4 Non-cationic polymers and lipids
Neutral liposomes encapsulate siRNAs, protect them from nuclease degradation, and deliver them to cells and tissue via membrane fusion or receptor-mediated endocytosis. Prominent examples include 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC)-based nanoliposomes (mean size <65 nm), which have been used for the in vivo delivery of siRNA sequences targeting EphA2, FAK,109 neutrophilin-2,110 or IL-8111 in various xenograft cancer models. These constructs have resulted in 10 to 30-fold increases in intratumoral siRNA levels compared to unconjugated DOTAP or naked siRNAs, and have induced substantial tumor shrinkage in the absence of toxicity in normal cell lineages, e.g., fibroblasts, bone marrow and hematopoietic cells. Preclinical studies of polyelectrolyte complex (PEC) micelles loaded with VEGFR-specific siRNAs showed similar results, including potent local or systemic delivery of siRNAs, robust intratumoral target knockdown, and suppression of tumor growth.112 Finally, DOTAP engineered with an outer lipid bilayer of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol-2000) and egg phosphatidylcholine (egg-PC) (PEG-DSPE) also document the potency of this platform, resulting in serum availability of siRNAs 20 hrs post injection.113 Additional non-cationic polymeric nanostructures include poly(isobutyl cyanoacrylate)-based liposomes with siRNAs targeting the EWSYFli1 transcriptional activator114 and poly(lactic-co-glycolic acid) (PLGA)-based liposomes loaded with Erc/mesothelin-specific siRNAs.115 In addition to encapsulation of siRNAs into liposomal structures, siRNAs can also be directly conjugated to polymer chains, including peptides, cholesterol, and aptamers. This strategy is exemplified by PBAVE, an amphipathic poly(vinyl ether) functionalized with siRNAs targeting hepatocellular apolipoprotein B (ApoB) and peroxisome proliferator-activated receptor alpha (PPARα).72
5.5 Metal nanoparticles
With approximately 10,000 articles published over the past decade, polyvalent gold nanoparticles (Au-NPs) densely functionalized with highly oriented DNA or siRNA oligonucleotides represent one of the most prominent nucleic acid-based nanoconjugates. After the introduction of DNA-Au-NPs in 1996 and demonstration of their relatively straightforward synthesis, their capacity to penetrate cells and their ability to silence gene expression via an antisense mechanism, several siRNA-Au-NP platforms were developed using gold-thiol chemistry or electrostatic Au-RNA interactions to decorate gold particles with RNAs (reviewed by Cutler et al116; see Figure 4 for a schematic overview of gold-RNAi nanoconjugates).
Fig. 4. Different RNAi-based gold nanoparticles based on gold-thiol chemistry and electrostatic interactions.

PEG, poly(ethylene glycol); PAMA, poly(alkyl methacrylate); F, folate; PbAE, poly (β-amino ester); PEI, poly(ethyleneimine); PAH-Cit, cis-aconitic anhydride-functionalized poly(allylamine).
Using thiol-functionalized siRNAs, 15 nm Au-NPs, together with PEG5000–PAMA7500 as passivating and stabilizing ligands, Oishi et al. reported the first siRNA-based gold nanoparticle, which triggered robust and persistent gene knockdown of luciferase expression in HuH-7.117 Giljohann et al. used a similar approach decorating 13 nm Au-NPs with thiolated siRNA and PEG400 (hereafter referred to as spherical nucleic acids, SNAs).118 Despite their large negative charge (zeta potential < −30 mV), these SNAs showed highly efficient cellular uptake, significant serum stability without the use of auxiliary transfection strategies or chemical modifications, and consequently efficient, specific, and persistent gene knockdown.118 Cellular uptake is mediated by the nucleic acid moiety of the particle, as coreless or iron oxide SNAs exhibit similar internalization rates.119 Uptake is reduced several fold when Au-NPs without DNA/RNA are passivated with BSA. It has been suggested that cellular uptake is mediated by scavenger receptor-driven endocytosis.120 This class of pattern-recognition receptors binds to serum proteins absorbed by the nucleic acid moiety of SNAs, which, upon their internalization and endosomal escape, accumulate within the cytoplasm to silence gene expression. Robust intracellular and serum stability of SNAs has been attributed to the high local sodium ion concentration, which potently deactivates nucleases.118, 121 Nucleic acid degradation is 4 times slower in aqueous solution compared to free duplexes, and serum stability is further increased by the aforementioned absorption of serum proteins, which limit access of nucleases to the gold-bound RNA. Due to high-level intracellular stability, SNAs are potent, and achieve highly persistent gene knockdown at picomolar concentrations.118 Due to potent penetration of SNAs into tissue in vivo, and minimal systemic toxicity and immunogenicity (as measured by quantification of β-IFN levels) in rodents, RNAi-functionalized gold nanoparticles represent a powerful therapeutic platform to combat cancers and other diseases of genetic etiology.
In addition, the stability of the nucleic acid shell allows for co-functionalization of SNAs with chemotherapeutics or corresponding pro-drugs, e.g., platinum(IV) prodrug, which upon internalization is reduced to a cytotoxic platinum(II) species, and released into the cytosol through reductive elimination of their axial ligands resulting in a Pt-SNA species that is more effective than cisplatin or the pro-drug. In addition, drug conjugation can increase the solubility of chemotherapeutic drugs such as paclitaxel, resulting in 4–10 fold lower IC50 values.122 The ability to generate these single-entity, highly effective hybrid (H)-SNAs capable of biotherapeutic gene silencing and chemotherapy-induced tumor cell killing illustrates the enormous potential of SNAs as anti-cancer compounds. Further harnessing the stability of the nucleic acid shell, along with efforts to co-functionalize SNAs with imaging agents such as gadolinium (Gd(III)), may aid in tracking intramural accumulation of particles via MRI.123 Most clinical studies rely on indirect measures to quantify tumor uptake of compounds such as SMIs or antibodies, e.g., drug responses or measurements of drug levels in body fluids. However, if a drug does not elicit a response, it is rarely determined if the drug was ineffective or if it simply failed to reach its intended target.
In addition to the 13 nm PEG400-SNAs discussed in detail above, a different conjugation strategy utilized 15 nm Au-NPs decorated with thiolated PEG1000-NH2, an siRNA corona attached via a disulfide crosslinker (SPDP) to the terminal amine group on the PEG, and further coated with poly(β-amino ester)s (PBAEs) to enhance cellular uptake and endosomal escape.124 The PBAE moiety appears to be critical to the capacity of the nano-conjugate to elicit significant gene knockdown, as particles without PBAEs are ineffective. It remains uncertain whether the ester conjugation triggers more efficient cellular uptake and endosomal escape, and/or impacts siRNA accessibility by the RISC complex.
In addition to using gold particles of various diameters, 40 nm hollow gold nanoshells (AuNSs) have emerged as yet another core structure for delivery of siRNAs into cells. Conjugation of AuNSs with SH–siRNA–PEG300 and SH–PEG300 resulted in constructs with >100 siRNA oligonucleotides per particle.125 Coating with transactivator of transcription (TAT) lipids to enable efficient internalization, and irradiation with near-infrared (NIR) light to trigger endosomal escape and decomplexation of the siRNA moiety, resulted in profound knockdown of a GFP reporter in C166 mouse endothelial cells. AuNSs co-functionalized with SH–siRNA and TA–PEG5000–F (TA, thioctic acid required for conjugation of siRNAs; F, folic acid required for cellular targeting), triggered potent knockdown of endogenous levels of the NFκB subunit p65 in HeLa cells.126 Gene silencing activity was strictly dependent on NIR irradiation and on the presence of the folate group. In vivo administration of these conjugates via tail vein injections resulted in knockdown of p65 in subcutaneous HeLa cell-derived tumors and increased sensitivity of tumor-bearing mice to irinotecan.126
Taking advantage of electrostatic interactions between negatively charged siRNAs and the Au-NP surface, several constructs have emerged that use 15 nm Au-NPs coated with layers of positively charged PEI and siRNA oligonucleotides.127 The resulting particles can have either a terminal RNA or a PEI layer, which impact cellular uptake and endosomal escape. Importantly, the surface density of siRNA oligonucleotides is significantly higher (780 vs. 30–50 oligonucleotides on Au-SH-NPs), likely due to the strong electrostatic interactions of the PEI and RNA moieties.127 Prominent constructs include Au-NPs decorated with layers of PEI, an anionic charge-reversal polyelectrolyte (PAH-Cit = cis-aconitic anhydride-functionalized poly(allylamine)), and siRNAs.128 During acidification within the endosomal compartment, PAH-Cit undergoes a charge reversal resulting in the release of the siRNAs. Additional constructs include (a) Au-NPs with PEI25000 chains replacing the PAH-Cit compound to create a positively charged Au-NP;129 (b) Au-NPs coated with a stabilizing and cationic block polymer poly(N-2-hydroxypropyl methacrylamide-block-N-[3-dimethylamino)propyl] methacrylamide P(HPMA70-b-DMAPMA24) and siRNA;130 (c) Au-NPs synthesized in the presence of cysteamine hydrochloride and functionalized with siRNA–PEG5000;131 and (d) cationic gold nanorods (AuNRs) without the addition of stabilizing PEG chains.132 All of these constructs triggered significant gene silencing in vitro, and pointed to specific ligand requirements for cellular uptake and endosomal escape; all are awaiting detailed in vivo efficacy studies.
5.6 Other Nanoconjugates
Additional nanoconjugates for RNAi delivery include hyaluronic acid (HA) nanoparticles, also referred to as nanogels, poly(d,l-lactic-co-glycolic) acid (PLGA) nanoparticles, and dendrimer-conjugated magnetofluorescent nanoworms (dendriworms), all of which can effectively penetrate cells and silence gene expression in vivo, and in the case of dendriworms, show prominent intracellular and intratumoral accumulation, which can be followed by fluorescent tracking (reviewed by Davis et al133).
5.7 Tumor-targeted nanoparticles
Materials on the nanoscale preferentially accumulate in tumor elements due to the EPR of a compromised tumor vasculature. Tumor vessels are characterized by poorly aligned endothelial cells with wide fenestration, which together with absent intratumoral lymphatic drainage results in highly distorted dynamics of molecular and fluid transport (reviewed by Hirsjarvi et al134). To further enhance tumor-specific uptake and tumoricidal activities, and to reduce systemic toxicity in non-tumors organ sites, a spectrum of nanoconstructs has been functionalized with ligands recognizing surface elements of cancerous tissue, foremost the transferrin, folate, and integrin receptors which display soaring overexpression in a variety of tumor tissues. Specifically, folate-conjugated cholesteryl-3-beta-carboxyamidoethylene-N-hydroxyethylamine, and PEG-distearoylphosphatidyl ethanolamine (DSPE) nanoparticles with Her2-specific siRNAs showed significant in vivo efficacy in xenograft models,135 and PEGylated transferrin receptor-targeted nanoparticles composed of a β-cyclodextrin-containing polycation loaded with siRNAs against the M2 subunit of ribonucleotide reductase triggered significant reduction in target mRNA in cell and animal models, and showed a dose-dependent accumulation in tumor cells of melanoma patients in early phase I clinical trials.136 Additional targeting ligands have included antibody–protamine fusion proteins for the targeted delivery of siRNAs to silence HIV-1 capsid gene (gag) to block HIV replication in primary T cells137, 138, anti-b7 integrin antibodies to deliver polymeric nanoparticles with cyclin D-specific siRNAs to leukocytes,139 short peptides derived from rabies virus glycoprotein (RVG) to enable siRNA to cross the blood-brain-barrier and accumulate within neurons,140 PEGylated RGD peptide targeting PEI-siRNA nanoconstructs to tumor vasculature to silence VEGF-2,141 aptamers recognizing prostate-specific membrane antigen (PSMA) to deliver siBcl-2 and siPLK1 oligonucleotides,142 and small molecules, e.g., lactose, and NAG to drive siRNA-mediated gene silencing in hepatocytes.72
Strikingly, targeted particles have yet to make the leap from promising concept to effective anti-cancer compound, as the vast majority of nanoconjugates in advanced preclinical testing and early phase clinical trials do not contain targeting ligands. The reasons for the apparent shortcoming of past efforts are complex: (1) Systematic auditing of biological and physico-chemical variables of targeted materials critical for cellular/tissue uptake and pharmacokinetical properties are often suboptimal. In addition, reproducible synthesis and consequently large-scale production of targeted conjugates is difficult; ligand density and activity often vary, impacting biodistribution and half-life (reviewed by Farokhzad and Langer143). (2) Surface molecule used as entry sides for targeted therapies via receptor-mediated endocytosis often display a highly heterogeneous intratumoral distribution, making robust tumor dissemination challenging. (3) Different receptors show different rates of endocytosis in different tissue. Thus, a detailed understanding of endosomal trafficking pathways in different cancers is required to choose the most optimal ligand/receptor system. (4) Intratumoral accumulation is a passive process, and requires the extravasation of nanodrugs through the structurally and functionally compromised tumor vasculature into the tumor mass. Thus, the improvement of biological, and physicochemical properties of the conjugate rather than the addition of a targeting ligand can increase circulating half-lives, and can lead to conjugates that tend to amass in cancerous tissue more effectively. While circulation and consequently intratumoral accumulation represent parameters that are independent of targeting ligands, retention and specific cellular uptake of conjugates, however, are processes driven by the targeting moiety of the nanoparticle, and can result in more potent anti-tumorigenic effects of targeted versus untargeted therapies.
6. Future Directions – toward individualized cancer nanomedicine
John F. Kennedy’s quote “The greater our knowledge increases the more our ignorance unfolds” summarizes the journey cancer genomics began more than 15 years ago. Cancer initiation, progression, dissemination, and therapy responses are driven by a vast and unpredictably complex spectrum of mutations. Copy number alterations, nucleotide changes, and translocations can alter protein abundance and protein function, or can create novel proteins. Collaborations between academic and industrial centers around the world (e.g., the TCGA, and the International Cancer Genome Consortium, ICGC) have resulted in the maturation and expansion of whole-genome, exome, and transcriptome sequencing, and have released partial cancer genome data sets pointing to more than 7,500 putative cancer genes (ICGC Dataset Version 6; http://www.icgc.org). Exemplified by the most recent introduction of ALK and BRAF inhibitors into clinical practice, this novel technological paradigm has already begun to revolutionize cancer medicine. Our advanced understanding of genomic perturbations has allowed us (a) to select specific patient populations for clinical trials testing specific target agents, e.g., trastuzumab for Her2-positive breast cancer, and (b) to predict clinical response, e.g., the ineffectiveness of EGFR mAb for the treatment of KRAS-mutated colon cancers (reviewed by Davis144), or the failure of Raf inhibitors to block progression of cancers with mutated Ras alleles (‘the Raf paradox’; reviewed by Cox and Der and by Weeraratna145, 146). Together with advances in the functional interrogation of cancer genomes and the development of RNAi-based nanotechnological strategies to target emerging driving and contributing oncogenes, the implementation of personalized cancer nanomedicine is an attainable and, most of all, ethically imperative goal. The rise of genomic medicine and nanotechnology, however, also point to significant challenges in translating genomic information and nanodrug development into clinical endpoints. Some of the most relevant questions for genomic and drug development pipelines are: Which are the most critical driving and contributing mutations, and how can they be distinguished from genomic noise? What is the genetic and tumor biological context of a given driving or contributing CCG? How can the efficacy, specificity, and biocompatibility of RNAi nanotherapeutics be improved? The equally important challenges for bioinformatics, pathology, imaging, biospecimen repositories, and clinical trial management to enable personalized medicine are summarized elsewhere in detail.147
6.1 Understanding the plasticity of cancer
The number of mutations in a cancer cell can range from thousands to hundreds of thousands. The majority of these perturbations result from genomic instability and DNA damage (i.e., passengers), with a relatively small number of aberrations having causal roles in disease progression (drivers and contributors). Among these, only a subset can be therapeutically targeted (‘druggable’) and/or has prognostic or diagnostic significance (‘actionable’) (reviewed by Dancey et al144). In addition, cancer is a highly heterogeneous disease with mutational profiles differing between cancer types, between tumors arising from the same cancer lineage, and between cells within a tumor. Cancer development and progression is also a highly dynamic process, as tumors can continuously acquire additional mutations due to increasing genomic instability (intrinsic selection), and due to extrinsic pressures conferred by therapy and environmental cues. Finally, CCGs do not function in linear pathways, but rather exist as part of a multidimensional circuitry, where the activity of one CCG is influenced by the mutational status and function of other CCGs. Such convoluted wiring of signaling pathways, together with heterotypic tumor-stroma interactions, and the overshadowing impact of patient-specific germline mutations on tumor development, add yet additional layers of complexity to an already intricate disease (reviewed by Chin and Gray and by Chin et al36, 45). In particular, a recent study by Todd Golub and colleagues revealed that that the tumor stroma significantly contributes to therapy susceptibility of cancer cells via non-tumor autonomous mechanisms. One specific mechanism identified in this study involved the stromal secretion of hepatocyte growth factor (HGF), subsequent activation of the HGF receptor MET, and downstream MAPK and PI3K pathways in BRAF mutant melanoma cells, triggering resistance of melanomas toward BRAF inhibitors.148
How can we make sense of the cancer genome? The continuous and rapid evolution of genomic technologies will likely unravel a complete list of cancer-relevant genes. These structural analyses have to be complemented by genome-wide, systematic and integrative approaches to characterize function of CCGs and their context dependencies (Figure 2). As described above, these efforts have already led to the identification of several novel cancer genes and pathways. It will require, however, further development and refinement of such functional assays to enable true genome-wide functionalization. Specifically, genome coverage of mammalian ORF, cDNA, and RNAi libraries is incomplete, splice variants are poorly characterized, one-dimensional readouts in most cases are only focused on cell survival, and screening is usually executed in a limited number of cell lines not reflecting the high-level heterogeneity seen in cancers. Consequently, larger collections of cell cultures have to be used in a wider spectrum of phenotypic assays, e.g., migration and invasion experiments, as critical cancer genes might not be involved in cellular growth, but may play important roles in other processes, e.g., metastasis. In addition, RNAi or cDNA complementation screens can produce false positives due to off-target effects. Here, the integration of functional data obtained from over- and underexpression screens in different cell lineages, derived from different cancer types, together with structural genomic information can provide additional levels of validation. Finally, the majority of HT screens have focused on the characterization of genes localizing to chromosomal regions with copy number variability. To study mutations and translocations, additional alleles encoding for mutant and fusion genes have to be generated to probe their impact on the biology of cancers. Once CCG lists have been created via HT gain- and loss-of-function screens, triangulated with structural genomic information, and analyzed in vitro for their impact on growth, cell death, migration, invasion, and angiogenesis, then sophisticated xeno- and syngeneic orthotopic models and GEMMs representing the final step in CCG validation can be conducted. As outlined below, these mouse models will not only aid in describing the role of a CCG in tumorigenesis, but will also serve as more refined in vivo testing platforms for novel therapeutics, and will provide important preclinical information preceding clinical trials. Importantly, such rigorous biological interrogation has to be an iterative process, where deep biological analyses or even early clinical trials have to inform structural and HT functional analyses. It is plausible that in-depth characterization of CCGs may contradict early-stage genomic and HT analyses, and may reveal that our definition of a given CCG as a driver or contributor is limited, has to be substantially revised, or is simply incorrect.
6.2 Preclinical validation of candidate nanodrugs – the mighty (xenografted) mouse and beyond
Much ink has been devoted to weighing the advantages and disadvantages of murine models for preclinical drug testing. Many cancer biologists and geneticists would agree that graft and GEM models have contributed considerably to our understanding of oncogenes and tumor suppressor function over the past decades. In addition, rodent models are the most tractable mammalian systems for pharmacokinetic, pharmacodynamic, and toxicology studies. Much disagreement, however, exists on the usefulness of xenograft models in cancer drug discovery. I argue that the poor correlation between drug efficacy in subcutaneous xenograft studies versus human clinical trials has proven that these models are largely ineffective for preclinical drug evaluation, and are better classified as human-in-mouse systems, in which established human cancer cells with limited genotypic resemblance to primary cancer specimens are grown in an immunocompromised host with the support of murine stroma and vasculature. Due to their cost-effectiveness, ease of generation, and suitability for HT applications, however, graft models are highly useful for drug testing, but have to be significantly refined (reviewed by Sharpless and Depinho55). First, patient-derived primary cells, e.g., tumor-initiating cells (TICs) enriched for cancer stem cells, should be used to generate xenogeneic grafts that more faithfully recapitulate genotypic and phenotypic hallmarks of the human disease compared to transformed cancer cell lines. Second, the numbers of genomically fully characterized cell lines have to be significantly increased, such that xenografts more adequately represent the genotypic diversity of human tumor specimens so that pharmacogenomic correlates of drug responses can be cataloged. Third, xenograft systems with more physiological tumor-stroma interactions have to be further developed by orthotopically injecting cells into the organ site of interest, by co-implanting tumor and stromal elements, or by using GEMM-derived cancer cells in syngeneic explant studies. Notwithstanding the continuing evolution of xenograft models, their inability to faithfully model tumor progression (i.e., the development of hyperplastic to dysplastic to more malignant stages), and to adequately represent the genetic diversity of human cancers limits their applicability, and point to GEMMs with spontaneously and stochastically developing tumors as superior drug testing platforms. In this regard, the costs and ease of generating and managing multi-allelic models, tumor latencies, penetrance, and faithful recapitulation of human tumor characteristics are critical parameters to consider. While short latencies and high penetrance of tumor development are preferred, rapidly evolving cancers driven by strong oncogenes with concomitant loss of potent tumor suppressors may not acquire stochastic secondary genetic or epigenetic lesions, but may develop multi-focal tumors that may not properly co-evolve with their stromal microenvironment. Consequently, we have to define the right balance between practicability and faithful recapitulation of cancer hallmarks. Although conditional gene targeting remains a lengthy and involved process, GEMMs can most certainly overcome shortcomings of explant models, as the tumor emerges in an immunocompetent environment, and displays functional stromal interactions. Simple breeding schemes can be developed to generate the desired multi-allelic cohort, as multiple controls, such as mice harboring individual mutant alleles, are typically not required for drug testing. High-penetrance and short latency allow for speedy, cost-effective HT-testing of drugs. Induction of conditional alleles is straightforward by administering tamoxifen to mice with somatically inducible Cre-ERT2 alleles. Finally, tumor development can be followed non-invasively by MR imaging, palpation, quantification of serological markers, or assessment of intratumoral luciferase activity by In vivo Imaging System (IVIS)-based analyses. These technological advancements will allow for the facile, yet conclusive preclinical evaluation of drugs. Several model systems for a variety of different cancer types are available through the Mouse Models of Human Cancer Consortium (MMHC) (reviewed by Sharpless and Depinho55). A recent study by Wong and colleagues demonstrated the predictive power of GEMM for clinical drug testing. They conducted synchronous ‘co-clinical’ trials in lung cancer patients and GEMMs harboring different, lung-cancer specific genetic signatures (KRas mutation with concomitant p53 and Lkb1 deletions). Drug testing in KRas-driven GEMMs successfully predicted which genetically defined patient populations would benefit from combinatorial treatment regimens consisting of docetaxel and the MEK inhibitor selumetinib (AZD6244).149
6.3 Further developing RNAi-based nanotherapeutics
The discovery and validation of novel oncogenes can immediately inform the design of RNAi nanomaterials to specifically target these cancer genes (Figure 5A). Several nanomaterials, such as SNAs, have emerged as fundamentally novel classes of therapeutics that can overcome the shortcomings of RNAi-based therapy, i.e., delivery, intracellular stability, off-target effects, systemic toxicity, and immunogenicity. In particular, SNAs with densely packed, highly oriented, siRNA oligonucleotides can be recognized and endocytosed by scavenger receptors, and following endosomal escape, can potently and persistently neutralize gene expression due to increased resistance toward nuclease-driven degradation (reviewed by Cutler et al116). However, further mechanistic and biological studies are required to fully understand some of the fundamental properties underlying cellular entry, tissue dissemination, low-level immunogenicity, and systemic toxicity.
Fig. 5. Integrative nano-drug development.
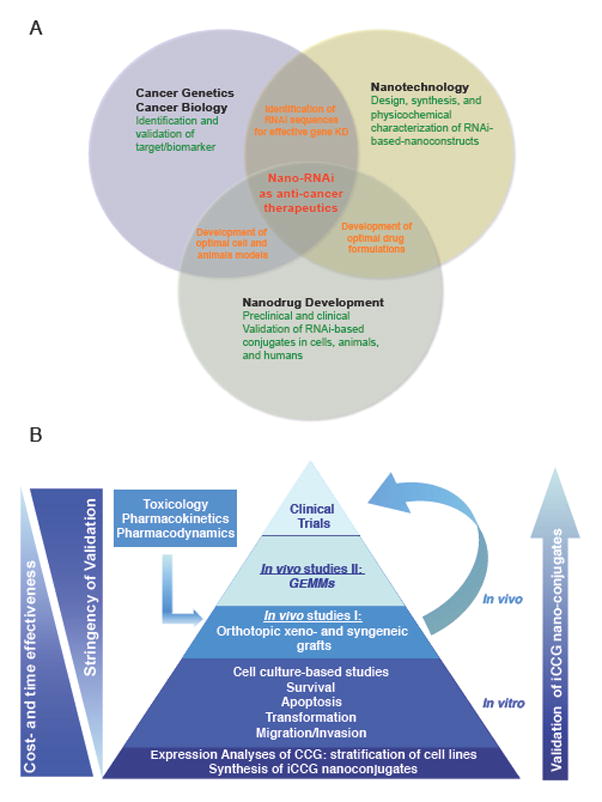
(A) Amalgamation of cancer biology/genetics, bioengineering, i.e., the design and synthesis of nanotechnological platforms, and nanodrug development to enable the preclinical and clinical development of RNA-based nanoconjugates. (B) Validation scheme for the (pre-)clinical characterization of iCCG nano-conjugates. Initial studies aim to assess CCG expression in a panel of cancer cells to stratify lineages with high, low, or absent expression for subsequent functional studies to assess siCCG-nanoconjugates. Animal studies, together with toxicology, pharmacokinetic and –dynamic studies will enable early phase 0/I/II clinical studies in humans. Of note, model systems to validate iCCG nanoconstructs show different degrees of cost- and time effectiveness, and complexity.
Furthermore, the modification of SNAs and other nano-RNAi conjugates with ligands or antibodies enabling more robust tumor-specific uptake beyond the EPR effect has to be optimized to further increase conjugate efficacy while reducing the potential for adverse side effects associated with systemic administration. Finally, targeted nano-constructs together with nanomaterials functionalized with multiple siRNA sequences enabling concomitant silencing of several oncogenes have to be enrolled into detailed in vivo validation, pharmacokinetic, pharmacodynamic, and toxicology studies in relevant graft and perhaps GEM models to drive these platforms toward early-phase clinical trials. Figure 5B exemplifies a validation scheme for RNAi-based nanoconstructs, starting with detailed expression analyses of CCGs to be targeted, followed by a detailed characterization of nano-RNAi-triggered knockdown in cells, animals, and humans.
Collectively, these nanoconstructs provide radically novel treatment options for cancer patients. Our capacity to design particles functionalized with multiple siRNA sequences targeting entire oncogenic signatures, together with the speed of nanodrug development and nanodrug testing in established xeno-, allo- and GEM models makes RNAi-based nanotechnology a highly attractive anti-cancer approach suitable for co-extinction strategies. The foreseeable progress in the development of targeting ligands through HT yeast, phage, and ribosomal display methodologies coupled with a better understanding of how particle size, geometry, and interactive forces between siRNA cargo and nanomaterial impact delivery, intracellular release, toxicity, and immunogenicity will most certainly drive the further implementation of nanopharmaceuticals into oncology practice.
6.4 Concluding remarks
With the signing of the National Cancer Act of 1971 by then U.S. President Richard Nixon, and a >200 billion USD investment in cancer research over the past decades, long-term survival of cancer patients has considerably improved; advanced-stage malignancies, however, still culminate in death within five years post diagnosis. Why did we fall short? Why are drug discovery and development processes so ineffective with a near 95% failure rate in gaining FDA approval? And why is the attrition rate of compounds most prominent in late-stage clinical trials, the most expensive phase of drug testing? Major contributing factors are the lack of stringent target gene identification and validation, ineffective preclinical drug testing in physiologically relevant model systems, and only moderate progress in enrolling novel therapeutic concepts, first and foremost nanodrugs, into preclinical and clinical pipelines. We have to move past the many proof-of-principle studies of nanoconstructs aimed at targeting an investigator’s favorite oncogene in standard cell and xenograft models, and move toward genome-scale, unbiased identification of rate-limiting oncogenic networks, and the systematic design of nano-RNAi conjugates targeting these oncogenic lesions. Furthermore, it will be critical to preclinically characterize nanomaterials neutralizing these networks in the most physiologically relevant cell and animal models. We have to develop smart combinatorial treatment regimes that combine the power of nano-RNAi constructs to neutralize virtually any oncogenic lesions, and the proven efficacy of conventional chemotherapy (e.g., DNA-damage-inducing agents) and targeted pharmaceuticals that inhibit critical driving oncogenes, such as RTKs. It will be critical to determine the molecular mechanisms that act as roadblocks preventing chemo- and RTK-targeted therapies from inducing tumor-specific apoptosis and regression. We then can target these roadblocks using RNAi-based nanomaterials, and can envision using hybrid conjugates, such as H-SNAs co-functionalized with chemotherapeutics and siRNA oligonucleotides to concomitantly target driving oncogenes and downstream anti-apoptotic roadblocks. Lastly, we have to integrate teams of specialists with expertise in the areas of cancer biology, clinical cancer sciences, and bioengineering to enable deep biological and clinical characterization of nanomaterials. Larger platform grants, such as Centers for Cancer Nanotechnology Excellence (CCNEs) will continue to be instrumental in driving this integration. Together with more effective collaborations of academia and industry, the establishment of novel academic constructs and infrastructures that combine multileveled biological and clinical validation with milestone-driven drug development may aid in overcoming the ‘valley of death’ separating bench from bed side.
The past few years have given us a glimpse of the potential of personalized cancer nanomedicine. Now, cancer geneticists and bioengineers are poised to build on these most recent successes, and to develop bold and ambitious plans to further translate basic genomic findings into clinical endpoints. It will be the amalgamation of chemistry and basic and clinical cancer research that will not only increase our knowledge, but also will also help us realize and overcome our ignorance to launch the most audacious attack on cancer yet.
Insight, innovation, integration.
High-throughput characterization and functional interrogation of cancer genomes has unraveled a complex landscape of genetic and epigenetic modifications, and has initiated the implementation of personalized cancer medicine into clinical practice. The sheer complexity of genomic information, and the difficulty to concomitantly modulate the action of multiple ‘undruggable’ targets, however, pose significant challenges to drug development. Here, I provide an overview of the current state and future challenges of genomics-driven cancer medicine. I discuss the role that nanotechnology may play in overcoming some of the most critical barriers to clinical progress, foremost the targeting of the undruggable oncogenome, and highlight the importance of integrative, cross-disciplinary approaches bridging cancer genetics, cancer biology, and bioengineering to enable personalized (nano)drug design.
Acknowledgments
This work was supported by grants from NIH (grants 5R00CA129172-05, U54CA151880), from American Cancer Society, from the Sidney Kimmel, Zell, Dixon and Coffman Foundations. A.H.S. is recipient of a James S. McDonnell 21st Century Science Initiative Research Award. A.H.S would like to thank C. Shad Thaxton for critical reading of the manuscript.
References
- 1.Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pegram MD, et al. Trastuzumab and chemotherapeutics: drug interactions and synergies. Semin Oncol. 2000;27(6 Suppl 11):21–5. discussion 92–100. [PubMed] [Google Scholar]
- 4.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 5.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 9.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 10.Yang JC, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349(5):427–34. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vredenburgh JJ, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25(30):4722–9. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 12.Vredenburgh JJ, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 13.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 14.Tsai J, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joseph EW, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107(33):14903–8. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verstovsek S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurzrock R, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660–6. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Courtney KD, et al. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–83. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Von Hoff DD, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361(12):1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 20.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 21.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5(12):1026–33. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 22.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 23.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 24.Van Raamsdonk CD, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2009;21(2):209–20. doi: 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figueroa ME, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delhommeau F, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 34.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin L, Gray JW. Translating insights from the cancer genome into clinical practice. Nature. 2008;452(7187):553–63. doi: 10.1038/nature06914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576(1–2):22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 38.Shayesteh L, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21(1):99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 39.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 40.Sweet-Cordero A, et al. An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat Genet. 2005;37(1):48–55. doi: 10.1038/ng1490. [DOI] [PubMed] [Google Scholar]
- 41.O’Neil J, et al. Activating Notch1 mutations in mouse models of T-ALL. Blood. 2006;107(2):781–5. doi: 10.1182/blood-2005-06-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim M, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125(7):1269–81. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 43.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125(7):1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodgson JG, et al. Copy number aberrations in mouse breast tumors reveal loci and genes important in tumorigenic receptor tyrosine kinase signaling. Cancer Res. 2005;65(21):9695–704. doi: 10.1158/0008-5472.CAN-05-0755. [DOI] [PubMed] [Google Scholar]
- 45.Chin L, et al. Making sense of cancer genomic data. Genes Dev. 2011;25(6):534–55. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boehm JS, Hahn WC. Towards systematic functional characterization of cancer genomes. Nat Rev Genet. 2011;12(7):487–98. doi: 10.1038/nrg3013. [DOI] [PubMed] [Google Scholar]
- 47.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108(30):12372–7. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brough R, et al. Functional Viability Profiles of Breast Cancer. Cancer Discov. 2011;1(3):260–73. doi: 10.1158/2159-8290.CD-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baldwin A, et al. Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. Proc Natl Acad Sci U S A. 2010;107(28):12463–8. doi: 10.1073/pnas.1007462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bommi-Reddy A, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci U S A. 2008;105(43):16484–9. doi: 10.1073/pnas.0806574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bandyopadhyay S, et al. Rewiring of genetic networks in response to DNA damage. Science. 2010;330(6009):1385–9. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horn T, et al. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods. 8(4):341–6. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- 55.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5(9):741–54. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 56.Akinc A, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26(5):561–9. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maser RS, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447(7147):966–71. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heyer J, et al. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10(7):470–80. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bachoo RM, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269–77. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 60.Bric A, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell. 2009;16(4):324–35. doi: 10.1016/j.ccr.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45(4):545–52. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 62.Chu GC, et al. Stromal biology of pancreatic cancer. J Cell Biochem. 2007;101(4):887–907. doi: 10.1002/jcb.21209. [DOI] [PubMed] [Google Scholar]
- 63.Hezel AF, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20(10):1218–49. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 64.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18(1):73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 65.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–92. [PubMed] [Google Scholar]
- 66.Maeda H, et al. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 67.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–27. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 68.Dillon CP, et al. Rnai as an experimental and therapeutic tool to study and regulate physiological and disease processes. Annu Rev Physiol. 2005;67:147–73. doi: 10.1146/annurev.physiol.67.040403.130716. [DOI] [PubMed] [Google Scholar]
- 69.Reischl D, Zimmer A. Drug delivery of siRNA therapeutics: potentials and limits of nanosystems. Nanomedicine. 2009;5(1):8–20. doi: 10.1016/j.nano.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371–8. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 71.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117(12):3623–32. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rozema DB, et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci U S A. 2007;104(32):12982–7. doi: 10.1073/pnas.0703778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heidel JD, et al. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc Natl Acad Sci U S A. 2007;104(14):5715–21. doi: 10.1073/pnas.0701458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bartlett DW, et al. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104(39):15549–54. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee H, et al. Target-specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels. J Control Release. 2007;119(2):245–52. doi: 10.1016/j.jconrel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 76.Kim WJ, Kim SW. Efficient siRNA delivery with non-viral polymeric vehicles. Pharm Res. 2009;26(3):657–66. doi: 10.1007/s11095-008-9774-1. [DOI] [PubMed] [Google Scholar]
- 77.Schaffert D, Wagner E. Gene therapy progress and prospects: synthetic polymer-based systems. Gene Ther. 2008;15(16):1131–8. doi: 10.1038/gt.2008.105. [DOI] [PubMed] [Google Scholar]
- 78.Midoux P, et al. Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br J Pharmacol. 2009;157(2):166–78. doi: 10.1111/j.1476-5381.2009.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shim MS, Kwon YJ. Acid-responsive linear polyethylenimine for efficient, specific, and biocompatible siRNA delivery. Bioconjug Chem. 2009;20(3):488–99. doi: 10.1021/bc800436v. [DOI] [PubMed] [Google Scholar]
- 80.Convertine AJ, et al. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release. 2009;133(3):221–9. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oliveira S, et al. Fusogenic peptides enhance endosomal escape improving siRNA-induced silencing of oncogenes. Int J Pharm. 2007;331(2):211–4. doi: 10.1016/j.ijpharm.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 82.Hatakeyama H, et al. A pH-sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA-containing nanoparticles in vitro and in vivo. J Control Release. 2009;139(2):127–32. doi: 10.1016/j.jconrel.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 83.Aigner A. Gene silencing through RNA interference (RNAi) in vivo: strategies based on the direct application of siRNAs. J Biotechnol. 2006;124(1):12–25. doi: 10.1016/j.jbiotec.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 84.Rao DD, et al. Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 2009;16(11):807–9. doi: 10.1038/cgt.2009.53. [DOI] [PubMed] [Google Scholar]
- 85.Rao DD, et al. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61(9):746–59. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 86.Wilson C, Keefe AD. Building oligonucleotide therapeutics using non-natural chemistries. Curr Opin Chem Biol. 2006;10(6):607–14. doi: 10.1016/j.cbpa.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 87.De Paula D, et al. Hydrophobization and bioconjugation for enhanced siRNA delivery and targeting. RNA. 2007;13(4):431–56. doi: 10.1261/rna.459807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Merkel OM, et al. Nonviral siRNA delivery to the lung: investigation of PEG-PEI polyplexes and their in vivo performance. Mol Pharm. 2009;6(4):1246–60. doi: 10.1021/mp900107v. [DOI] [PubMed] [Google Scholar]
- 89.Merkel OM, et al. Stability of siRNA polyplexes from poly(ethylenimine) and poly(ethylenimine)-g-poly(ethylene glycol) under in vivo conditions: effects on pharmacokinetics and biodistribution measured by Fluorescence Fluctuation Spectroscopy and Single Photon Emission Computed Tomography (SPECT) imaging. J Control Release. 2009;138(2):148–59. doi: 10.1016/j.jconrel.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 90.Crombez L, et al. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol Ther. 2009;17(1):95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crombez L, et al. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009;37(14):4559–69. doi: 10.1093/nar/gkp451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eguchi A, et al. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotechnol. 2009;27(6):567–71. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Malek A, et al. PEG grafting of polyethylenimine (PEI) exerts different effects on DNA transfection and siRNA-induced gene targeting efficacy. J Drug Target. 2008;16(2):124–39. doi: 10.1080/10611860701849058. [DOI] [PubMed] [Google Scholar]
- 94.Zintchenko A, et al. Simple modifications of branched PEI lead to highly efficient siRNA carriers with low toxicity. Bioconjug Chem. 2008;19(7):1448–55. doi: 10.1021/bc800065f. [DOI] [PubMed] [Google Scholar]
- 95.Shim MS, Kwon YJ. Controlled delivery of plasmid DNA and siRNA to intracellular targets using ketalized polyethylenimine. Biomacromolecules. 2008;9(2):444–55. doi: 10.1021/bm7007313. [DOI] [PubMed] [Google Scholar]
- 96.Andersen MO, et al. RNAi using a chitosan/siRNA nanoparticle system: in vitro and in vivo applications. Methods Mol Biol. 2009;555:77–86. doi: 10.1007/978-1-60327-295-7_6. [DOI] [PubMed] [Google Scholar]
- 97.Sun TM, et al. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29(32):4348–55. doi: 10.1016/j.biomaterials.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 98.Tietze N, et al. Induction of apoptosis in murine neuroblastoma by systemic delivery of transferrin-shielded siRNA polyplexes for downregulation of Ran. Oligonucleotides. 2008;18(2):161–74. doi: 10.1089/oli.2008.0112. [DOI] [PubMed] [Google Scholar]
- 99.Hoon Jeong J, et al. Reducible poly(amido ethylenimine) directed to enhance RNA interference. Biomaterials. 2007;28(10):1912–7. doi: 10.1016/j.biomaterials.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 100.Jere D, et al. Poly(beta-amino ester) as a carrier for si/shRNA delivery in lung cancer cells. Biomaterials. 2008;29(16):2535–47. doi: 10.1016/j.biomaterials.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 101.Tseng SJ, Tang SC. Development of poly(amino ester glycol urethane)/siRNA polyplexes for gene silencing. Bioconjug Chem. 2007;18(5):1383–90. doi: 10.1021/bc060382j. [DOI] [PubMed] [Google Scholar]
- 102.Semple SC, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28(2):172–6. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 103.Love KT, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci U S A. 2010;107(5):1864–9. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barros SA, Gollob JA. Toxicity profile of RNAi nanomedicines. Adv Drug Deliv Rev. 2012 Jun 22; doi: 10.1016/j.addr.2012.06.007. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 105.Lens SM, et al. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10(12):825–41. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 106.Strumberg D, et al. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int J Clin Pharmacol Ther. 2011;50(1):76–8. doi: 10.5414/cpp50076. [DOI] [PubMed] [Google Scholar]
- 107.Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–30. doi: 10.1146/annurev-bioeng-071910-124709. [DOI] [PubMed] [Google Scholar]
- 108.Schroeder A, et al. Lipid-based nanotherapeutics for siRNA delivery. J Intern Med. 2010;267(1):9–21. doi: 10.1111/j.1365-2796.2009.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shahzad MM, et al. Dual targeting of EphA2 and FAK in ovarian carcinoma. Cancer Biol Ther. 2009;8(11):1027–34. doi: 10.4161/cbt.8.11.8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gray MJ, et al. Therapeutic targeting of neuropilin-2 on colorectal carcinoma cells implanted in the murine liver. J Natl Cancer Inst. 2008;100(2):109–20. doi: 10.1093/jnci/djm279. [DOI] [PubMed] [Google Scholar]
- 111.Merritt WM, et al. Effect of interleukin-8 gene silencing with liposome-encapsulated small interfering RNA on ovarian cancer cell growth. J Natl Cancer Inst. 2008;100(5):359–72. doi: 10.1093/jnci/djn024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim SH, et al. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J Control Release. 2008;129(2):107–16. doi: 10.1016/j.jconrel.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 113.Yagi N, et al. A nanoparticle system specifically designed to deliver short interfering RNA inhibits tumor growth in vivo. Cancer Res. 2009;69(16):6531–8. doi: 10.1158/0008-5472.CAN-08-3945. [DOI] [PubMed] [Google Scholar]
- 114.Toub N, et al. Efficacy of siRNA nanocapsules targeted against the EWS-Fli1 oncogene in Ewing sarcoma. Pharm Res. 2006;23(5):892–900. doi: 10.1007/s11095-006-9901-9. [DOI] [PubMed] [Google Scholar]
- 115.Imamura O, et al. siRNA-mediated Erc gene silencing suppresses tumor growth in Tsc2 mutant renal carcinoma model. Cancer Lett. 2008;268(2):278–85. doi: 10.1016/j.canlet.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 116.Cutler JI, et al. Spherical nucleic acids. J Am Chem Soc. 2012;134(3):1376–91. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- 117.Oishi M, et al. Smart PEGylated gold nanoparticles for the cytoplasmic delivery of siRNA to induce enhanced gene silencing. Chemistry Letters. 2006;35(9):1046–7. [Google Scholar]
- 118.Giljohann DA, et al. Gene regulation with polyvalent siRNA-nanoparticle conjugates. J Am Chem Soc. 2009;131(6):2072–3. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cutler JI, et al. Polyvalent nucleic acid nanostructures. J Am Chem Soc. 2011;133(24):9254–7. doi: 10.1021/ja203375n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Patel PC, et al. Scavenger receptors mediate cellular uptake of polyvalent oligonucleotide-functionalized gold nanoparticles. Bioconjug Chem. 2010;21(12):2250–6. doi: 10.1021/bc1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seferos DS, et al. Polyvalent DNA nanoparticle conjugates stabilize nucleic acids. Nano Lett. 2009;9(1):308–11. doi: 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang XQ, et al. Strategy for increasing drug solubility and efficacy through covalent attachment to polyvalent DNA-nanoparticle conjugates. ACS Nano. 2009;5(9):6962–70. doi: 10.1021/nn201446c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Song Y, et al. Multimodal gadolinium-enriched DNA-gold nanoparticle conjugates for cellular imaging. Angew Chem Int Ed Engl. 2009;48(48):9143–7. doi: 10.1002/anie.200904666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee JS, et al. Gold, poly(beta-amino ester) nanoparticles for small interfering RNA delivery. Nano Lett. 2009;9(6):2402–6. doi: 10.1021/nl9009793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Braun GB, et al. Laser-Activated Gene Silencing via Gold Nanoshell-siRNA Conjugates. ACS Nano. 2009;3(7):2007–15. doi: 10.1021/nn900469q. [DOI] [PubMed] [Google Scholar]
- 126.Lu W, et al. Tumor site-specific silencing of NF-kappaB p65 by targeted hollow gold nanosphere-mediated photothermal transfection. Cancer Res. 2010;70(8):3177–88. doi: 10.1158/0008-5472.CAN-09-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Elbakry A, et al. Layer-by-layer assembled gold nanoparticles for siRNA delivery. Nano Lett. 2009;9(5):2059–64. doi: 10.1021/nl9003865. [DOI] [PubMed] [Google Scholar]
- 128.Guo S, et al. Enhanced gene delivery and siRNA silencing by gold nanoparticles coated with charge-reversal polyelectrolyte. ACS Nano. 2010;4(9):5505–11. doi: 10.1021/nn101638u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Song WJ, et al. Gold nanoparticles capped with polyethyleneimine for enhanced siRNA delivery. Small. 2009;6(2):239–46. doi: 10.1002/smll.200901513. [DOI] [PubMed] [Google Scholar]
- 130.Kirkland-York S, et al. Tailored design of Au nanoparticle-siRNA carriers utilizing reversible addition-fragmentation chain transfer polymers. Biomacromolecules. 2010;11(4):1052–9. doi: 10.1021/bm100020x. [DOI] [PubMed] [Google Scholar]
- 131.Lee SH, et al. Amine-functionalized gold nanoparticles as non-cytotoxic and efficient intracellular siRNA delivery carriers. Int J Pharm. 2008;364(1):94–101. doi: 10.1016/j.ijpharm.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 132.Bonoiu AC, et al. Nanotechnology approach for drug addiction therapy: gene silencing using delivery of gold nanorod-siRNA nanoplex in dopaminergic neurons. Proc Natl Acad Sci U S A. 2009;106(14):5546–50. doi: 10.1073/pnas.0901715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Davis ME, et al. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–82. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 134.Hirsjarvi S, et al. Passive and active tumour targeting with nanocarriers. Curr Drug Discov Technol. 2011;8(3):188–96. doi: 10.2174/157016311796798991. [DOI] [PubMed] [Google Scholar]
- 135.Yoshizawa T, et al. Folate-linked lipid-based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur J Pharm Biopharm. 2008;70(3):718–25. doi: 10.1016/j.ejpb.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 136.Davis ME, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464(7291):1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Song E, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9(3):347–51. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 138.Vornlocher HP. Antibody-directed cell-type-specific delivery of siRNA. Trends Mol Med. 2006;12(1):1–3. doi: 10.1016/j.molmed.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 139.Song E, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23(6):709–17. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 140.Kumar P, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448(7149):39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 141.Schiffelers RM, et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004;32(19):e149. doi: 10.1093/nar/gnh140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McNamara JO, 2nd, et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24(8):1005–15. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 143.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 144.Dancey JE, et al. The genetic basis for cancer treatment decisions. Cell. 2012;148(3):409–20. doi: 10.1016/j.cell.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 145.Cox AD, Der CJ. The RAF inhibitor paradox revisited. Cancer Cell. 2012;21(2):147–9. doi: 10.1016/j.ccr.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Weeraratna AT. RAF around the edges--the paradox of BRAF inhibitors. N Engl J Med. 2011;366(3):271–3. doi: 10.1056/NEJMe1111636. [DOI] [PubMed] [Google Scholar]
- 147.Wistuba, et al. Methodological and practical challenges for personalized cancer therapies. Nat Rev Clin Oncol. 2011;8(3):135–41. doi: 10.1038/nrclinonc.2011.2. [DOI] [PubMed] [Google Scholar]
- 148.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012 doi: 10.1038/nature11183. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483(7391):613–7. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]