

Summary
Staphylococcus aureus is a pathogen that infects multiple anatomical sites leading to a diverse array of diseases. Although vertebrates can restrict the growth of invading pathogens by sequestering iron within haem, S. aureus surmounts this challenge by employing high-affinity haem uptake systems. However, the presence of excess haem is highly toxic, necessitating tight regulation of haem levels. To overcome haem stress, S. aureus expresses the detoxification system HrtAB. In this work, a transposon screen was performed in the background of a haem-susceptible, HrtAB-deficient S. aureus strain to identify the substrate transported by this putative pump and the source of haem toxicity. While a recent report indicates that HrtAB exports haem itself, the haem-resistant mutants uncovered by the transposon selection enabled us to elucidate the cellular factors contributing to haem toxicity. All mutants identified in this screen inactivated the menaquinone (MK) biosynthesis pathway. Deletion of the final steps of this pathway revealed that quinone molecules localizing to the cell membrane potentiate haem-associated superoxide production and subsequent oxidative damage. These data suggest a model in which membrane-associated haem and quinone molecules form a redox cycle that continuously generates semiquinones and reduced haem, both of which react with atmospheric oxygen to produce superoxide.
Keywords: Staphylococcus aureus, menaquinone, haem toxicity, small colony variants, bacterial respiration, oxidative stress
Introduction
Staphylococcus aureus is an important human pathogen
S. aureus is a Gram-positive coccus that is ubiquitous among the human population owing to the fact that the skin and anterior nares of more than one quarter of all individuals are asymptomatically colonized by this organism (DeLeo et al., 2010, Wertheim et al., 2004). However, S. aureus is armed with an arsenal of toxins, host-modulating factors, and other virulence determinants that enable this bacterium to infect virtually every organ and cause numerous diseases including pneumonia, endocarditis, toxic shock syndrome, and sepsis (Gordon & Lowy, 2008). Because of these characteristics, staphylococcal infections can be either hospital- or community-acquired and result in high rates of mortality and morbidity (DeLeo et al., 2010, Wertheim et al., 2004, Klevens et al., 2007). Compounding this problem is the widespread existence of antibiotic resistant S. aureus, necessitating the identification of new targets for therapeutic intervention.
S. aureus elaborates haem acquisition systems to overcome host-induced nutrient limitation
During infection, vertebrates sequester essential nutrients from invading pathogens in order to restrict microbial growth in a defense tactic known as nutritional immunity (Kehl-Fie & Skaar, 2010, Wakeman & Skaar, 2012). Bioavailable iron concentrations within the host are particularly limiting due to a number of factors: a physiological pH that results in low iron solubility, storage of iron within iron-binding proteins, and intracellular localization of iron (Crichton, 2001, Bullen & Griffiths, 1999). The net result of these mechanisms creates an environment virtually devoid of free iron with greater than 90% of iron residing within host cells. The bulk of host iron is contained within the tetrapyrrole ring of haem, which is further bound within the protein haemoglobin and sequestered inside erythrocytes (Hammer & Skaar, 2011). In order to gain access to this abundant iron source, S. aureus secretes haemolysins to lyse erythrocytes, liberating free haemoglobin. S. aureus is able to acquire haem-iron from haemoglobin using the iron-regulated surface determinant (Isd) system (Skaar et al., 2004, Reniere et al., 2007).
Haem and menaquinone (MK) are required for S. aureus respiration
Haem biosynthesis and acquisition are important to the survival of S. aureus. In addition to being the most abundant iron source available within vertebrates, haem is also utilized as a cofactor in cytochromes and is thus required for bacterial respiration. During respiration, electrons are passed between various carriers to a terminal electron acceptor, which is oxygen in the case of aerobic respiration. The function of respiration is to create a proton motive force (PMF) that can be harnessed to generate ATP. Therefore, much of the electron transport cascade is carried out at the membrane. Electrons are extracted during the catabolism of carbon sources and passed along the electron transport chain via diffusible electron carriers such as NADH to membrane-associated quinones (Simon et al., 2008). While multiple types of quinone molecules exist in nature, the only quinone known to be utilized for respiration in S. aureus is menaquinone (MK) (Bentley & Meganathan, 1982). Quinones subsequently donate electrons to the haem molecules located within cytochromes. S. aureus, like the Gram-positive model organism Bacillus subtilis, possesses two classes of these haem-containing protein complexes, the cytochrome bd and cytochrome aa3 quinol-oxidases (Zamboni & Sauer, 2003).
Various genetic mutations can yield bacterial strains that are deficient in respiration. These strains exhibit a slowed growth phenotype and are thus known as small colony variants (SCV). This type of mutation is of clinical interest because SCVs of S. aureus and various other pathogens are commonly isolated from patients experiencing chronic infections (Proctor et al., 2006). The respiration deficiency of SCV strains provides increased resistance to aminoglycoside antibiotics because this therapeutic class requires a membrane potential in order to enter the cell (Baumert et al., 2002). The most common disease-associated SCVs are defective in either MK or haem biosynthesis (Proctor et al., 2006).
Excess haem is toxic to bacteria
Despite the beneficial aspects of haem, this molecule becomes extremely toxic to S. aureus and other organisms when present in excess (Anzaldi & Skaar, 2010). The nature of this toxicity is not fully understood and possibly multi-faceted. However, it is likely that this phenomenon is, at least in part, the result of cellular damage due to the reactivity of the iron atom within haem. Previous studies have demonstrated that haem can induce oxidative damage to DNA and proteins in vitro (Aft & Mueller, 1983, Aft & Mueller, 1984). Haem-induced DNA damage has also been observed in vivo in both S. aureus and Escherichia coli (Nir et al., 1991). Additionally, recent studies have demonstrated that, while invading pathogens can utilize haem-containing host proteins such as haemoglobin as nutrient sources, the host can also employ these proteins as a form of innate immunity to generate reactive oxygen species (Jiang et al., 2007, Du et al., 2010). Interestingly, Gram-positive microorganisms tend to be more sensitive to haem toxicity than Gram-negative bacteria (Anzaldi & Skaar, 2010, Nitzan et al., 1994). S. aureus in particular is known to be exquisitely sensitive to haem stress (Ladan et al., 1993). This observation, combined with the fact that the hallmark changes in protein expression associated with oxidative damage (Gaupp et al., 2012) are absent in haem-exposed S. aureus, suggests that additional mechanisms of haem toxicity may exist in this pathogen (Friedman et al., 2006).
Haem toxicity can be overcome in S. aureus
To cope with haem stress, S. aureus expresses a putative efflux pump known as HrtAB (Torres et al., 2007, Stauff et al., 2008). HrtAB was identified by a proteomics analysis screening for staphylococcal proteins regulated in response to variations in the concentration of exogenous haem (Friedman et al., 2006). The two genes most highly up-regulated in response to increasing levels of haem were named haem-regulated transport proteins HrtA and HrtB. hrtA and hrtB are located in an operon and encode a putative ATPase and permease, respectively. The expression of the HrtAB putative efflux pump is positively regulated by a two-component system known as the haem sensor system HssRS (Torres et al., 2007, Stauff et al., 2007). Deletion of either hssRS or hrtAB results in increased haem sensitivity (Stauff & Skaar, 2009, Stauff et al., 2007, Torres et al., 2007). Orthologs of this haem detoxification system have been identified in a number of other Gram-positive microorganisms including Bacillus anthracis, Enterococcus faecalis, Corynebacterium diphtheriae, Lactococcus lactis, Listeria monocytogenes, and Streptococcus agalactiae (Torres et al., 2007, Bibb & Schmitt, 2010, Fernandez et al., 2010, Lechardeur et al., 2012), indicating that the need to resist haem stress is widespread among bacteria. Work performed in L. lactis indicates that the HrtAB putative efflux pump detoxifies haem by exporting excess haem from the cell (Lechardeur et al., 2012, Pedersen et al., 2008), a mechanism that is likely employed by the HrtAB homologs of other microorganisms.
Results
A genetic screen implicates menaquinone (MK) biosynthesis in contributing to S. aureus haem stress
In an effort to uncover gene products required to facilitate haem toxicity, a ΔhrtA S. aureus strain that is highly susceptible to haem stress was subjected to random transposon mutagenesis via transformation with the pTV1 vector carrying the Tn917 transposon (Youngman et al., 1983). Haem resistant mutants were selected based on growth upon medium infused with 30–50 μM haem. The resulting mutants were analyzed to identify the site of the transposon insertion, and all of the identified sites mapped to genes encoding products that are either members of the MK biosynthesis pathway or are responsible for the production of substrates that feed into this pathway (Fig. 1). The exact sites of the transposon insertions and the operonic arrangements of the genes within the MK pathway can be visualized in Fig. S1. Mutants were isolated containing disruptions of aroB, aroK, and aroC. These genes are part of the phenylalanine, tyrosine, and tryptophan biosynthetic pathway and encode 3-dehydroquinate synthase, shikimate kinase, and chorismate synthase, respectively. AroB, AroK, and AroC are required for the production of chorismate, the first precursor in the MK biosynthesis pathway. Additional mutants were isolated with insertions in menC (encoding 2-succinylbenzoate synthase), menE (encoding 2-succinylbenzoate-CoA ligase), menB (encoding dihydroxynaphthoic acid synthetase), and an unnamed gene denoted as NWMN_1381. NWMN_1381 is located within an operon containing the MK biosynthetic genes, ubiE and hepT. UbiE is predicted to be an S-adenosylmethionine:2-DMK methyltransferase responsible for catalyzing the final step of MK biosynthesis in which a methyl group is attached to demethylmenaquinone (DMK) (Wissenbach et al., 1992). NWMN_1381 and HepT are homologues of the heptaprenyl diphosphate synthase subunits of B. subtilis responsible for the production of the isoprenyl lipid chain that allows MK to be inserted into the cell membrane (Leatherbarrow et al., 1998, Zhang et al., 1997). All of these gene products are predicted to be directly involved in MK biosynthesis, establishing the putative MK biosynthetic pathway of S. aureus. These data suggest that a component of MK biosynthesis contributes to haem stress in S. aureus.
Fig. 1. Transposon screen reveals that mutations in menaquinone (MK) biosynthesis protect a haem-sensitive Staphylococcus aureus strain from haem toxicity.

This figure depicts a diagrammatic representation of the MK pathway adapted from the KEGG database (http://www.genome.ad.jp/kegg) showing the sites in which transposon insertions allowed a ΔhrtA S. aureus strain to grow in the presence of high haem concentrations. The transposon insertions that conferred haem-resistance to ΔhrtA disrupted the genes highlighted by gray boxes. MK, the final product of this pathway is marked (*). Dotted lines denote intermediate reactions that are not shown in the diagram. SEPHCHC is 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate. SHCHC is (1R,6R)-2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate.
Biochemical analyses of non-polar mutations in the MK biosynthesis pathway yield insight into previously uncharacterized aspects of S. aureus respiration
In order to assess the impact of MK biosynthesis on haem stress, we generated non-polar deletions in the genes encoding the final three annotated enzymes of this pathway: menB, menA, and ubiE. These genes were deleted in both the background of wild-type (WT) S. aureus and the ΔhrtB S. aureus strain lacking a functional HrtAB system. Although the transposon screen was performed in an ΔhrtA background, the non-polar deletions were constructed in the ΔhrtB background because ΔhrtA cells have a defect in membrane stability due to dysregulated HrtB permease function, which is unrelated to the role that HrtA plays in protection against haem stress (Attia et al., 2010). If the MK biosynthetic pathway predicted by the genomic screen is correct, deletion of menB should result in a blockage of MK biosynthesis at the formation of 2-succinylbenzoyl-CoA, deletion of menA should yield 1,4-dihydroxy-2-naphthoate as the final MK biosynthetic product, and deletion of ubiE should result in a buildup of demethylmenaquinone (DMK) (Fig. 1). As expected, all strains containing deletions in menA and menB possessed an SCV phenotype when streaked onto agar plates, indicating that respiration was no longer occurring in these mutants (Fig. 2a). The respiration deficiency of these SCV strains was further confirmed by their inability to generate a PMF or to oxidize NADH to the levels of their MK-producing parental strains (Fig. 3a and Fig. S2). Because SCV strains cannot respire, they resort to energy production via fermentation which results in an accumulation of L- and D-lactate (Fig. 3b). Of note, several disease-associated SCV isolates were recently found to have mutations mapping specifically to the menB locus (Lannergard et al., 2008). Interestingly, the ΔubiE strains, unlike the ΔmenA and ΔmenB strains, maintained normal colony morphology (Fig. 2a), indicating that both DMK and MK can function within the electron transport chain of S. aureus. This finding is not entirely surprising given that DMK possesses a redox potential midway between that of MK and ubiquinone and this molecule has previously been shown to function as an electron carrier in other bacteria (Holländer, 1976).
Fig. 2. Non-polar deletions of the final enzymatic steps of the S. aureus MK biosynthesis pathway can be generated and chemically characterized.

A. WT and various mutants of S. aureus were plated on agar to highlight differences in colony morphology for certain strains when grown on plain TSA medium versus growth in the presence of 12.5 μM exogenous menaquinone-4 (MK4) or 2.5 μM of the MK precursor, menadione. Pictures were taken after 48 hours of growth at 37 °C. B. MK and DMK were extracted from various S. aureus strains, resolved by thin layer chromatography (TLC) adjacent to a commercially available MK standard (MK4), and visualized by UV shadowing. C. MK and DMK extracts isolated from the WT and ΔubiE S. aureus strains were further resolved by mass spectrometry, revealing the number of isoprenyl groups (highlighted by brackets in the inset chemical structures) present on each of these molecules. MK and DMK were detected using parent ion scans for m/z 187 and m/z 173, respectively as previously described (Geyer et al., 2004).
Fig. 3. A MK-deficient strain is incapable of aerobic respiration and, therefore, must generate energy via fermentation.

A. Proton motive force (PMF) was measured as the mean ratio of red:green fluorescence of WT and mutant strains of S. aureus grown to mid-exponential phase and incubated with 30 μM of the dye 3,3-diethyloxacarbocyanine iodide (DiOC2). High levels of red fluorescence relative to green fluorescence indicate the presence of a PMF. Each PMF data set was generated from biological triplicates. B. D- and L-lactate production was measured in WT and SCV strains of S. aureus. Increased lactate production is indicative of an increase in the fermentation pathway. D- and L-lactate measurements were performed three times on three separate days. Additionally, technical triplicates were performed on each day.
The respiration deficiency of the menB mutant can be rescued through the addition of menadione, a synthetic analogue of 1,4-dihydroxy-2-naphthoate (Fig. 2a). The SCV phenotype of menA mutants cannot be reversed by the presence of exogenous menadione but can be chemically complemented by the addition of MK4, a commercially-available MK possessing a lipid tail comprised of four isoprenyl groups (Fig 2a). These observations support the annotated function of menA as being the enzyme responsible for the addition of the polyprenyl lipid tail to the quinone molecule.
Mutants of the MK biosynthesis pathway were further verified to be deficient in MK production through the use of thin layer chromatography (TLC) and mass spectrometry. MK extracts from WT, ΔubiE, ΔmenA, and ΔmenB were resolved adjacent to MK4 on TLC plates (Fig. 2b). The MK band from WT cells resolved at a slightly higher position on the plate than MK4, indicating that the polyprenyl tail of MK from S. aureus is longer than that of the commercially-available MK. The DMK band observed in the ΔubiE extract resolved at a slightly lower position than the WT MK band possibly due to the lack of a methyl group in this molecule. No MK or DMK bands could be observed in the ΔmenA or ΔmenB extracts.
In order to prove that the bands observed by TLC were MK and DMK and to determine the number of prenyl groups comprising the lipid tails of these molecules, the extracts were also analyzed by mass spectrometry (Fig. 2c). Mass spectrometry of WT extracts revealed that the predominant MK species of these cells possessed eight isoprenyl groups (MK8) but MK with seven isoprenyl groups (MK7) and nine isoprenyl groups (MK9) were also present. These results support prior findings which indicated that MK8 is the major MK produced by S. aureus during aerobic growth (Jeffries et al., 1967, Hammond & White, 1969). Mass spectrometry of ΔubiE extracts demonstrates that these cells are incapable of producing MK and instead accumulate DMK. The bulk of this DMK has eight isoprenyl groups (DMK8) but DMK7 and DMK9 isoprenologues can also be detected. These results establish the S-adenosylmethionine:2-DMK methyltransferase function of UbiE in S. aureus.
Growth analyses confirm that MK biosynthesis is required to potentiate haem toxicity
To verify that disruption of MK biosynthesis protects S. aureus from haem toxicity, WT and mutant S. aureus strains were streaked onto agar plates in the absence or presence of 40 μM haem to mimic the conditions of the transposon mutagenesis selection (Fig. 4a). As expected, no growth was observed for the ΔhrtB strain upon exposure to high levels of exogenous haem. Haem toxicity was not eliminated in a ΔhrtB strain lacking ubiE, the gene encoding for the final enzyme in MK biosynthesis. However, ΔhrtB strains that were also deficient in menA and menB grew as well as the ΔmenA and ΔmenB strains with functional HrtAB systems, indicating that the haem toxicity associated with the lack of HrtAB could be completely reversed by eliminating biosynthetic steps prior to the formation of DMK. When respiration was restored in the ΔmenA and ΔmenB strains through the addition of MK4 to the growth media, the ΔhrtB ΔmenA and ΔhrtB ΔmenB mutants became susceptible to haem stress.
Fig. 4. Deletions of MK biosynthesis resulting in small colony variant (SCV) phenotypes protect S. aureus from haem toxicity.

A. WT and various mutants of S. aureus were grown on agar plates to highlight differences in haem susceptibility of certain strains. S. aureus strains were streaked out on TSA, TSA containing 40 μM haem, and TSA containing 40 μM haem plus 12.5 μM MK4 in order to demonstrate which strains are haem-resistant and whether chemical complementation of MK biosynthesis reverses the haem-resistant phenotype. Pictures were taken after 48 hours of growth at 37 °C. B. The growth of S. aureus ΔhrtB and ΔhrtB ΔmenB strains was monitored hourly by measuring optical density at 600 nm of cells in TSB in the presence or absence of 6 μM haem. C. Quantification of the fraction growth of haem-treated S. aureus strains relative to untreated culture after 8 hours of growth. D. The growth of S. aureus ΔhrtB ΔmenB was monitored hourly in the presence or absence of 6 μM haem and 12.5 μM MK4. E. Quantification of the fraction growth of haem-treated S. aureus strains relative to untreated culture after 8 hours of growth in the presence or absence of 12.5 μM MK4. Growth assays were performed at least three times on separate days. Additionally, technical triplicates were performed on each day. Error bars represent SEM of the biological replicates. * indicates p < 0.05 as determined by a two-tailed Student’s t test.
The fact that non-polar deletion of ubiE does not counter haem toxicity indicates that the protective effect of the transposon insertion disrupting the ubiE-containing operon results from the deletion of the NWMN_1381 and hepT polyprenyl biosynthetic genes. Deletion of NWMN_1381 and hepT yields a blockage in MK biosynthesis equivalent to the disruption of menA, the enzyme responsible for attaching the polyprenyl chain to the quinone molecule (Fig. 1).
We next sought to determine the effects of MK-deficiency on haem toxicity in liquid culture (Fig. 4b-e, Fig. S3a-b). Because haem toxicity increases in liquid media, these cells were grown in the presence or absence of 6 μM haem as compared to the 40 μM haem used in the agar plate assay. After 8 hours of haem exposure, WT haem-treated cells grew almost as well as untreated cells (Fig. 4c). This result was in marked contrast to the haem-treated ΔhrtB cells which displayed virtually no growth. This haem-induced growth inhibition was almost completely reversed in the ΔhrtB ΔmenA and the ΔhrtB ΔmenB double mutant strains. However, as observed on solid medium, it was evident that the ΔhrtB ΔubiE mutant was as susceptible to haem stress as the ΔhrtB strain, indicating that strains containing DMK experience as much haem toxicity as strains producing MK. The protective effect of ΔmenA and ΔmenB deletion against haem toxicity was completely reversed upon restoration of aerobic respiration with the addition of MK4 to the growth medium (Fig. 4d-e, Fig. S3b). Since only respiration-deficient MK-biosynthesis mutants are protected against haem stress, we sought to test the possibility that any disruption of aerobic respiration would counteract haem toxicity.
In order to assess the contribution of aerobic respiration to haem-induced stress, we tested haem toxicity in a strain lacking both of the S. aureus cytochromes. This strain has a non-polar deletion of the qoxB gene and a transposon insertion in the cydB gene, which encode subunits of cytochrome aa3 and cytochrome bd, respectively. The ΔqoxB ΔcydB strain is completely deficient in aerobic respiration (N. D. Hammer, unpublished). Because the ΔqoxB ΔcydB strain possesses an intact HrtAB haem detoxification system, we compared its haem susceptibility to that of WT S. aureus and the ΔmenB mutant. The growth of these strains was assessed in the presence or absence of 10 μM haem in order to induce sufficient haem toxicity in these HrtAB-expressing strains (Fig. 5a-b, Fig. S3c). While deletion of menB increased resistance of S. aureus to haem stress, the ΔqoxB ΔcydB strain was not protected from haem toxicity and actually appeared to be more susceptible to haem exposure than WT cells.
Fig. 5. General defects in respiration do not protect S. aureus strains against haem stress.

A. The growth of two respiration-deficient S. aureus strains, ΔqoxB ΔcydB and ΔmenB, was monitored hourly in the presence or absence of 10 μM haem. B. Quantification of the fraction growth of haem-treated S. aureus strains relative to untreated culture after 5 hours of growth. Growth assays were performed in triplicate on three separate days. Additionally, technical triplicates were performed on each day. Error bars represent SEM of the biological replicates. * indicates p < 0.05 as determined by a two-tailed Student’s t test.
Together, these data indicate that aerobic respiration is not directly potentiating haem stress. Instead, it appears that quinone molecules such as DMK and MK possessing lipid tails and thus the ability to associate with cell membranes are contributing to the toxic effects of haem. These data also reveal that deletion of the haem-containing cytochromes actually increases haem stress. It is possible that by eliminating natural cellular haem reservoirs such as cytochromes, haem toxicity is increased because displaced haem is being trafficked to inappropriate locations. Alternatively, deletion of the cytochromes may alter the overall redox status of cellular MK pools to a more toxic state.
MK deficiency does not impair haem uptake
While the growth analyses clearly demonstrate a link between MK biosynthesis and haem stress, these results could potentially be attributed to a general defect in haem uptake. In order to confirm that haem is still imported by MK-deficient strains, cell membranes were isolated from ΔhrtB and ΔhrtB ΔmenA S. aureus strains grown in the presence or absence of exogenous haem. Spectrophotometric analysis of the haem contents of these strains revealed that significant haem accumulation can be observed in membranes from cells with or without the ability to synthesize MK (Fig. 6). These data suggest that the MK-dependent toxic effects of haem can be attributed to a direct interaction between these two molecules.
Fig. 6. MK-biosynthesis is not required for haem uptake.
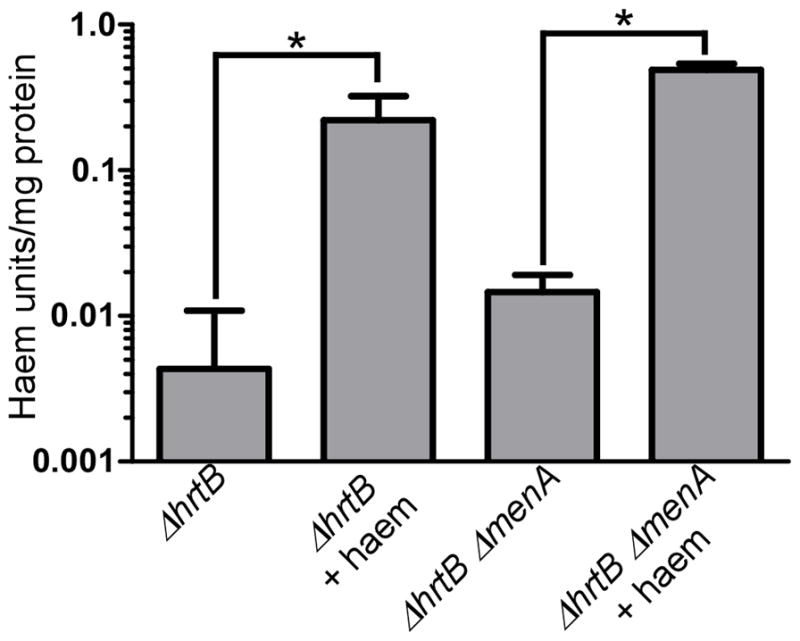
Haem contents were measured in S. aureus ΔhrtB and ΔhrtB ΔmenA cells grown in the presence or absence of 5 μM haem. Both strains demonstrate significant accumulation of membrane-associated haem when exogenous haem is supplemented into the medium. Error bars represent SEM of four biological replicates. * indicates p < 0.05 as determined by a two-tailed Student’s t test.
Disruption of MK biosynthesis alleviates haem-associated oxidative stress
Previous reports have indicated that haem is capable of inducing DNA damage in S. aureus, which is likely a result of oxidative stress (Nir et al., 1991). Therefore, we assessed the presence of haem-induced oxidative protein damage using an OxyBlot™ kit (Millipore) (Fig. 7a). Oxidative damage to proteins results in the formation of carbonyl groups that can be chemically modified and detected through standard immunoblotting methods. Bacterial cultures were grown in the presence or absence of 5 μM haem and assessed for oxidative stress. WT S. aureus cells displayed virtually no haem-induced protein damage after 8 hours of haem exposure. However, increased protein carbonylation could be observed for the ΔhrtB mutant grown in the presence of haem as evidenced by high levels of antibody staining throughout the entire haem-treated ΔhrtB lane. The increased haem-induced protein damage associated with the loss of hrtB was dramatically reversed by disruption of either menA or menB. The antibody staining for individual lanes was quantified and graphed as percent staining in haem-treated culture relative to untreated samples (Fig. 7b). While significant increases in haem-induced protein damage were observed in ΔhrtB as compared to WT, no significant differences were detected between ΔmenA and ΔhrtB ΔmenA or ΔmenB and ΔhrtB ΔmenB strains, indicating that MK is contributing to haem-induced oxidative stress. The role of MK in haem-induced protein damage was further confirmed through chemical complementation. When grown in the presence of exogenous MK4, the ΔhrtB ΔmenA strain accumulated high levels of haem-associated protein carbonylation comparable to the damage observed in the respiration-competent ΔhrtB and ΔhrtB ΔubiE strains (Fig. 7c-d).
Fig. 7. Haem induces increased oxidative protein damage in HrtAB-deficient S. aureus, and this phenomenon is reversed by disruption of MK biosynthesis.

A. Total protein was extracted from the protoplasts of S. aureus strains that were grown in the presence or absence of 5 μM haem. A representative immunoblot is shown containing protein samples that have been derivatized with 2,4-dinitrophenylhydrazine (DNP) to modify sites of oxidative damage. Blots were probed with antibodies specific to the DNP moiety on the proteins. The symbols “-”and “+” denote the absence or presence of 5 μM exogenous haem in the growth media. The ladder shown in the image is the Precision Plus Protein™ All Blue Standard (BioRad). B. Quantification of data from part A representing the percent oxidative protein damage in haem-treated samples as compared to untreated samples. C. Representative immunoblot demonstrating that the protective effect of the ΔhrtB ΔmenA mutation can be reversed through addition of 12.5 μM MK4 to the growth media. D. Quantification of data from part C. Graphs represent the average of at least six independent immunoblots from samples prepared on separate days. Error bars represent SEM. * denotes p < 0.05 as determined by a two-tailed Student’s t test.
Haem-associated oxidative damage localizes to cell membranes
Exogenously acquired haem is known to accumulate in the cell membranes of S. aureus strains grown under iron-replete conditions (Skaar et al., 2004). Additionally, the haem stress-associated quinones, MK and DMK, are predicted to localize to the phospholipid bilayer due to the fact that these molecules possess lipophilic polyprenyl tails. Therefore, it seemed likely that haem-induced oxidative damage was originating within and possibly confined to the membrane. In order to determine which cellular components are most severely impacted by haem stress, cells were fractionated and these fractions analyzed for the presence of protein carbonylation (Fig. 8a-b). Significant antibody staining was observed in the haem-treated membrane fractions of ΔhrtB cells but virtually no haem-induced protein damage could be measured in cytoplasmic fractions. These data demonstrate that haem-induced oxidative damage is primarily localized to the cell membrane.
Fig. 8. Haem-induced oxidative damage localizes primarily to the cell membrane.

A. Cell membranes and cytoplasmic fractions were isolated from S. aureus strains grown in the presence or absence of 5 μM haem and analyzed for the presence of protein carbonylation by immunoblotting. The symbols “-” and “+” denote the absence or presence of 5 μM exogenous haem in the growth media. B. Quantification of immunoblots performed in triplicate representing the percent oxidative protein damage in haem-treated samples as compared to untreated samples. Error bars represent SEM. * denotes p < 0.05 as determined by a two-tailed Student’s t test.
Endogenous MK biosynthesis and exogenous haem excess result in superoxide production
In order to elucidate the nature of the MK- and haem-associated oxidative damage, S. aureus strains were analyzed by electron paramagnetic resonance (EPR) and spin trapping technique. Increased levels of various transient free radical species can be detected upon incubation with the spin trap molecule 5-ethoxycarbonyl-5-methyl-1-pyrroline N-oxide (EMPO) which results in the formation of stable EMPO-free radical adducts. These EMPO adducts can be identified based on their unique EPR spectra. EPR was used to assess ΔhrtB and ΔhrtB ΔmenA samples cultured in the presence or absence of exogenous haem (Fig. 9a). These analyses revealed EPR signals that were only present in the haem-treated ΔhrtB cultures. The hyperfine splitting constants of these spectra were indicative of the EMPO-OH adduct (Zhang et al., 2000). However, the EMPO-OH adduct can be generated either through the presence of hydroxyl radicals or through the decay of the superoxide EMPO adduct (Locigno et al., 2005) (Fig. 9b). In order to distinguish between these possibilities, samples were treated with either superoxide dismutase (SOD) or dimethyl sulfoxide (DMSO) to scavenge superoxide or hydroxyl radicals, respectively (Fig. 9c). The specific inhibition of EMPO-OH signal by the superoxide scavenger SOD demonstrates that enhanced superoxide production is occurring in haem-treated ΔhrtB S. aureus.
Fig. 9. Endogenous MK biosynthesis and exogenous haem exposure result in enhanced superoxide production.

A. Using the spin trap 5-ethoxycarbonyl-5-methyl-1-pyrroline N-oxide (EMPO), only a MK-producing S. aureus strain (ΔhrtB) displays visible EPR signal upon haem exposure whereas a MK-deficient S. aureus strain (ΔhrtB ΔmenA) shows no peaks above background levels in either the presence or absence of exogenous haem. * denotes hyperfine splitting constants aN=14.5 G and aH=13.6 G which are consistent with an EMPO-OH radical adduct (Zhang et al., 2000). B. Schematic diagramming the production of the EMPO-OH adduct by either superoxide or hydroxyl radicals. The distinction between superoxide- and hydroxyl radical-generated EMPO-OH signal can be made through inhibition using superoxide dismutase (SOD) or dimethyl sulfoxide (DMSO). C. Haem-induced EPR peaks are quenched only by the presence of exogenous SOD, indicating that the EMPO-OH adduct is formed by superoxide. Data are representative of biological triplicates.
Discussion
A model for the role of MK in potentiating haem toxicity
An explanation for the role of MK in facilitating haem stress derives from the function of this molecule within the electron transport chain. During typical aerobic respiration, membrane-associated quinones such as MK and DMK donate electrons to cytochrome-associated haem to promote the generation of a PMF (Simon et al., 2008). MK and DMK can possess up to two electrons when fully reduced. Loss of a single electron results in the formation of a semiquinone (SQ) which can react with molecular oxygen to generate superoxide (Barja, 1999) (Fig. 10a). While SQs are produced to some extent during aerobic respiration, there are cellular mechanisms in place to limit their formation and toxicity (McMillan et al., 2012, Osyczka et al., 2005).
Fig. 10. A model for MK- and haem-associated superoxide production.

A. Under standard growth conditions, a certain level of biosynthesized haem exists within S. aureus cytochromes in order to support aerobic respiration. During the process of respiration, a portion of the cellular MK or DMK pool – labeled as (D)MK for the purposes of this figure – can lose a single electron, resulting in the formation of the highly reactive semiquinone (SQ) species. SQs can react with molecular oxygen to generate superoxide, but the levels of SQs and superoxide formed during typical respiration are kept in check by various cellular factors. B. Under conditions of haem excess, haem molecules accumulate in the cell membrane within an unknown haem reservoir. Upon interaction with atmospheric oxygen, haem can autoxidize into haemin, producing superoxide in the process. C. In the presence of excess haem, cellular pools of reduced MK continuously redox cycle haemin to regenerate haem. This process likely results in the formation of SQs, which react with oxygen to produce superoxide.
Upon exposure to excess exogenous haem, haem molecules accumulate in the bacterial membrane within an as yet unidentified haem reservoir (Skaar et al., 2004). Like SQs, reduced haem molecules are known for their ability to react with atmospheric oxygen to generate superoxide and haemin (Rifkind et al., 2004) (Fig. 10b). Haemin is thought to be relatively inert; however, just as MK is able to donate electrons to cytochrome-associated haemin, MK should also possess the ability to reduce other membrane-localized haemin molecules. The reduction of haemin by MK and DMK should allow the superoxide-generating haem autoxidation to continuously occur, resulting in high levels of superoxide production. Additionally, the reduction of haemin by MK and DMK should result in increased levels of superoxide-generating SQs, leading to even greater haem stress in MK-producing cells. (Fig. 10c).
In WT S. aureus, haem-catalyzed superoxide production is minimized due to the presence of the HrtAB haem detoxification system. The fact that the HrtAB system is minimizing haem-associated oxidative damage is likely the reason that no oxidative stress response proteins were identified in previous screens analyzing haem-responsive protein expression (Friedman et al., 2006). In this work, we have shown that both ΔqoxB ΔcydB and ΔhrtB S. aureus experience increased haem stress as compared to WT cells. It is possible that this phenomenon is due to an increase in haem trafficking to an unknown membrane-associated reservoir. Because there are no cytochromes to appropriately house haem molecules in the ΔqoxB ΔcydB strain, cellular haem might be diverted to inappropriate sites which are more prone to haem autoxidation and/or SQ production. An alternative explanation for the increase in haem stress identified within the ΔqoxB ΔcydB strain is that the overall redox status of the cellular MK pool is altered, resulting an overabundance of reduced MK. Greater levels of reduced MK might be capable of promoting more rapid rates of redox cycling within the membrane-associated haem molecules and, therefore, a more rapid accumulation of superoxide. The haem stress of the ΔhrtB mutant is even greater than that observed in the ΔqoxB ΔcydB strain because ΔhrtB lacks the HrtAB detoxification system. Without a detoxification system, the cell membrane becomes saturated with superoxide-generating haem. However, haem stress is significantly alleviated by eliminating MK biosynthesis in the ΔhrtB strain because, in the absence of MK or DMK, the membrane-associated haemin cannot be redox-cycled to its toxic form.
This hypothesis for the connection between MK production and haem toxicity is supported by previous literature in which quinone molecules were used as reducing agents to potentiate the formation of reactive oxygen species in vitro (Yu & Anderson, 1997, Duesterberg & Waite, 2007). Other researchers have shown that quinones can play an in vivo role in the generation of free radicals (Gutteridge et al., 1984, Stohs & Bagchi, 1995, Huycke et al., 2001, Medina et al., 2006, Korshunov & Imlay, 2006) and, in fact, deletion of MK biosynthesis can protect against copper-mediated oxidative stress in L. lactis (Rezaïki et al., 2008).
Implications
Because S. aureus and other pathogens encounter haem during infection of a vertebrate host, it is possible that these organisms experience haem stress within certain niches of the host environment. In support of this hypothesis, haem detoxification systems are present in a number of Gram-positive microorganisms that regularly come in contact with vertebrate blood such as B. anthracis, E. faecalis, C. diphtheriae, L. lactis, L. monocytogenes, and S. agalactiae (Torres et al., 2007, Bibb & Schmitt, 2010, Fernandez et al., 2010, Lechardeur et al., 2012). Of these organisms, it appears evident that B. anthracis experiences haem toxicity during infection as demonstrated by the up-regulation of the HrtAB haem detoxification system in a murine infection model (Stauff & Skaar, 2009). This finding likely applies to S. aureus, a related Gram-positive pathogen of the Bacillales order. While it is known that disruption of hssRS or hrtAB alters the virulence of S. aureus (Attia et al., 2010, Torres et al., 2007), the impact of haem toxicity during staphylococcal infection remains to be fully explored. If S. aureus does experience haem stress within the host environment, the haem-susceptibility of this pathogen is an area of potential interest that could be exploited as a target for therapeutic intervention.
Prolonged staphylococcal infections can induce genetic mutations that result in a phenotype known as small colony variants (SCV) (Proctor et al., 2006). These strains are characterized by decreased growth rate due to an impaired respiratory chain as well as an ability to reside within eukaryotic cells (von Eiff et al., 1997a, von Eiff et al., 1997b). SCVs are associated with osteomyelitis, device-related infections, and cystic fibrosis and are often isolated from patients experiencing persistent infections (von Eiff et al., 1997b, von Eiff et al., 1997a, von Eiff et al., 2006, Seifert et al., 1999, Becker et al., 2004). SCV isolates are thought to arise because of their increased resistance to aminoglycoside antibiotics, a therapeutic class that requires a PMF to enter the cell (Baumert et al., 2002, Musher et al., 1977). SCVs isolated from patients often have a respiration defect associated with MK biosynthesis pathways (Proctor et al., 2006). While studies have shown that aminoglycoside exposure can induce the formation of SCVs in vitro (Musher et al., 1977), there have been cases in which SCVs arise without prior aminoglycoside treatment, indicating that there may be additional factors contributing to SCV selection (Proctor et al., 2006). Our research has demonstrated that mutations in MK biosynthesis protect against haem toxicity. Therefore, it is possible that resistance to haem stress could be a factor in the selection for SCV formation during infection.
Experimental procedures
Chemicals and oligonucleotides
All chemicals used in this study were purchased from Sigma-Aldrich unless otherwise specified. DNA oligonucleotides were purchased from Integrated DNA Technologies. The DNA oligonucleotides used in these studies are shown in Table S1.
Bacterial strains and growth conditions
All experiments in this study were performed in the S. aureus clinical isolate Newman (Duthie & Lorenz, 1952). Isogenic mutants lacking hrtA and hrtB were previously described (Attia et al., 2010, Torres et al., 2007). S. aureus cultures were grown on tryptic soy broth (TSB) solidified with 1.5 % agar at 37 °C or in TSB at 37 °C with shaking at 180 rpm. Growth curves were performed in 96-well round-bottomed plates containing 100 μl of culture per well. 5 ml bacterial cultures were grown in 15 ml conical tubes, and 20 ml bacterial cultures were grown in 50 ml conical tubes. All cultures were grown in the dark to avoid the compounding issue of singlet oxygen that can be generated by haem in the presence of ambient light.
S. aureus ΔhrtA transposon mutagenesis and selection
S. aureus ΔhrtA was transformed with the Tn917-containing plasmid pTV1 (Youngman et al., 1983) and plated onto TSA containing 10 μg ml-1 chloramphenicol at 30 °C in order to promote replication of the temperature-sensitive plasmid. A single colony was inoculated into 5 ml TSB containing 10 μg ml-1 chloramphenicol, and the culture was shaken at 30 °C until turbid. Cells were centrifuged, washed twice in fresh TSB, suspended in 1 ml TSB, and 1:10, 1:100, and 1:1000 serial dilutions were made. 400 μl of cell suspension from each dilution were spread onto separate haemin selection medium (TSA, 10 μg ml-1 erythromycin, 30–50 μM haemin) and warmed to 42.5 °C, a non-permissive temperature for the pTV1 plasmid which promotes transposon insertion in order to maintain antibiotic resistance. Plates were incubated at 42.5 °C for 3 to 5 days until colonies appeared. Colonies from the plate harboring a low density of mutants were streaked for single colonies onto fresh haemin selection medium and grown at 37 °C for 5 days.
Transposon integration site determination
Genomic DNA was prepared from 5 ml cultures of ΔhrtA transposon mutants using a Wizard® Genomic DNA Purification Kit (Promega) according to the manufacturer’s instructions. Two μg of genomic DNA was digested with DraI (New England Biolabs) overnight at 37 °C followed by incubation at 65 °C for 20 minutes to inactivate DraI. Intermolecular ligation reactions containing 200 ng of digested DNA and T4 DNA ligase (Promega) were assembled and incubated at room temperature for 20 minutes. 20 ng ligated DNA was used as a template in PCR reactions containing primers annealing within tn917 and amplifying in opposite directions, transposon primer 1 and transposon primer 2. PCR products were analyzed by agarose gel electrophoresis and treated with exonuclease I (NEB) and SAP (Promega) according to manufacturer’s instructions. PCR-amplified DNA was column purified using a PCR Purification Kit (Qiagen) and sequenced using transposon primer 2. The sequence flanking the tn917 inverted repeat was used to interrogate the S. aureus Newman genome sequence to determine the tn917 integration site.
Construction of ΔubiE, ΔmenA, and ΔmenB mutants
Established methods to inactivate genes in S. aureus were used to construct a marked in-frame mutation in ubiE encoding the menaquinone biosynthesis methyltransferase and the unmarked mutations in the menB and menA genes encoding dihydroxynaphthoic acid synthetase and 1,4-dihydroxy-2-naphthoate octaprenyltransferase, respectively (Bae & Schneewind, 2006, Lukomski et al., 2000). Briefly, approximately 500–1000 bp regions upstream and downstream of the targeted genes were amplified. The flanking primers incorporated attB1 and attB2 recombinase sites. The downstream and upstream fragments were either fused through PCR sewing or sequential cloning into pCR2.1 (Invitrogen). The marked deletion for the ubiE gene was generated by inserting the non-polar spectinomycin resistance cassette from pSL60-1 (Lukomski et al., 2000) in-frame in an XmaI site between the two flanking regions. The knock-out constructs were then recombined in the pKOR1 vector (Bae & Schneewind, 2006). The resultant vectors (pKOR1-ΔmenA, pKOR1-ΔmenB, and pKOR1-ΔubiE::spc) were transformed into the S. aureus restriction-negative modification-positive strain RN4220 (Novick, 1991) followed by electroporation into strain Newman or the ΔhrtB strain generated in a Newman background, and the mutants were obtained as previously described (Bae & Schneewind, 2006). Primers AA502, AA505, AA508, and AA509 were used to generate the ubiE deletion. Primers CAW430, CAW431, CAW437, and CAW438 were used to generate the menA deletion. Primers DLS365, DLS366, DLS367, and DLS368 were used to generate the menB deletion.
MK extraction
After 16 hours of growth, 20 ml bacterial cultures (OD600 = 3.2) were pelleted at 3200 × g for 10 minutes at 4 °C and washed in TSM (100 mM Tris pH 7; 500 mM sucrose; 10 mM MgCl2). Cell pellets were stored frozen at −80 °C prior to MK extraction. Quinone extraction was performed using cold acetone in a manner similar to previously described protocols (Rezaïki et al., 2008). Whenever possible, extraction procedures were performed at 4 °C in the dark. Cell pellets were suspended in 5 ml of ice-cold acetone and 1 ml acid-washed glass beads. Samples were vortexed for 2 minutes and sonicated in an ice-cooled water bath for 15 minutes. Cell debris was pelleted at 13,000 × g and the supernatant was retained. Another 5 ml of cold acetone was added to the cell debris and the vortexing, sonication, and pelleting steps were repeated. Finally, 10 ml of cold acetone were added to the remaining cell debris and the vortexing, sonication, and pelleting steps were repeated. The supernatants from each extraction were combined and evaporated under vacuum at 30 °C. Evaporated pellets were suspended in 1 ml heptane and loaded onto a preparative silica gel column [SPE DSC-Si silica tube 1 ml, 100 mg, Supelco]. The sample-loaded column was washed once with 1 ml heptane and quinones were eluted with the addition of 1 ml of 3% diethyl ether/97% heptane. Samples were dried under nitrogen at 37 °C. The dried quinone extracts were suspended in 20 μl ethanol for subsequent analysis.
Thin layer chromatography
Thin layer chromatography (TLC) was performed using 5 × 10 cm silica gel aluminum sheets (60 F254, EMD) in a manner similar to previously described methods (Rezaïki et al., 2008). 10 μl of the quinone extract was loaded adjacent to 10 μl of a migration standard consisting of 0.5 mM MK-4 and resolved using heptane-diethyl ether (85:15, v/v) as the mobile phase. Quinones were visualized by UV exposure, and the plates were photographed using an AlphaImager (Alpha Innotech).
Mass spectrometry
MK extracts were subjected to high-pressure liquid chromatography (HPLC) mass spectrometry (MS) using an Acquity Ultra Performance LC system (Waters) interfaced with a Finnigan TSQ Quantum Ultra Triple Quadrupole mass spectrometer (Thermo Scientific). Ten μl of sample was injected and HPLC separation was achieved using an Xterra C18 column (Waters) using a gradient of (A) methanol:H2O (95:5, v/v) and (B) methanol:isopropanol (30:70, v/v). Gradient conditions were as follows: 0–1 min, A=100%; 1–9 min, B=0–100%; 9–10 min, B=100%; 10–11 min, A=0–100%; 11–14 min, A-100%. The column temperature was maintained at 50 °C. Eluted molecules were subjected to positive ion MS using APCI as the ionization interface. MK and DMK molecules were detected using parent ion scans for m/z 187 and m/z 173, respectively as previously described (Geyer et al., 2004).
Measurement of proton motive force (PMF)
Following the manufacturer’s instructions, the Baclight Bacterial Membrane Potential Kit (Invitrogen) was used to monitor the PMF of S. aureus mutants. Briefly, bacterial cultures were grown to mid-exponential phase and added to a 1 ml PBS solution containing 30 μM 3,3-diethyloxacarbocyanine iodide (DiOC2). Suspensions were incubated for 30 minutes at 37° C prior to analysis by the Vanderbilt Flow Core. For each sample, the membrane decoupler, carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 5 μM), was used a positive control for loss of the PMF.
Measurement of L- and D-lactate production
Bacterial cultures grown overnight were diluted 1:100 into fresh TSB. These cultures were grown for 15 hours at 37° C. The cells were centrifuged and the supernatant was collected. Following manufacturer’s instructions, D- and L-lactate was monitored in culture supernatants using the Boehringer Mannheim enzymatic UV-method (Roche).
Measurement of haem content
In order to assay haem uptake by S. aureus strains, cells were cultured overnight in plain TSB and diluted to an OD600 of 0.5 in the morning. The diluted cultures were grown in 5 mL TSB for 6 hours in the presence or absence of 5 μM haem. Cultures were pelleted at 3200 × g for 10 minutes at 4 °C, and pellets were washed in TSM (100 mM Tris pH 7; 500 mM sucrose; 10 mM MgCl2). Pellets were suspended in 500 μl TSM supplemented with 100 μg lysostaphin and incubated for 30 minutes at 37 °C. The resulting protoplasts were sedimented by centrifugation at 20,000 × g for 5 minutes. Protoplasts were suspended in 200 μl TKM (50 mM Tris HCl pH 7; 60 mM KCl; 10 mM MgCl2) and lysed by sonication for 10 seconds. Cell membranes were sedimented by centrifugation at 100,000 × g for 30 minutes. Membranes were suspended in TKM, and protein content was measured by absorbance at 280 nm. The haem content of concentration-matched samples was assayed similarly to previously described methods (Lombardo et al., 2005). Briefly, membrane fractions were diluted to a final volume of 550 μL in TKM containing 300 mM NaCl and 24% DMSO. Samples were then acidified through the addition of 0.2 mL 50 mM glycine-HCl, pH 1 and haem was subsequently extracted by adding 0.2 mL chloroform and vortexing several times. Spectral absorbance readings were taken of the chloroform phase at wavelengths ranging from 300–500 nm. Haem units were determined using the formula Ac= 2A388-(A450+A330) and concentration was graphed as haem units/mg protein.
Analysis of oxidative protein damage
Five mL bacterial cultures were grown for 8 hours in TSB in the presence or absence of 5 μM haem. These cultures were pelleted at 3200 × g for 10 minutes at 4 °C, and pellets were washed in TSM (100 mM Tris pH 7; 500 mM sucrose; 10 mM MgCl2). Pellets were suspended in 500 μl TSM supplemented with 100 μg lysostaphin and incubated for 30 minutes at 37 °C. The resulting protoplasts were sedimented by centrifugation at 20,000 × g for 5 minutes. Protoplasts were suspended in 200 μl lysis buffer (50 mM Tris HCl pH 7; 60 mM KCl; 10 mM MgCl2; 2% β-mercaptoethanol) and lysed by sonication for 10 seconds. For experiments in which cells were fractionated, cell lysates were further processed by centrifugation at 100,000 × g for 30 minutes. The supernatant was retained as the cytoplasmic fraction, and the pellet was suspended in 200 μl lysis buffer and retained as the membrane fraction. Proteins affected by oxidative damage were detected using the OxyBlot™ Protein Oxidation Detection Kit (Millipore). Following manufacturer’s instructions, the oxidatively-damaged proteins were derivatized with 2,4-dinitrophenylhydrazine (DNP). DNP-derivatized proteins were detected via standard immunoblotting methods using a primary antibody specific to the DNP moiety on the damaged proteins (Millipore) and a goat anti-rabbit IgG (Alexa Fluor 680-conjugated) secondary antibody (Invitrogen). Immunoblots were visualized using an Odyssey® Imager (LI-COR Biosciences). Staining intensity of individual sample lanes was quantified using the Odyssey® software.
Electron paramagnetic resonance (EPR) study
Bacterial cultures were grown overnight and diluted to OD600 = 0.1 in 20 mL of TSB. Diluted cultures were grown at 37 °C for 30 minutes in the presence or absence of 5 μM haem. Cultures were pelleted at 3200 × g for 10 minutes at 4 °C, and pellets were washed in ice-cold KHB (99.01 mM NaCl, 4.69 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 1.03 mM K2HPO4, 20 mM Na-HEPES, and 5.6 mM D-glucose, pH 7.35). Bacterial pellets were suspended in KHB at a concentration of 6 × 108 cfu/ml. Prior to EPR analysis, 30 μM diethylene triamine pentaacetic acid (DTPA) and 50 mM 5-ethoxycarbonyl-5-methyl-1-pyrroline N-oxide (EMPO, Enzo Life Sciences) were added to 200 μL of sample. Samples were incubated at 37 °C for 5 minutes before being subjected to EPR analyses at the Vanderbilt Free Radicals in Medicine core facility. When applicable, pegylated superoxide dismutase (SOD, 25 units) or dimethyl sulfoxide (DMSO, 5% final volume) was included in the EPR samples.
Supplementary Material
Acknowledgments
We would like to thank members of the Skaar laboratory for critical reading of this manuscript. Mass spectrometry and flow cytometry core services were performed through Vanderbilt University Medical Center’s Digestive Disease Research Center supported by NIH grant P30DK058404. EPR analyses were performed at the Vanderbilt University Free Radicals in Medicine core facility. Work in the Skaar laboratory is supported by grant numbers AI069233, AI073843, and AI091771 from the National Institutes of Health. CAW was supported by grant number T32-HL094296 from the National Heart, Lung, and Blood Institute and grant number F32-AI100535 from the National Institute of Allergy and Infectious Diseases. NDH is supported by the National Institute of Health Ruth L. Kirschstein fellowship F32-AI091244-01. LLA was supported by grant T32-HL069765 from the National Institute of Allergy and Infectious Diseases. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute, the National Institute of Allergy and Infectious Diseases, or the National Institutes of Health.
References
- Aft RL, Mueller GC. Hemin-mediated DNA strand scission. Journal of Biological Chemistry. 1983;258:12069–12072. [PubMed] [Google Scholar]
- Aft RL, Mueller GC. Hemin-mediated oxidative degradation of proteins. Journal of Biological Chemistry. 1984;259:301–305. [PubMed] [Google Scholar]
- Anzaldi LL, Skaar EP. Overcoming the heme paradox: Heme toxicity and tolerance in bacterial pathogens. Infection and Immunity. 2010;78:4977–4989. doi: 10.1128/IAI.00613-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia AS, Benson MA, Stauff DL, Torres VJ, Skaar EP. Membrane damage elicits an immunomodulatory program in Staphylococcus aureus. PLoS Pathogens. 2010;6:e1000802. doi: 10.1371/journal.ppat.1000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. Journal of bioenergetics and biomembranes. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- Baumert N, von Eiff C, Schaaff F, Peters G, Proctor RA, Sahl HG. Physiology and antibiotic susceptibility of Staphylococcus aureus small colony variants. Microbial Drug Resistance. 2002;8:253–260. doi: 10.1089/10766290260469507. [DOI] [PubMed] [Google Scholar]
- Becker K, Harmsen D, Mellmann A, Meier C, Schumann P, Peters G, von Eiff C. Development and evaluation of a quality-controlled ribosomal sequence database for 16S ribosomal DNA-based identification of Staphylococcus species. Journal of clinical microbiology. 2004;42:4988–4995. doi: 10.1128/JCM.42.11.4988-4995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley R, Meganathan R. Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiological Reviews. 1982;46:241–280. doi: 10.1128/mr.46.3.241-280.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb LA, Schmitt MP. The ABC transporter HrtAB confers resistance to hemin toxicity and is regulated in a hemin-dependent manner by the ChrAS two-component system in Corynebacterium diphtheriae. Journal of Bacteriology. 2010;192:4606–4617. doi: 10.1128/JB.00525-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen JJ, Griffiths E. Iron and infection. In: Griffiths JJBaE., editor. Molecular, Physiological and Clinical Aspects. 2. New York: John Wiley & Sons; 1999. [Google Scholar]
- Crichton R. Inorganic biochemistry of iron metabolism. In: Crichton R, editor. Molecular Mechanisms to Clinical Consequences. 2. West Sussex, England: John Wiley & Sons, Ltd; 2001. [Google Scholar]
- DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. The Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Ho B, Ding JL. Rapid reprogramming of haemoglobin structure-function exposes multiple dual-antimicrobial potencies. The EMBO journal. 2010;29:632–642. doi: 10.1038/emboj.2009.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesterberg CK, Waite TD. Kinetic modeling of the oxidation of p-hydroxybenzoic acid by Fenton’s reagent: implications of the role of quinones in the redox cycling of iron. Environmental science & technology. 2007;41:4103–4110. doi: 10.1021/es0628699. [DOI] [PubMed] [Google Scholar]
- Duthie ES, Lorenz LL. Staphylococcal coagulase: Mode of action and antigenicity. Journal of General Microbiology. 1952;6:95–107. doi: 10.1099/00221287-6-1-2-95. [DOI] [PubMed] [Google Scholar]
- Fernandez A, Lechardeur D, Derré-Bobillot A, Couvé E, Gaudu P, Gruss A. Two coregulated efflux transporters modulate intracellular heme and protoporphyrin IX availability in Streptococcus agalactiae. PLoS Pathogens. 2010;6:e1000860. doi: 10.1371/journal.ppat.1000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Stauff DL, Pishchany G, Whitwell CW, Torres VJ, Skaar EP. Staphylococcus aureus redirects central metabolism to increase iron availability. PLoS Pathogens. 2006;2:e87. doi: 10.1371/journal.ppat.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaupp R, Ledala N, Somerville GA. Staphylococcal response to oxidative stress. Frontiers in cellular and infection microbiology. 2012;2:33. doi: 10.3389/fcimb.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer R, Peacock AD, White DC, Lytle C, Van Berkel GJ. Atmospheric pressure chemical ionization and atmospheric pressure photoionization for simultaneous mass spectrometric analysis of microbial respiratory ubiquinones and menaquinones. Journal of Mass Spectrometry. 2004;39:922–929. doi: 10.1002/jms.670. [DOI] [PubMed] [Google Scholar]
- Gordon RJ, Lowy FD. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clinical Infectious Diseases. 2008;46:S350–S359. doi: 10.1086/533591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge JM, Quinlan GJ, Wilkins S. Mitomycin C-induced deoxyribose degradation inhibited by superoxide dismutase. A reaction involving iron, hydroxyl and semiquinone radicals. FEBS Letters. 1984;167:37–41. doi: 10.1016/0014-5793(84)80828-5. [DOI] [PubMed] [Google Scholar]
- Hammer ND, Skaar EP. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annual Review of Microbiology. 2011;65:129–147. doi: 10.1146/annurev-micro-090110-102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RK, White DC. Formation of vitamin K2 isoprenologues by Staphylococcus aureus. Journal of Bacteriology. 1969;100:573–578. doi: 10.1128/jb.100.2.573-578.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holländer R. Correlation of the function of demethylmenaquinone in bacterial electron transport with its redox potential. FEBS Letters. 1976;72:98–100. doi: 10.1016/0014-5793(76)80821-6. [DOI] [PubMed] [Google Scholar]
- Huycke MM, Moore D, Joyce W, Wise P, Shepard L, Kotake Y, Gilmore MS. Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Molecular Microbiology. 2001;42:729–740. doi: 10.1046/j.1365-2958.2001.02638.x. [DOI] [PubMed] [Google Scholar]
- Jeffries L, Harris M, Price SA. Atypical menaquinone pattern in a strain of. Staphylococcus aureus Nature. 1967;216:808–809. doi: 10.1038/216808a0. [DOI] [PubMed] [Google Scholar]
- Jiang N, Tan NS, Ho B, Ding JL. Respiratory protein-generated reactive oxygen species as an antimicrobial strategy. Nature immunology. 2007;8:1114–1122. doi: 10.1038/ni1501. [DOI] [PubMed] [Google Scholar]
- Kehl-Fie TE, Skaar EP. Nutritional immunity beyond iron: a role for manganese and zinc. Current Opinion in Chemical Biology. 2010;14:218–224. doi: 10.1016/j.cbpa.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK f. t. A. B. C. s. M. Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA: The Journal of the American Medical Association. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- Korshunov S, Imlay JA. Detection and quantification of superoxide formed within the periplasm of Escherichia coli. Journal of Bacteriology. 2006;188:6326–6334. doi: 10.1128/JB.00554-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladan H, Nitzan Y, Malik Z. The antibacterial activity of haemin compared with cobalt, zinc and magnesium protoporphyrin and its effect on pottassium loss and ultrastructure of Staphylococcus aureus. FEMS Microbiology Letters. 1993;112:173–177. doi: 10.1111/j.1574-6968.1993.tb06444.x. [DOI] [PubMed] [Google Scholar]
- Lannergard J, von Eiff C, Sander G, Cordes T, Seggewiss J, Peters G, Proctor RA, Becker K, Hughes D. Identification of the genetic basis for clinical menadione-auxotrophic small-colony variant isolates of Staphylococcus aureus. Antimicrobial agents and chemotherapy. 2008;52:4017–4022. doi: 10.1128/AAC.00668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherbarrow AJH, Yazdi MA, Curson JP, Moir A. The gerC locus of Bacillus subtilis, required for menaquinone biosynthesis, is concerned only indirectly with spore germination. Microbiology. 1998;144:2125–2130. doi: 10.1099/00221287-144-8-2125. [DOI] [PubMed] [Google Scholar]
- Lechardeur D, Cesselin B, Liebl U, Vos MH, Fernandez A, Brun C, Gruss A, Gaudu P. Discovery of intracellular heme-binding protein HrtR, which controls heme efflux by the conserved HrtB-HrtA transporter in Lactococcus lactis. Journal of Biological Chemistry. 2012;287:4752–4758. doi: 10.1074/jbc.M111.297531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locigno EJ, Zweier JL, Villamena FA. Nitric oxide release from the unimolecular decomposition of the superoxide radical anion adduct of cyclic nitrones in aqueous medium. Organic & biomolecular chemistry. 2005;3:3220–3227. doi: 10.1039/b507530k. [DOI] [PubMed] [Google Scholar]
- Lombardo ME, Araujo LS, Ciccarelli AB, Batlle A. A spectrophotometric method for estimating hemin in biological systems. Analytical biochemistry. 2005;341:199–203. doi: 10.1016/j.ab.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Lukomski S, Hoe NP, Abdi I, Rurangirwa J, Kordari P, Liu M, Dou SJ, Adams GG, Musser JM. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infection and Immunity. 2000;68:535–542. doi: 10.1128/iai.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DG, Marritt SJ, Butt JN, Jeuken LJ. Menaquinone-7 is specific cofactor in tetraheme quinol dehydrogenase CymA. The Journal of biological chemistry. 2012;287:14215–14225. doi: 10.1074/jbc.M112.348813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina LF, Hertz PF, Stefani V, Henriques JA, Zanotto-Filho A, Brandelli A. Aminonaphthoquinone induces oxidative stress in Staphylococcus aureus. Biochemistry and cell biology. 2006;84:720–727. doi: 10.1139/o06-087. [DOI] [PubMed] [Google Scholar]
- Musher DM, Baughn RE, Templeton GB, Minuth JN. Emergence of variant forms of Staphylococcus aureus after exposure to gentamicin and infectivity of the variants in experimental animals. The Journal of infectious diseases. 1977;136:360–369. doi: 10.1093/infdis/136.3.360. [DOI] [PubMed] [Google Scholar]
- Nir U, Ladan H, Malik Z, Nitzan Y. In vivo effects of porphyrins on bacterial DNA. Journal of Photochemistry and Photobiology B: Biology. 1991;11:295–306. doi: 10.1016/1011-1344(91)80035-g. [DOI] [PubMed] [Google Scholar]
- Nitzan Y, Wexler HM, Finegold SM. Inactivation of anaerobic bacteria by various photosensitized porphyrins or by hemin. Current Microbiology. 1994;29:125–131. doi: 10.1007/BF01570752. [DOI] [PubMed] [Google Scholar]
- Novick RP. Genetic systems in staphylococci. Methods in Enzymology. 1991;204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- Osyczka A, Moser CC, Dutton PL. Fixing the Q cycle. Trends in biochemical sciences. 2005;30:176–182. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Pedersen MB, Garrigues C, Tuphile K, Brun C, Vido K, Bennedsen M, Mollgaard H, Gaudu P, Gruss A. Impact of aeration and heme-activated respiration on Lactococcus lactis gene expression: identification of a heme-responsive operon. Journal of Bacteriology. 2008;190:4903–4911. doi: 10.1128/JB.00447-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nature Reviews Microbiology. 2006;4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- Reniere M, Torres V, Skaar E. Intracellular metalloporphyrin metabolism in Staphylococcus aureus. BioMetals. 2007;20:333–345. doi: 10.1007/s10534-006-9032-0. [DOI] [PubMed] [Google Scholar]
- Rezaïki L, Lamberet G, Derré A, Gruss A, Gaudu P. Lactococcus lactis produces short-chain quinones that cross-feed Group B Streptococcus to activate respiration growth. Molecular Microbiology. 2008;67:947–957. doi: 10.1111/j.1365-2958.2007.06083.x. [DOI] [PubMed] [Google Scholar]
- Rifkind JM, Ramasamy S, Manoharan PT, Nagababu E, Mohanty JG. Redox reactions of hemoglobin. Antioxidants & redox signaling. 2004;6:657–666. doi: 10.1089/152308604773934422. [DOI] [PubMed] [Google Scholar]
- Seifert H, von Eiff C, Fatkenheuer G. Fatal case due to methicillin-resistant Staphylococcus aureus small colony variants in an AIDS patient. Emerging infectious diseases. 1999;5:450–453. doi: 10.3201/eid0503.990319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J, van Spanning RJM, Richardson DJ. The organisation of proton motive and non-proton motive redox loops in prokaryotic respiratory systems. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2008;1777:1480–1490. doi: 10.1016/j.bbabio.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. Iron-source preference of Staphylococcus aureus infections. Science. 2004;305:1626–1628. doi: 10.1126/science.1099930. [DOI] [PubMed] [Google Scholar]
- Stauff DL, Bagaley D, Torres VJ, Joyce R, Anderson KL, Kuechenmeister L, Dunman PM, Skaar EP. Staphylococcus aureus HrtA is an ATPase required for protection against heme toxicity and prevention of a transcriptional heme stress response. Journal of Bacteriology. 2008;190:3588–3596. doi: 10.1128/JB.01921-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauff DL, Skaar EP. Bacillus anthracis HssRS signalling to HrtAB regulates haem resistance during infection. Molecular Microbiology. 2009;72:763–778. doi: 10.1111/j.1365-2958.2009.06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauff DL, V, Torres J, Skaar EP. Signaling and DNA-binding activities of the Staphylococcus aureus HssR-HssS two-component system required for heme sensing. Journal of Biological Chemistry. 2007;282:26111–26121. doi: 10.1074/jbc.M703797200. [DOI] [PubMed] [Google Scholar]
- Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free radical biology & medicine. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui Kelsi J, Anderson L, Dunman PM, Joyce S, Skaar EP. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host & Microbe. 2007;1:109–119. doi: 10.1016/j.chom.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eiff C, Bettin D, Proctor RA, Rolauffs B, Lindner N, Winkelmann W, Peters G. Recovery of small colony variants of Staphylococcus aureus following gentamicin bead placement for osteomyelitis. Clinical Infectious Diseases. 1997a;25:1250–1251. doi: 10.1086/516962. [DOI] [PubMed] [Google Scholar]
- von Eiff C, Heilmann C, Proctor RA, Woltz C, Peters G, Götz F. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. Journal of Bacteriology. 1997b;179:4706–4712. doi: 10.1128/jb.179.15.4706-4712.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eiff C, Peters G, Becker K. The small colony variant (SCV) concept -- the role of staphylococcal SCVs in persistent infections. Injury. 2006;37(Suppl 2):S26–33. doi: 10.1016/j.injury.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Wakeman CA, Skaar EP. Metalloregulation of Gram-positive pathogen physiology. Current Opinion in Microbiology. 2012;15:169–174. doi: 10.1016/j.mib.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HFL, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JAJW, van Keulen PHJ, Vandenbroucke-Grauls CMJE, Meester MHM, Verbrugh HA. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. The Lancet. 2004;364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- Wissenbach U, Ternes D, Unden G. An Escherichia coli mutant containing only demethylmenaquinone, but no menaquinone: effects on fumarate, dimethylsulfoxide, trimethylamine N-oxide and nitrate respiration. Archives of microbiology. 1992;158:68–73. doi: 10.1007/BF00249068. [DOI] [PubMed] [Google Scholar]
- Youngman PJ, Perkins JB, Losick R. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proceedings of the National Academy of Sciences. 1983;80:2305–2309. doi: 10.1073/pnas.80.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TW, Anderson D. Reactive oxygen species-induced DNA damage and its modification: a chemical investigation. Mutation research. 1997;379:201–210. doi: 10.1016/s0027-5107(97)00141-3. [DOI] [PubMed] [Google Scholar]
- Zamboni N, Sauer U. Knockout of the high-coupling cytochrome aa3 oxidase reduces TCA cycle fluxes in Bacillus subtilis. FEMS Microbiology Letters. 2003;226:121–126. doi: 10.1016/S0378-1097(03)00614-1. [DOI] [PubMed] [Google Scholar]
- Zhang H, Joseph J, Vasquez-Vivar J, Karoui H, Nsanzumuhire C, Martasek P, Tordo P, Kalyanaraman B. Detection of superoxide anion using an isotopically labeled nitrone spin trap: potential biological applications. FEBS Letters. 2000;473:58–62. doi: 10.1016/s0014-5793(00)01498-8. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Koyama T, Ogura K. Two cistrons of the gerC operon of Bacillus subtilis encode the two subunits of heptaprenyl diphosphate synthase. Journal of Bacteriology. 1997;179:1417–1419. doi: 10.1128/jb.179.4.1417-1419.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.