

Abstract
Porous silicon microparticles presenting pathogen-associated molecular patterns mimic pathogens, enhancing internalization of the microparticles and activation of antigen presenting dendritic cells. We demonstrate abundant uptake of microparticles bound by the TLR-4 ligands LPS and MPL by murine bone marrow-derived dendritic cells (BMDC). Labeled microparticles induce concentration-dependent production of IL-1β, with inhibition by the caspase inhibitor Z-VAD-FMK supporting activation of the NLRP3-dependent inflammasome. Inoculation of BALB/c mice with ligand-bound microparticles induces a significant increase in circulating levels of IL-1β, TNF-α, and IL-6. Stimulation of BMDC with ligand-bound microparticles increases surface expression of co-stimulatory and MHC molecules, and enhances migration of BMDC to the draining lymph node. LPS-microparticles stimulate in vivo C57BL/6 BMDC and OT-1 transgenic T cell interactions in the presence of OVA SIINFEKL peptide in lymph nodes, with intact nodes imaged using two-photon microscopy. Formation of in vivo and in vitro immunological synapses between BMDC, loaded with OVA peptide and LPS-microparticles, and OT-1 T cells are presented, as well as elevated intracellular interferon gamma levels in CD8+ T cells stimulated by BMDC carrying peptide-loaded microparticles. In short, ligand-bound microparticles enhance 1) phagocytosis of microparticles; 2) BMDC inflammasome activation and up-regulation of co-stimulatory and MHC molecules; 3) cellular migration of BMDC to lymphatic tissue; and 4) cellular interactions leading to T cell activation in the presence of antigen.
Keywords: microparticle, vaccine, dendritic cell, migration, phagocytosis, atomic force microscopy, LPS, monophosphoryl lipid A
Introduction
Dendritic Cells (DC) and macrophages/monocytes are the body’s sentinel gatekeepers, tasked with guarding against foreign antigens, pathogens, and damaged cells. Differentiation of DC from bone marrow (BM) cells is influenced by cytokines, with different cytokines activating alternative pathways and producing different DC types and subtypes with altered functional capabilities 1. Both humans and mice have two major DC types, classical and plasmacytoid 2. Classical DC in mice are further divided into lymphoid (CD8α+CD11b−CD11c+DEC205+) and myeloid (CD8α−CD11b+ CD11c+), which broadly induce T helper (Th)1 (CD8+ T cell immunity) and Th2 (CD4+ T cell immunity) responses, respectively 2–3. In the immature state, DCs are highly phagocytic and are characterized by low levels of surface major histocompatibility complex (MHC) class II molecules, co-stimulatory molecules, chemokine receptors (e.g. CCR7 which triggers migration), and cytokine secretion 4. Microbial products, known as pathogen-associated molecular patterns (PAMPs), stimulate DC maturation through toll-like receptors (TLR) or other pattern recognition receptors (PRRs), promoting expression of co-stimulatory molecules and cytokines, and stimulating migration of DC to the draining lymph node 5. For example, Escherichia coli lipopolysaccharide (LPS) stimulates DC via TLR-4, inducing an IL-12 dependent Th1 response, while LPS from Porphyromonasgingivalis activates DC through TLR-2, inducing an IL-10 mediated Th2 response 6. Early TLR responses in DC include membrane ruffling, macopinocytosis and phagocytosis, and phagosome maturation 7. The increased expression of actin network regulators leads to the distinct DC “dendrite-rich” morphology and enhances the ability of DC to interact with T cells.
Nanoparticles are being investigated as vehicles to co-deliver antigens and adjuvant for vaccine development 8. Labeling nanoparticles with ligands that enhance uptake by DC and stimulate cellular activation is one approach to creating active vaccine delivery vehicles. Monophosphoryl lipid A (MPL), a non-toxic derivative of LPS, is reported to be a Th1-baised adjuvant 9. MPL is approved for commercial use in Europe and is used in a variety of clinical vaccine trials 9. Similar to LPS, MPL binds to TLR-4 and activates a pro-inflammatory signaling cascade 10. Both LPS and MPL are reported to enhance DC phagocytosis, up-regulate activation markers, and stimulate maturation and migration to the draining lymph nodes 11. Presentation of antigen in the presence of TLR ligands has been shown to enhance antigenic presentation, leading to positive immune responses7b. LPS-labeled beads are reported to induce both external and internal TLR signaling through surface and endosomal TLR ligation, with endosomal TLR engagement inducing more potent cellular stimulation 7a. In addition, LPS-modified poly(lactic-co-glycolic acid) (PLGA) nanoparticles activate the intracellular nucleotide-binding domain and leucine-rich repeat containing receptor protein (NLRP)3 inflammasome in an endocytosis-dependent manner 12, leading to activation of caspase 1 and production of the pro-inflammatory cytokine interleukin (IL)-1β and immune activation 13.
Here we introduce TLR-ligand-bound porous silicon (pSi) microparticles as a vaccine vehicle. We have previously demonstrated that pSi microparticles are biocompatible with respect to endothelial and macrophage cell viability, morphology, mitosis and cell cycle 14. Due to their porous nature, the microparticles are degradable, with kinetics dependent on the degree of porosity. pSi microparticles can be loaded directly or using nanoparticles as carriers for biologically active agents 15. With respect to vaccines, cargo could include antigens and immuno-modulatory agents. Release kinetics from the primary and secondary carriers would add an additional level of control with respect to antigen release kinetics, intracellular trafficking, and duration of immune stimulation. We have shown that the microparticles are internalized by macrophages into phagosomes, which mature into late endosomes and lysosomes16, which would permit antigen processing and presentation in association with MHC class II molecules. We have also shown that the presence of chitosan enhances endosomal escape of pSi microparticle-delivered nanoparticles 17, which may be beneficial for cytoplasmic expression of virally-expressed antigens and increased cross presentation of antigens with MHC class I molecules.
In this study, we examined the impact of cytokines and LPS on APC phenotype and its impact on microparticle internalization. We also examined the impact of microparticle-bound LPS and MPL on APC maturation, internalization of pSi microparticles, cellular viability, and migration to the draining lymph node. Atomic force microscopy (AFM) was used to characterize LPS and MPL binding to silicon. Scanning Electron (SE) and two-photon microscopy were used to image lymph nodes and lymphocyte-rich regions within the node. Two-photon microscopy was also used to visualize migration and localization of microparticle-laden DC and T cells within the lymph node and to study the influence of T cell recognition of MHC class I presented peptide on co-localization of T cells and BMDC in the lymph node. C57BL/6-Tg(TcraTcrb) (a.k.a. OT-1) mice were chosen as a source of T cells based on the presence of a transgenic T cell receptor that recognizes ovalbumin (OVA) residues 257–264 (i.e. SIINFEKL peptide) in association with MHC class I molecules. Transmission electron (TE) microscopy was used to demonstrate peptide and microparticle-driven BMDC-T cell interactions and intracellular interferon gamma (IFN-γ) levels in CD8+ T cells were used as a measure of T cell activation by peptide-loaded microparticle-stimulated BMDC. The overall goal of the study was to optimize our vaccine vehicle by maximizing factors positively influencing uptake, activation, and migration of APC to T cell-rich zones of the lymph node.
Experimental Section
Materials
LPS from Escherichia coli and MPL from Salmonella enteric serotype Minnesota RE 595 were purchased from Sigma-Aldrich (St. Louis, MO). DyLight products for labeling microparticles were obtained from Invitrogen (Grand Island, NY). TNF-α, IL-6, IL-1β ELISA kits were purchased from R&D Systems, Inc. (Minneapolis, MN). CellTracker™ Orange, CellTrace™ Carboxyfluorescein succinimidyl ester (CSFE), and PKH26 Red Fluorescent Cell Linker were obtained from Invitrogen and Sigma-Aldrich. Alexa Fluor 488-conjugated annexin V and Oregon Green® 488 phalloidin were purchased from Invitrogen.
pSi microparticle preparation, chemical conjugation and surface modification
pSi microparticles were made by the Methodist Hospital Research Institute (TMHRI) NanoEngineering Core as previously described 18. Microparticles were treated with piranha solution (1 volume H2O2 and 2 volumes of H2SO4) to chemically oxidize the surface. The suspension was heated to 110–120 °C for 2 hr, centrifuged and washed in deionized water repeatedly. Amine surface modification was achieved using a 2% (v/v) 3-aminopropyl triethoxy-silane (APTES) (Sigma-Aldrich) solution in 95% isopropanol and 5% water for 2 hr at 35°C with agitation at 1300 RPM. For labeling particles with fluorescent dye, APTES-modified particles were conjugated to DyLight 594 or DyLight 488 N-hydroxysulfosuccinimide (NHS)-ester activated fluorescent dyes according to the manufacturer’s protocol (Invitrogen). TLR ligands (unlabeled or FITC; 0.6 mg/ml final concentration) were conjugated to pSi microparticles using ethyl-dimethylaminopropyl carbodiimide (EDC) according the manufacturer’s protocol (Pierce, Rockford, IL). Adsorption was done by immersing the APTES-microparticles in a 1 mg/ml aqueous solution of ligand followed by two wash steps. The amount of ligand conjugated to microparticles was determined by measuring the fluorescent intensity of LPS-FITC on a solution of labeled microparticles and calculating concentration based on a standard curve generated with known quantities of free LPS-FITC. Zeta potential measurements were performed in 10 mM phosphate buffer using a Malvern Zetasizer (Malvern Instruments Inc, Westborough, MA). Chromogenic Limulus amebocyte lysate (LAL) tests (GenScript, Piscataway, NJ) were used to determine if unlabeled microparticles were free of endotoxin. The binding density and conformation of bound MPL and LPS was determined using AFM analysis as described below.
AFM analysis of ligand binding to silicon was performed using a Veeco MultiMode Nanoscope IIIA (Bruker AXS, Madison, WI). Silicon chips were oxidized in piranha solution and APTES-modified with a 2% solution (30 min). For adsorption studies, silicon was immersed in a 1 mg/ml aqueous solution of TLR ligand for 1 hr at 4°C, followed by 2 washes and drying in nitrogen. For covalent modification, a 1 mg/ml aqueous solution of LPS (400 μg) was mixed with ethyl-dimethylaminopropyl carbodiimide (270 μg) (final volume 500 μl), then introduced to APTES-modified silicon for 2 hr at 30°C. Scanning was performed in tapping mode in air using rotated tapping mode etched silicon probe cantilevers (fo=320 kHz, k=42 Nm−1). Image analysis was performed using Veeco NanoScope Analysis software version 1.20.
Animals
BALB/c and C57BL/6 (6–10 wk old) mice were obtained from Charles River Laboratories, Inc, Wilmington, MA) and C57BL/6-Tg(TcraTcrb) mice from The Jackson Laboratory (Bar Harbor, Maine). All procedures were performed in accordance with protocols reviewed and approved by the Institutional Animal Care and Use Committee at TMHRI.
BMDC culture
BMDCs were prepared, as previously described, with some modifications (25, 26). Briefly, bone marrow cells were harvested from the femurs and tibias and filtered through a cell strainer (BD Biosciences, San Jose, CA). RBC was lysed with ACK lysis buffer (Quality Biologicals, Inc., Gaithersburg, MD), and cells were washed and cultured in six-well plates at 106 cells/ml (3 ml/well) in RPMI 1640 (Sigma-Aldrich) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA), 100 U/ml penicillin/streptomycin (Fishers Scientific, Waltham, MA), 50 μM 2-ME (Sigma-Aldrich), and mouse GM-CSF (20 ng/ml; Pierce), in the presence or absence of IL-4 (100 ng/ml; Peprotech, Rocky Hill, NJ) for 5–7 days. Half of the media was replaced every 2–3 days with fresh media and cytokines.
Particle uptake by BMDC - SE Microscopy
BMDC cells were plated in 24 well plates containing 5 × 7 mm Silicon Chip Specimen Supports (Ted Pella, Inc., Redding, CA) at 1 × 105 cells per well. The next day, cells were incubated with TLR ligand-conjugated or unconjugated pSi microparticles for 1–3 hr at 37°C at a 1:25 cell to microparticle ratio. Cells were processed for SEM imaging as previously described 14a, 16. Specimens were mounted on SEM stubs (Ted Pella, Inc.) using conductive adhesive tape (12 mm OD PELCO Tabs, Ted Pella, Inc.), and sputter-coated with a 4 nm layer of platinum/palladium (80:20) using a Cressington Sputter Coater 208 HR (Ted Pella, Inc.). SEM images were acquired under high vacuum, at 10–15 kV, spot size 3.0, using a Nova™ NanoSEM Scanning Electron Microscope (FEI Company, Hillsboro, OR). Images have been pseudo-colored and gamma levels adjusted made to enhance image contrast and brightness.
Particle uptake by BMDC - Confocal imaging pSi microparticles, control and co-labeled with DyLight 594 and TLR ligands (LPS and MPL), were incubated with BMDC grown on 4-well chamber slides seeded at 5×104 cells/well for 1–3 hr at 37°C at a 1:25 cell to microparticle ratio. Cells were then washed and fixed with 2.5% paraformaldehyde, then permeabilized with 0.1% Triton X-100 for 5 min. Samples were then blocked with 2% BSA in PBS for 20 min followed by Oregon Green® 488 phalloidin labeling for 20 min. Slides were then washed and mounted with coverslips using DAPI-containing Vectashield mounting media (Vector Laboratories, Burlingame, CA). Images were obtained using a Nikon A1 confocal microscope equipped with a 60x oil immersion objective.
Flow cytometry – APC phenotyping and microparticle uptake
BMDC expression of phenotype and maturation markers, as well as microparticle association, were analyzed by flow cytometry. Mouse (C57BL/6) bone marrow cells (5×106) were treated with cytokines (GM-CSF or GM-CSF/IL-4 for 5–7 days), followed by either TNF-α (50 ng/ml; Peprotech) or LPS (1 μg/ml) for 24 hrs. For cell internalization experiments, microparticles (Dylight 488-labeled, 1.25×107) were introduced for 24 hrs. Cells [adherent and non-adherent (the latter released by scrapping)] were washed twice with FACS wash buffer (PBS containing 0.1% sodium azide and 1% BSA) and labeled with antibodies or analyzed directly. The following antibodies were used: FITC-conjugated anti-mouse CD8, CD14, CD40, CD80, CD86 (BD Biosciences), APC-conjugated anti-mouse MHC Class II (I-A/I-E; C57BL/6) and Ly-6C (eBiosciences, San Diego, CA), PE-conjugated MHCI (H2Kd; BALB/c), MHCII (I-Ad; BALB/c), CD11b, and CD11c (BD Biosciences), and PerCP-Cy5.5 conjugated CCR7 (BD Biosciences). Isotype controls used in the study were purchased from BD Biosciences. Samples were analyzed using a FACS FORTESSA Flow Cytometer (BD Biosciences, Mountain View, CA) using FACSDIVA software.
Measurement of cytokine release
To study BMDC-stimulated cytokine release in vitro, either free ligand (1 μg/ml × 2 ml/well) or microparticle-conjugated [0.125, 0.63, or 1.25 μg LPS/5, 25, 50 (×106) microparticles/well] LPS or MPL was incubated with 1×106 BMDC/well for 24 hr or as indicated at 37°C. Human acute monocytic (THP-1) cells were stimulated with phorbol myristate acetate (PMA) (1 μg/ml) for 48 hr, followed by treatment with control or LPS-microparticles (1:20; 0.2 μg LPS equivalent) or free LPS (0.2 μg) for 3 and 24 hr. The following reagents were included in the cell culture media as indicated: ATP (5 μM), cytochalasin B (10 μg/ml), and Z-Val-Ala-Asp fluoromethyl ketone (Z-VAD-FMK) caspase inhibitor (10 μg/ml). Cell media was analyzed for IL-1β, TNF-α, and IL-6 using commercial ELISA kits, according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN; human IL-1β ELISA from BD Biosciences).
To study in vivo cytokine secretion, approximately 5×108 pSi microparticles [control or TLR-4 ligand (10 μg) conjugated] were injected into BALB/c mice (n=2) subcutaneously. Blood samples were collected 24 hr post injection via cardiac puncture and blood serum was prepared by centrifugation at 2500 g for 10 min. Serum samples were stored at −80°C and analyzed by ELISA.
Intracellular interferon assay
BMDC were cultured in GM-CSF for 5 days, and then incubated with OVA peptide loaded LPS-microparticles for 1 hr before co-culturing with splenic T cells at a ratio of one BMDC to five T cells for 4 hr. LPS-microparticles were loaded with peptide by capillary action using a 10 μg/ml solution of SIINFEKL peptide (Genscript, Piscataway, NJ). Flow cytometry was used to measure intracellular interferon gamma levels in CD8+ T cells (PE-conjugated anti-IFN-γ and FITC-conjugated anti-CD8 antibodies; BD Biosciences).
Annexin V/propidium iodide analysis of cellular apoptosis
BMDC were incubated with 3 doses of pSi microparticles (control or TLR ligand-conjugated at a cell to microparticle ratio of 1:10, 1:25 and 1:50) for 24 hr. BMDC were then washed with ice-cold PBS and incubated with Alexa Fluor 488 Annexin V (5 μl/100 μl) and propidium iodide (1 μl of 100 μg/ml solution/100 μl) for 15 min at 4°C, followed by washing in PBS and analysis by flow cytometry.
In Vivo BMDC Migration Assay
BMDC were incubated with control or TLR-4 ligand-conjugated pSi microparticles (1:25 cell to microparticle ratio, containing an equivalent of 12.5 mg LPS) for 3 hr at 37°C. Following microparticle uptake, BMDC were labeled with either PKH26, CellTracker Orange, or CSFE and 3–5×106cells were injected subcutaneously via hock injection (n=5 per group) 19. Control vehicle, unlabeled pSi microparticles, and microparticle-free BMDC were administered as controls. The popliteal and inguinal lymph nodes were collected 24 hr after injection. A cell suspension was created by passing the lymph node tissue through a cell strainer. Cell populations were examined by flow cytometry using a FACS LSRII (Becton-Dickinson, Mountain View, CA). Data were analyzed using either FACSDIVA (Becton-Dickinson) or FCS Express software.
Two-photon microscopy
To study DC migration and co-localization with T cells, BMDC were incubated with control or LPS-conjugated (0.625 mg LPS/kg) pSi microparticles and OVA peptide (SIINFEKL; 10 μg/ml), followed by labeling with CellTracker™ Orange (2μM) according to the manufacturer’s protocol. Control (n=2 mice) or microparticle (n=3 mice) treated C57BL/6 BMDC (5×106) were injected into the footpad in 50 μl of PBS, followed at 6 hr post-injection with CellTrace™ CFSE -labeled T cells (1×107), derived from C57BL/6-Tg(TcraTcrb) spleen and enriched by depletion of MHC class II expressing cells using magnetic beads (Miltenyi Biotech Inc. Auburn, CA) by intravenous inoculation. Intact popliteal and inguinal lymph nodes were imaged 18 hours later using an Olympus BX61WI upright multi-photon microscope. Images were acquired using an Olympus 40x water-immersion objective (NA, 0.8). A mode-locked Ti:sapphire laser (Mai Tai; Spectra-Physics, Santa Clara, CA) generated two-photon excitation at 820 nm, and emitted fluorescence collected in the range of 495–540 nm and 575–630 nm. 3D Z-stacks were obtained at a resolution of 2 pixels μm−1 in 1 μm steps (100 slices). Each xy plane image was an average of 2 frames. 2D images shown in this paper are maximum intensity projections of the 3D z-stacks.
Tissue TE Microscopy
GM-CSF stimulated C57BL/6 BMDC were cultured on poly-l-lysine glass slides and then pulsed with LPS-bound porous silicon microparticles (1:20 ratio) in the presence of 10 μg/ml SIINFEKL peptide for 3 hr at 37°C. C57BL/6-Tg(TcraTcrb) splenic T cells were introduced in fresh media at a ratio of 2:1 (BMDC to T cells) ratio for an additional 1 hr, followed by washing and fixation in 2% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4. After washing with cacodylate buffer, cells were incubated in a mixture of osmium tetroxide and 1% potassium ferrocyanate in 0.1M cacodylate buffer for 30 min at 48°C. Cells were then dehydrated in increasing concentrations of ethanol and embedded with a mixture of epon and araldite. After polymerization at 60°C, the coverslips were removed from the resin by freezing in liquid nitrogen. Ultrathin sections were counter-stained with uranyl acetate and imaged using a JEOL 1210 TE microscope equipped with an AMT Imaging System. Gamma adjustments were made to the micrographs to enhance image contrast and brightness.
Statistics
Statistical comparisons were performed using a two-sample, equal variance, two-tailed T test. Fold-increases were calculated using sample means. The error bars in graphical presentations represent standard deviations.
Results
BMDC association with unlabeled pSi microparticles
Discoidal pSi microparticles were created from silicon wafers using photolithography and electrochemical etching as previously described 20. The resulting microparticles had dimensions of 1000×400 nm. SE micrographs showing high and low porosity faces of the microparticle are shown in Figure 1A. The diameter of the pore openings are approximately 50 nm.
Figure 1. Association of APC with discoidal porous silicon microparticles.

A) Scanning electron micrographs show three perspectives of the 1000 × 400 nm discoidal silicon microparticles, emphasizing microparticle geometry and pore size, with an average pore diameter of 50 nm (left: 160kx mag; 7.5keV; right: 250kx mag; 7.5keV; bars 500 nm). B-C) Pseudo-colored scanning electron micrographs show microparticle association with GM-CSF-stimulated APC at the adherence (B) and initiation (C) stages of uptake (left: 15kx mag; right: 40kx mag; 15 kV; bars 1000 nm). D) Flow cytometry was used to measure association of fluorescent microparticles with both adherent and non-adherent populations of bone marrow-derived APC cultured in the presence of various combinations of GM-CSF, IL-4, TNF-α, and LPS (populations significantly greater than GM-CSF treated cells, *p<0.005, **p<0.04, ***p<0.0006, ****p<0.003). The gamma levels were adjusted on the electron micrographs to enhance contrast and brightness.
Visualization of microparticle adhesion and early uptake are seen in pseudo-colored SE micrographs that show adherent GM-CSF-stimulated murine bone marrow cells after incubation with microparticles for 1 hr (Figure 1B,C). The cells have veiled surfaces, characteristic of phagocytic antigen presenting cells. The high magnification image shows cellular protrusions forming an actin cup to engulf the microparticle.
The influence of cytokines on microparticle association with BMDC was determined by incubating GM-CSF-stimulated bone marrow cells with LPS or TNF-α for 24 hr, followed by incubation with DyLight® 488-labeled microparticles for 3 hr at 37°C (Figure 1D). Since adherent and non-adherent BM cells are reported to have macrophage and DC phenotypes, respectively, we compared microparticle uptake by each population. Treatment of cells with GM-CSF, with or without TNF-α, resulted in 62% of the non-adherent cells associating with microparticles. Addition of IL-4 to the culture stimulated an increase in microparticle uptake (GM-CSF: 73%, p<0.004; GM-CSF/TNF-α: 72% p<0.02). Addition of LPS to the GM-CSF culture induced even higher levels of uptake, with 85% of GM-CSF/LPS (p<0.04) and 83% (p<0.003) of GM-CSF/IL-4/LPS treated cells associating with microparticles. MFI data also supports greater microparticle association per cell in the presence of IL-4 and LPS, with a 43% increase with IL-4 and a 30% increase with IL-4 and LPS. Microparticle association by adherent cells was more similar among the treatment groups, with 67–73% of cells associating with microparticles across all groups. In summary, maturation of pre-DCs with LPS enhanced microparticle phagocytosis, and non-adherent DCs were more phagocytic than their adherent counterparts.
Preparation and characterization of TLR-ligand bound microparticles
Since LPS favored microparticle association with BMDC and is known to stimulate uptake of bacteria, we surface-labeled microparticles with LPS or MPL to mimic pathogens. Oxidation of silicon microparticles creates a functional hydroxylated surface, modifiable by silylation and standard EDC conjugation chemistries. As expected, the surface potential of oxidized microparticles was negative, while APTES silylation created a positive surface potential due to the addition of free amine units to the microparticle surface (Table 1). A mean of approximately 0.025 μg of LPS per million microparticles was achieved (25 femtograms per microparticle), as determined based on the fluorescent intensity of bound LPS FITC against a standard curve (Figure S2). Modification of the microparticle surface with either MPL or LPS, which are electronegative, reversed the positive APTES surface potential (Table 1). There was no significant difference in particle size based on dynamic light scattering (DLS) measurements.
Table 1. Microparticle characterization.
The surface potential and size, based on zeta potential and dynamic light scattering, respectively, of microparticles is presented.
| Zeta Potential (mV) | DLS (nm) | |
|---|---|---|
| Oxidized | −30.9 + 1.0 | |
| APTES | 4.9+0.8 | 692+48 |
| APTES-MPL (pSi-MPL) | −19.1+1.3 | 675+47 |
| APTES-LPS (pSi-LPS) | −9.1+0.5 | 697+45 |
Characterization of TLR ligand binding to silicon
TLR ligand grafting density and conformation when bound to silicon were studied by AFM analysis of ligand bound silicon chips. Binding by adsorption was compared to that achieved by using covalent crosslinkers. While it has been reported that LPS O-antigens bind to silanol groups through hydrogen-bonds 21, LPS binding to oxidized silicon was poor (data not shown), so therefore APTES-modified silicon was used for all studies. High resolution imaging showed tight packing of LPS for both adsorbed and covalently bound sample (Figure 3), with an average vertical height at 2.9±0.3 nm for both presentations (Figure S4). Surface roughness for bound LPS was 1.4 and 2.7 nm, for adsorbed and covalently bound LPS, respectively, with enhanced roughness in the latter due to a greater abundance of surface-bound LPS aggregates. In contrast, MPL binding was vastly different for the two approaches, with large rises and dents for adsorbed MPL and low surface roughness for covalently bound MPL (6.3 and 0.8 nm, respectively). Introduction of MPL in the presence of EDC was unique in that it led to the formation of a homogeneous monolayer (1.7±0.9 nm vertical height) across the silicon surface. The presence of abundant ‘liposome-like’ structures in both MPL samples, albeit at much higher frequency in the adsorbed sample, indicates formation of large aggregates prior to surface binding, consistent with reports that MPL exists in aggregates in aqueous solution due to its amphiphillic nature and presence of relatively large hydrophobic regions 22. The formation of these ‘liposome-like’ entities functions to exclude the hydrophobic regions from the aqueous surrounding. Due to the presence of lipid monolayers for both LPS and MPL microparticles prepared in the presence of EDC, all studies presented in the manuscript, unless otherwise stated, used microparticles labeled in the presence of crosslinkers. The resulting average number of MPL and LPS units bound per μm2 of silicon was estimated by measuring the lateral dimensions of bumps (protrusions) in 2D images (Figure S5). Limits to the lateral resolution did not permit detection of individual molecules, but allowed us to estimate the average number of ligand units (clusters) per measured area. The average unit diameter was similar for MPL (23±7) and LPS (21±7), as was the number of ligand units per μm2.
Figure 3. Impact of microparticle-bound TLR ligand on cellular uptake and apoptosis.

A) Pseudo-colored scanning electron micrographs show control and MPL- or LPS-microparticle stimulated BMDC 1–3 hr following the introduction of microparticles (top: 8kx mag, bar 10 μm; bottom: 25kx mag; bar 1 μm). B) Confocal images show BMDC labeled with Oregon Green® 488 Phalloidin, DAPI, and incubated with Wheat Germ Agluttinin Alexa Fluor® 594 and TLR ligand-conjugated microparticles for 3 hr. C) Microparticle uptake by BMDC is shown for each population based on the average intensity of particle signal relative to DAPI signal for various ROIs (*p<0.00002). D) Flow cytometry was used to determine the percentage of live BMDC (annexin V−PI−) relative to a non-treatment control following a 24 hr incubation with control or TLR ligand-bound microparticles. Microparticles were introduced at ratios of one cell to every 10, 25 and 50 microparticles (*p<0005, **p<0.03; relative to low dose control microparticles).
Cellular Internalization of TLR-ligand bound microparticles
To study the impact of microparticle-bound MPL and LPS on cellular engulfment of the microparticles, control or ligand-bound microparticles were incubated with BMDC and imaged by SE and confocal microscopy. SE micrographs provided a qualitative assessment of microparticle association at the cell surface (Figure 3A). Representative images of BMDC are shown at low (7000x) and high (25,000x) magnification for each treatment group. The high magnification images show microparticles embedded in DC membrane veils for both control and labeled microparticles. While microparticles are visible on the surface of all groups, protrusions/bumps beneath the plasma membrane, representing internalized microparticles, are also visible, predominately in the MPL-treated group. MPL-microparticles induced the formation of classical DC dendrites. The images were pseudo-colored to highlight the location and interactions of microparticles with the cell surface.
To further support internalization of the microparticles, treated BMDC were imaged by confocal microscopy (Figure 3B). For these experiments, microparticles were simultaneously conjugated with Dylight 594 and either MPL or LPS. Actin labeling of treated cells with Oregon Green® 488 phalloidin is shown in the left column in Figure 3B; DAPI was used as a nuclear dye (blue) and the microparticles are shown in red. The confocal images show high levels of uptake of unlabeled microparticles, and even higher uptake of MPL and LPS labeled microparticles. Using NIS Elements software, the amount of fluorescence per cell (Alexa Fluor 594/DAPI intensity per ROI, n=3–9 ROI/group) was estimated to be 211, 387, and 281 for unlabeled, MPL, and LPS labeled microparticles (Figure 3C). The presence of MPL on the microparticle surface significantly increased uptake compared to control microparticles. The lower density of cells in the LPS-treated group, and the lower levels of uptake compared to the MPL-treated group, are most likely due to LPS-microparticle cytotoxicity in the presence of large numbers of microparticles.
Biocompatibility of LPS bound microparticles
Cytotoxicity of LPS bound to microparticles was assessed by incubating BMDC with 3 different doses of microparticles for 24 hr, followed by labeling both adherent and non-adherent cells with Alexa Fluor 488 annexin V and propidium iodide (Figure 3D). Cells treated with all microparticle presentations displayed an equivalent increase in double positive cells at a dose of 10 microparticles per cell, presented in Figure 3D as percent live cells relative to the no treatment control (blue bars, mean 71+4.8%). However, at a dose of 1:25, LPS-bound microparticles expressed significantly more double positive cells than control and MPL-bound microparticles. While there was no change in the live cell population for control (78%) and MPL-bound (69%) microparticles, there was a large decrease in cell viability in the LPS-microparticle group (39%, p<0.0005). At the high dose of 50 microparticles per cell, all cell populations had a significant decrease in live cells (p<0.03), with BMDC consuming excessive quantities of microparticles. The percentage of live cells was 31%, 12% and 12% for control, MPL and LPS microparticle groups, respectively.
Impact of ligand presentation on DC maturation
To study the impact of ligand binding to silicon on DC maturation, we prepared pre-DCs by culturing murine bone marrow cells in GM-CSF (CD11b+ with 50% of the population expressing CD11c; Figure S1). BMDC were treated with either free (1 μg/ml × 2 ml/well) or microparticle-bound TLR-4 ligands (0.25 μg LPS/1×107 microparticles/well) for 24 hr and surface expression of CD40, CD80, CD86, and MHC class I and II molecules was determined (Figure 4A,B). With respect to surface expression of MHC molecules, microparticle-bound LPS was superior to free LPS (p<0.01) at inducing BMDC surface expression of MHC class II molecules, despite higher levels of free compared to bound LPS (Figure 4A). Both free and bound MPL microparticles stimulated similar levels of MHC class II surface expression (p<0.37), which was significantly greater than that expressed by untreated BMDC (p<0.009) and BMDC treated with control pSi microparticles (p<0.015). Free and microparticle-bound LPS and MPL all stimulated expression of equivalent levels of surface MHC class I molecules, which were significantly greater than control and control microparticle-treated cells.
Figure 4. Comparison of pre-DC stimulation by free and microparticle-bound ligand.
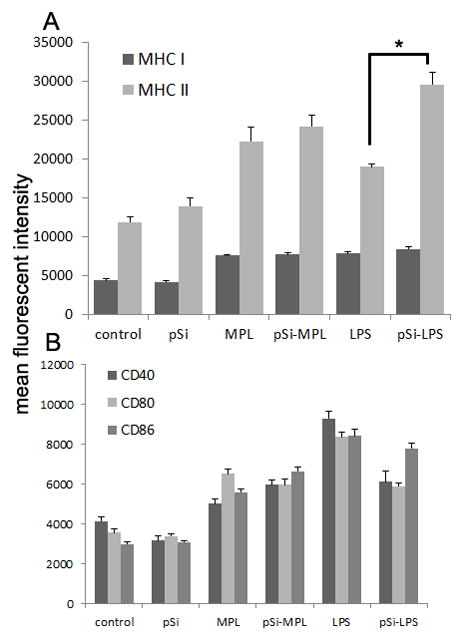
Murine bone marrow cells were incubated with GM-CSF for 5–7 days, then treated with free or microparticle-bound MPL or LPS for 24 hrs. Expression of MHC (A) and co-stimulatory molecules (B) was determined using flow cytometry (*p<0.01).
Based on mean fluorescent intensity, free LPS elicited the highest levels of expression of CD40 and CD80 (p<0.018, p<0.007, respectively) with LPS-labeled microparticles inducing similar high levels of CD86 expression (p<0.18), despite higher levels of free-LPS (8-fold) over bound-LPS (Figure 4B). Both MPL- and LPS-conjugated microparticles induced similar levels of CD40 and CD80, being superior (p<0.05) or equivalent (p<0.18) to free MPL, respectively. Control microparticles did not increase surface expression of CD40, CD80, or CD86.
Pro-inflammatory impact of ligand bound microparticles
The impact of microparticles on DC and macrophage activation was examined by measuring secretion of pro-inflammatory cytokines at various time points after in vitro introduction of microparticles (Figure 5). In addition to TNF-α and IL-6, IL-1β was measured as an indication of NLRP3-dependent inflammasome activation 23. Initial studies in human THP-1 macrophages showed that LPS-microparticles stimulated an equivalent increase in IL-1β at 3 and 24 hr (Figure 5A). The increase in IL-1β secretion was inhibited by the pan caspase inhibitor Z-VAD-FMK, supporting activation of the NLRP3-dependent inflammasome by the ligand-bound microparticles. Surprisingly, inhibition of microparticle uptake by cytochalasin B (10 μg/ml) did not reduce IL-1β secretion in response to LPS-microparticles, in fact, it increased it, indicating that intracellular TLR-4 signaling from within the endosome was not fully responsible for the strong IL-1β secretion in response to LPS-microparticles. Cytochalasin B blocked microparticle phagocytosis in THP-1 cells (and BMDC) in a dose-dependent manner, with 45% inhibition at 10 μg/ml (Figure S7). A similar increase in IL-1β secretion was seen when cytochalasin B was added to cells treated with free LPS (Figure S6). The increase was larger at 3 hr (p<3×10−6), but also present at 24 hr (p<0.04). Treatment of cells with Z-VAD-FMK, cytochalasin B or unlabeled microparticles alone did not increase cytokine levels (Figure S6).
Figure 5. Activation of the inflammasome with TLR ligand-conjugated microparticles.

A) Secretion of IL-1β by human THP-1 macrophages was measured in cell culture media 3 and 24 hr after introduction of LPS-microparticles (pSi-LPS; 1:20; *p<3×10−11 compared to no treatment controls). The influence of the caspase inhibitor Z-VAD-FMK (*p<3×10−10, **p<7×10−11 compared to pSi-LPS) or cytochalasin B (Cyto; *p<3×10−6, **p<0.04 compared to pSi-LPS) on IL-1β levels was determined. ATP was used as a positive control for inflammasome activation (*p<2×10−8, **p<6×10−8 compared to control levels; n=6 per group). B-C) Secretion of IL-1β by GM-CSF-stimulated bone marrow cells was studied at B) various time points after introduction of free, adsorbed, or conjugated LPS (1:25; *p<0.05; **p<0.005; ***p<0.02 compared to equivalent free LPS), or C) different concentrations of control or ligand-bound microparticles at 24 hr (*p<0.01; **p<0.007; ***p<0.02, compared to same concentration control microparticles). D–E) Secretion of TNF-α and IL-6 by GM-CSF-stimulated bone marrow cells 24 hr after introduction of microparticles titrated at 3 concentrations (*,*p<0.04 1:5 MPL-pSi compared to untreated cells and control microparticles; **p<0.0005; ***p<0.006 LPS-pSi compared to same concentration pSi). F) Serum cytokine levels were measured in mice 24 hr after subcutaneous injection with control or ligand-bound microparticles (10 μg LPS equivalent for 5 × 108 LPS-microparticles per mouse; TNF-α *p<0.05; IL-6 *p<0.03; **p<0.003; ***p<0.001; IL-1β * p<0.0002. **p<0.01 compared to basal levels). G–I) Cytokine production was compared in cells treated with free ligand or microparticle-bound ligand (covalent) at 5 microparticles per cell (see C–E for cytokine levels at higher numbers of microparticles). Free MPL and LPS were presented at 1μg/ml in 2 ml media (2 μg ligand per well).
(5-fold,p<0.04; and 2-fold, p<0.02, respectively
The impact of LPS presentation on IL-1β secretion in BMDC was determined for free LPS and LPS either covalently attached or adsorbed to the microparticle surface. All three presentations lead to a dose dependent increase in IL-1β, with bound LPS (covalent or adsorbed) leading to significantly greater (p<0.005 and p<0.02, respectively) increases in IL-1β compared to free LPS at 24 hr (Figure 5B). LPS covalently bound to the microparticle also produced significantly more IL-1β at 3 hrs (p<0.05). The delay in the differentiation between free compared to bound LPS is most likely due to the time it takes microparticles to make contact with the cells compared to freely diffusible LPS. An IL-1β dose response was seen for control and TLR4 ligand-conjugated microparticles at 24 hr, with microparticle bound LPS inducing the highest levels at all doses (Figure 5C). Control microparticles (1×108) were free of endotoxin based on a Limulus amebocyte lysate assay (Figure S3).
Control microparticles did not induce secretion of TNF-α or IL-6 in BMDC at any of the doses tested and the most significant increase in either cytokine by MPL-microparticles was at the low MPL-microparticle dose (5-fold,p<0.04; and 2-fold, p<0.02, respectively) (Figures 5D,E). LPS-microparticles stimulated large increases in both IL-6 and TNF-α at all doses, with the lowest dose leading to the largest increase in IL-6 secretion
Microparticles [5×108; LPS dose equivalent 0.63 mg/kg (12.5 μg)] were administered to BALB/c mice (n=2/group) by subcutaneous injection and the serum was collected 24 hr post-injection for cytokine analysis (Figure 5F). LPS microparticles induced an increase in serum levels of TNF-α (5-fold, p<0.05), IL-6 (60–fold, p<0.001), and IL-1β (15–fold, p<0.01). MPL-microparticles lead to small, yet significant, increases in IL-1β (3-fold, p<0.0002; 9.5 pg/ml compared to 3.2 pg/ml for control mice) and IL-6 (4-fold, p<0.003; 13 pg/ml compared to 3.2 pg/ml for control mice). While control microparticles caused a slight increase in IL-6 (1.7 fold, p<0.03), there was no change in TNF-α or IL-1β serum levels.
A comparison of free and bound LPS and MPL with respect to cytokine production showed that bound LPS, at all microparticle doses, was a stronger stimulator of IL-1β than free LPS (57-fold higher), despite being present at 15–150 times lower dose of LPS per well (Figure 5G). IL-1β levels were identical for free LPS and the low dose of MPL-microparticles, with MPL-microparticles surpassing free LPS induction of IL-1β at higher microparticle doses (6.5–15 times higher). Free MPL did not increase IL-1β production, while a 33–76-fold increase over control levels was seen for bound MPL (p<0.02). While presentation in the microparticle-bound form increased TNF-α secretion for MPL (4.5 times higher, p<0.04), bound and free LPS induced similar levels of TNF-α. MPL was a weak stimulator of IL-6 production, both in the free and bound states, while LPS induced high levels of IL-6 secretion regardless of presentation, with higher levels seen for free LPS.
Enhanced BMDC migration to the lymph node following treatment with ligand-bound microparticles
In addition to stimulating DC maturation, the ability of TLR-4 ligand-bound microparticles to stimulate DC migration to the draining lymph node was studied. Control or ligand-bound pSi microparticles were incubated with GM-CSF-stimulated BALB/c BMDC for 3 hr, followed by labeling with Cell Tracker™ Orange, PKH26, or CellTrace™ Green. BMDC were then injected subcutaneously into the hind leg via hock injection into a syngeneic mouse and the ipsilateral inguinal and popliteal lymph nodes were collected 24 hr post injection (n=5). Cell suspensions were analyzed using flow cytometry (Figure 6). The combined results of two independent experiments (n=5 mice per group), show that 3.2% of the cells in the combined inguinal and popliteal lymph nodes were newly migrated control BMDC (Figure 6A). The percent of newly migrated BMDC decreased 2-fold for BMDC treated with control microparticles (1.4%), but increased by 2.8- and 2.3-fold for BMDC treated with MPL (9.1%) and LPS (7.5%) microparticles, respectively.
Figure 6. Migration of BMDC to the draining lymph node following subcutaneous injection of microparticle-loaded cells.

A) The percent of combined inguinal and popliteal lymph node cells that are positive for injected (CellTrace™ Green- or PKH26-labeled) GM-CSF-stimulated BMDC 24 hr after injection of mice with 3–5 × 106 control or microparticle-laden BMDC via ipsilateral hock injection (n=5 per group; *p<0.001 compared to BMDC, **p<0.0007 compared to no treatment controls). B) Comparison of microparticle-laden BMDC migration to the popliteal and inguinal lymph nodes following hock injection (*p<0.03, **p<0.04 for BMDC-pSi-MPL compared to non-treated controls). C) Fluorescent contour plots showing inguinal lymph node cell labeling for injected CellTracker™ Orange-labeled BMDC and LPS-FITC bound microparticles (preloaded into the BMDC prior to injection) 24 hr after injection of BALB/c mice with 5 0 × 106 BMDC via hock injection.
For half of the mice included in the migration study, inguinal and popliteal lymph nodes were analyzed independently. BMDC that was pre-loaded with MPL-bound microparticles had significantly more BMDC in both the inquinal (p<0.02) and popliteal (p<0.04) lymph nodes compared to animals treated with BMDC pre-loaded with control microparticles. There were similar amounts of migrating BMDC in the inquinal and popliteal lymph nodes from mice treated with MPL-microparticles.
To verify that injected BMDC within the draining lymph node were loaded with microparticles, CellTracker™ Orange labeled BMDC were loaded with LPS-FITC-bound microparticles and the fluorescent intensity in the green channel of lymph node resident BMDC was determined by flow cytometry (Figure 6C). For mice injected with control BMDC, 11.4% of the lymph node cells in the selected scatter gate were positive for the CellTracker™ Orange label. Mice injected with LPS-FITC-pSi microparticle-laden BMDC showed higher levels of BMDC within the draining lymph node (37.8%). The shift of BMDCs towards the green channel (374 compared to 190 for control BMDC) supported the presence of labeled microparticles within the injected BMDCs. Only 4% of the gated lymph node population was positive for labeled BMDC in a control mouse injected with unlabeled microparticle-laden BMDC (data not shown).
Imaging the lymphatic microenvironment using scanning electron microscopy
Scanning electron micrographs show the size and morphology of an inguinal lymph node and its surrounding tissue (Figure 7A, top left). The excised and processed murine lymph node had dimensions of approximately 2 × 3 mm. A higher magnification scanning electron micrograph in the bottom left image illustrates a lymphocyte-rich region of the lymph node, with a higher magnification image on its right showing a single blue pseudo-colored lymphocyte.
Figure 7. Imaging lymphocyte-rich regions of the lymph node.

A) Top left: Scanning electron micrograph showing a cross-section of the inquinal lymph node (BALB/c; 44x mag, 10 kV). Top right: compiled two-photon micrographs showing T cell rich regions of the popliteal lymph node 24 hr after intravascular injection of CellTrace™ Green-labeled T cells (1×107). Bottom left: lymphocyte-rich region within the lymph node (5000x mag, 30 kV). Bottom right: high magnification micrograph highlighting a single lymphocyte pseudo-colored in blue (15,000x mag, 10 kV). B) Two-photon microscopy was used to image an intact BALB/c popliteal lymph node 24 hr post subcutaneous injection of mice with 5×106 ex-vivo-bone marrow-derived APC (GM-CSF cultured; CellTracker™ Orange) and 18 hr after intravenous injection with 1×107 splenetic T cells (CellTrace™ Green). The top left image is an enlargement of the boxed region in the image to the right. C) 2D micrographs of the ipsilateral popliteal lymph node from a C57BL/6 mouse injected (via footpad) with OVA peptide, LPS-microparticle primed CellTracker™ Orange-labeled BMDC (C57BL/6) and CellTrace™ Green-labeled T cells (OT1-derived) showing separate and merged channels 24 hr after injection. D) A channel-merged 3D z-stack showing the cortex of a C57BL/6 popliteal lymph node from a mouse injected as described in “C”. E) Single and merged channels of a C57BL/6 popliteal lymph node from a mouse injected with OVA peptide, LPS-microparticle primed orange-labeled BMDC (C57BL/6; 24 hr) and green-labeled T cells (OT1-derived; 18 hr). The boxed region in the lower left image in enlarged in the image to the right.
T cell zones within the lymph node were mapped by injecting BALB/c mice (intravenous) with syngeneic splenic T cells labeled with CellTrace™ CSFE. 24 hr post injection, the intact popliteal lymph node was imaged using two-photon microscopy. Images were taken using a 40x objective, then aligned to map out the cortex and paracortical regions of the lymph node. T cells were found in clusters, with T cell-independent regions representing B cell follicles.
Two-photon imaging of newly migrated BMDC and T cells in the intact lymph node
CellTracker™ Orange-labeled BALB/c-derived, GM-CSF-stimulated BMDC were injected into the footpad of a syngeneic mouse, followed 6 hr post injection by an intravenous injection of spleen-derived T cells labeled with CellTrace™ CSFE. Two photon imaging of the draining popliteal lymph node 24 hours following the second injection shows migration of both T cells and BMDC into similar regions of the lymph node (Figure 7B). While the infiltrating immune cells are in similar locations, there is little overlap in fluorescent signals, supporting little interaction between naive T cells and non-primed BMDC. However, co-localization in T cell zones supports the potential for inducing T cell-mediated immune responses in the presence of appropriate antigens.
To determine if antigen-primed BMDC and T cells expressing the corresponding TCR co-localize following injection, CellTracker™ Orange C57BL/5 BMDC, primed with OVA SIINFEKL peptide and LPS-microparticles, were injected subcutaneously into C57BL/6 mice, followed 6 hr post-injection with an intravenous injection of CellTrace™ Green T cells derived from the spleen of C57BL/6-Tg(TcraTcrb) mice. Two-photon, merged and unmerged images of the draining lymph node, 24 hr after the first injection, are shown in Figure 7C. Figure 7D is a 3D composite of the cortex showing independent BMDC (red) and T cells (green), however, the majority of the cells appear yellow, supporting in vivo interaction between BMDC presenting OVA peptide in association with MHC class I molecules and TCR transgenic T cells from OT-1 mice. Confocal imaging of a histological section from the popliteal lymph node from a unique C57BL/6 mouse similarly injected with peptide, LPS-microparticle treated BMDC and OT-1 T cells shows co-localization of treated BMDC and T cells, and formation of immunological synapses (i.e. BMDC surrounded by T cells; Figure 7E).
Impact of microparticles on cellular interactions and activation
Continuous cross-talk exists between immune cells and is mediated in part by membrane-to-membrane protein interactions. Another form of communication, and a means of sharing intracellular signals, is via what are known as ‘tunneling nanotubes’ (TNT) 24. An array of TNT is shown connecting two BMDC in the scanning electron micrograph shown in Figure 8A. TNT are very common among phagocytic cells, and we have previously documented cellular communication through TNT by endothelial cells 25. The frequency of TNT occurrence is greater for APC than endothelial cells and is present in most SEM images of macrophages and dendritic cells. A red pseudo-colored microparticle can be seen on the surface of the lower cell one hour after introducing particles to the cell culture media, supporting continued cellular communication in the presence of microparticles. Figure 8B is a pseudo-colored micrograph of a lymphocyte-rich region of a lymph node emphasizing in vivo cellular interactions between DC and lymphocytes.
Figure 8. Cellular interactions among immune cells.

A) SE micrograph showing in vitro cell-to-cell interactions (i.e. Tunneling NanoTubes) between adjacent dendritic cells after treatment with microparticles (left; a silicon microparticle is pseudo-colored red; 8400x mag; 15 kV; bar 5 μm). B) SE micrograph showing in vivo cell-to-cell communication by means of pseudopods in a lymphocyte-rich region of the inguinal lymph node (DC pseudo-colored in blue and a lymphocyte in green (5800x mag; 8 keV; bar 5 μm). C–D) TE micrographs show two OVA peptide, LPS-microparticle primed BMDC (C57BL/6; 3 hr) and adjacent OT-1 transgenic T cells following 1 hr of co-culture at two magnification levels (1200x and 4000x mag; bars 1 μm). C) Arrows indicate the presence of 4 intracellular silicon microparticles in the BMDC. D) An immunological synapse is seen in the enlarged (bottom) image. E) Flow cytometry analysis of intracellular IFN-γ levels in OT-1 CD8+ T cells following stimulation of C57BL/6 BMDC with free or microparticle-presented OVA peptide and LPS BMDC for 4 hr.
C57BL/6 BMDC, pulsed with OVA SIINFEKL peptide and LPS-microparticles, were incubated with OT-1 splenic T cells for one hour at 37°C. The presence of intracellular silicon microparticles in BMDC (see arrows in Figure 7C, bottom image) and cellular interactions with T cells, as shown in TE images (Figure 8C, D), supports MHC-peptide presentation by BMDC to T cells. Two sets of interacting BMDC and T cells are shown (Figure 8C, D), with an example of an immunological synapses presented in Figure 8D. BMDC are identified by the presence of dendrites protruding from the cells, while the smaller T cells are distinguished by the presence of more prominent nuclei.
In vitro activation of OT-1 CD8+ T cells by C57BL/6 BMDC cells treated with OVA SIINFEKL peptide-loaded microparticles was investigated by measuring intracellular IFN-γ levels in T cells following a 4 hr incubation with BMDC (Figure 8E). Peptide-pulsed control and LPS-matured BMDC stimulated 2.5- and 5-fold increases in IFN-γ levels, respectively. Unloaded control and LPS-microparticles induced none or minimal changes (1.7-fold increase) in IFN-γ levels, respectively. Peptide-loaded microparticles, with and without LPS, stimulated 3- and 10-fold increases in intracellular IFN-γ, respectively, supporting microparticle-based delivery of peptide and LPS microparticle-driven activation of immune cells.
Discussion
pSi microparticles are ideal for delivering complex payloads, such as free or nanoparticle-loaded antigens and adjuvant. Here we introduce a microparticle-based delivery vehicle that presents PAMP molecules, enhancing uptake, activation, and migration of APCs to the draining lymph node. Previously, Falo et al. 26 reported that solid inert beads with surface adsorbed antigen and a diameter of 1 μm were optimal for activation of CD8+ T cell responses, with particles less than 0.5 μm producing weaker response. Kanchan and Panda 27 reported that polylactic acid microparticles (2–8 μm) promote humoral responses, while nanoparticles (200–600 nm) promoted cellular Th1 type responses. However, while this group reported that polylactic acid particles larger than 0.5 μm are poorly internalized, in this study, 1 μm pSi microparticles are abundantly internalized by BMDC, with LPS and MPL enhancing uptake.
Kobayashi et al. 28 reported that cytokine (GM-CSF, IL-4, and TNF)-induced maturation of DCs decreased their phagocytic potential by 13% and enhanced their migration to mesenteric lymph nodes. Conversely, we found that addition of TNF-α to GM-CSF-supplemented media did not significantly alter the capacity of BMDC to phagocytose pSi microparticles, while both IL-4 and LPS increased microparticle association (10% and 20%, respectively). Since non or loosely adherent DCs are considered ‘qualified’ DCs 29, we also compared microparticle uptake between adherent and non-adherent cells and found that non-adherent cells, treated with either LPS or IL-4, had significantly greater (p<0.04) uptake than their adherent counterparts. Phenotypic markers on the two populations were similar, with a larger representation of CD14 and Ly6C on the adherent population. Consistent with reports that TLR engagement stimulates pathogen internalization 30, we also demonstrated that LPS- and MPL-bound microparticles have greater BMDC internalization than control microparticles.
Using AFM to study the nature of LPS and MPL binding to silicon, we found uniform packing of LPS for both covalent and adsorbed presentations. Identical vertical heights for bound LPS indicated the same molecular orientation on the silicon surface. Based on the thickness of the LPS layer (2.9±0.3 nm), which is roughly half that reported for an LPS bilayer (7.0 nm) 31, LPS most likely exists as a bound monolayer sporadically fringed with LPS lamellar vesicles. AFM imaging of MPL-bound silicon revealed a closely packed, 2-D solid-like organization of lipid A molecules, similar to that reported for liposomal DPPC monolayers 32. The vertical height of the lipid layer was 1.7±0.9 nm, similar to the 2.3 nm height estimated for the lipid A component of LPS based on the presence of C14 acyl chains 33. Similar numbers of molecular entities for EDC-bound MPL and LPS supports similar numbers of molecules bound per unit area of silicon.
Chitta et al. 34 reported that human peripheral blood monocytes cultured in GM-CSF acquire a veiled morphology characteristic of immature DC. We similarly found that stimulation of murine bone marrow cells with GM-CSF establishes cells with ample membrane extensions, creating a veiled morphology. Activation of the resulting BMDC with MPL-labeled microparticles triggered elongation of membrane extensions, leading to the formation of classical DC dendrons, supporting MPL-microparticle-induced maturation of BMDC.
West et al. 35 reported that simultaneous exposure to antigens and LPS increases antigen presentation on both class I and II MHC molecules. Others have reported that LPS favors differentiation towards M1-like macrophages, favoring IL-12 driven Th1 responses with enhanced up-regulation of MHC class I molecules (54, 61, 62). We found that association of LPS and MPL, either free or microparticle-bound, increased surface expression of both class I and II MHC molecules on murine BMDC in vitro, with microparticle-bound LPS being superior to free LPS with respect to up-regulation of surface MHC class II molecules. Surprisingly, microparticle-bound MPL was also superior to free LPS with respect to enhancing surface MHC class II molecule expression (p<0.04). Our findings showing increased levels of surface MHC molecules following treatment with MPL- and LPS-bound microparticles are consistent with claims that LPS shuts down MHC complex endocytosis 36, extending the half-life of MHC complexes on the cell surface 37. Surface expression of CD40, CD80, and CD86 were increased in the presence of free and microparticle-bound TLR-4 ligands, with free LPS having the greatest impact.
pSi microparticles, including control microparticles, stimulated secretion of IL-1β from macrophages in a time and dose-dependent manner, with a 40-fold greater induction by LPS-microparticles compared to free LPS. Blockade of IL-1β secretion by the caspase inhibitor Z-VAD-FMK supports activation of the NLRP3-dependent inflammasome by TLR-4 ligand-bound microparticles. Both covalent and adsorbed LPS induced large increases in IL-1β, with similar efficiencies owing to similar binding densities and presentation.
We found that LPS-microparticle internalization was not fully responsible for enhanced cytokine secretion. In fact, treatment of macrophages with cytochalasin B stimulated higher levels of IL-1β secretion compared to that achieved with LPS-microparticles alone. Arend et al. 38 also reported that cytochalasin B enhances LPS induced IL-1 secretion. It has also been demonstrated that IL-1β transcripts are associated with actin microfilaments in the cell periphery, and that cytochalasin B disrupts the microfilaments, releasing the transcripts 39. LPS is reported to enhance IL-1β translation by redistributing mRNA to polysomes 39, thereby supporting a collaborative effort of LPS and cytochalasin B on stimulating IL1-β expression. In addition, we have previously demonstrated that cytochalasin B results in “frustrated phagocytosis” of pSi microparticles by endothelial cells, locking the microparticles on the cell surface in actin cups. It is possible that continued adhesion of microparticles to the cell surface could stimulate sustained extracellular activation of TLR-4, further contributing to enhanced inflammasome activation in BMDC in the presence of cytochalasin B.
LPS-bound microparticles also induced secretion of the pro-inflammatory cytokines TNF-α and IL-6, both in vitro and in vivo. While MPL-microparticles induced a large increase in IL-1β secretion (76-fold), there was no increase in IL-6 and only a 4-fold increase in TNF-α secretion, suggesting a strong mechanism to activate DC without stimulating a potential toxic inflammatory response. While extremely potent at activating BMDC, LPS-bound microparticles were also fairly cytotoxic, inducing high levels of cytokine production and concentration-dependent cell death.
Harrel et al. mapped lymphatic drainage of the hind leg in mice and found that unlike rats, in which the hind foot drains primarily through the popliteal to the iliac lymph node 40, in mice, the hind foot drains equally to the popliteal and inguinal lymph nodes 41. In this study, MPL-microparticle-laden BMDC injected subcutaneously into the hind-leg hock resulted in BMDC migration to both the popliteal and inguinal lymph nodes. The percentage of newly migrated BMDC in the draining lymph nodes increased by more than 200% following BMDC uptake of either MPL- or LPS-microparticles.
While dendritic cells are migratory cells and can travel from peripheral organs to lymphoid nodes via afferent lymphatics and high endothelial venules 42, T cells require intravenous administration and enter the paracortex through the high endothelial venules 43. Here, we first demonstrated that intravenous injection of T cells leads to paracortical and cortical localization, distinct from areas occupied by B cell follicles. We then co-introduced labeled BMDC (subcutaneous) and T cells (intravenous), and imaged their distribution and prevalence in the paracortex, and found that co-administration of BMDC and T cells led to migration of the cells to similar regions in the lymph node, with discrete fluorescent signals coming from non-associating BMDC and T cells. To stimulate in vivo interactions between BMDC and T cells, we inoculated C57BL/6 mice with antigen-pulsed BMDC (i.e. OVA peptide and LPS-microparticle treated) and splenic OT-1 CD8+ TCR transgenic T cells. This led to spectral overlap in signals from BMDC and T cells within the draining lymph node, indicating in vivo cellular associations between antigen-primed BMDC and TCR transgenic T cells. In addition, immunological synapses were imaged both in vitro and in vivo following stimulation of BMDC with antigen and LPS-microparticles. We demonstrated functionality of the synapse by showing that BMDC, loaded with antigen and LPS microparticles, stimulated CD8+ T cells, leading to production of intracellular IFN-γ.
In summary, the presence of TLR-4 ligands on pSi microparticles greatly enhances microparticle uptake by BMDC and leads to cellular activation, both with respect to presentation of surface co-stimulatory and MHC molecules and production of cytokines, including IL-1β, supporting activation of the inflammasome. Both MPL- and LPS-bound microparticles enhance BMDC migration to the draining lymph node, with the addition of antigen stimulating BMDC-T cell interactions. In addition, incubation of CD8+ T cells with BMDC carrying antigen-loaded microparticles induced production of intracellular IFN-γ, supporting antigen presentation leading to T cell activation. The clinical benefit of using microparticles to present PAMPs to APC is presentation of immune stimulating factors to the same cells that are to be presented with antigen, creating a more localized response that occurs in the context of the desired antigenic target. An additional benefit of using particles for vaccine development is that once optimized, the vehicle can be loaded with diverse antigens, permitting vaccine development for a broad range of diseases. We are currently working on creating particle-based vaccines for HIV therapy and on the development of therapeutic cancer vaccines.
Supplementary Material
Figure 2. Characterization of ligand binding to silicon.
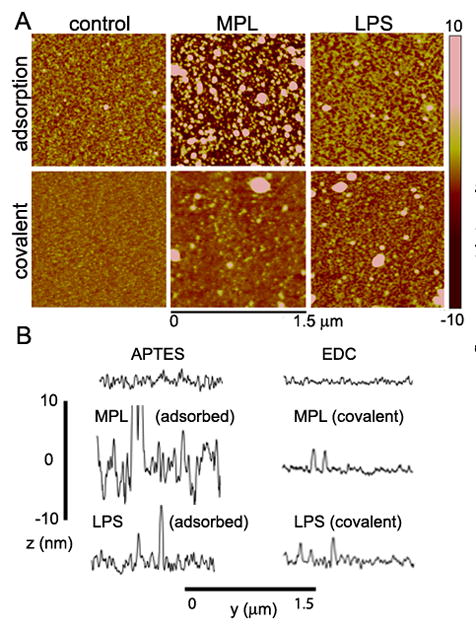
A) AFM images show MPL and LPS grafting densities and conformation on APTES-modified silicon chips following introduction by adsorption or covalent cross-linking. B) Line sections are shown for each AFM sample to illustrate the thickness and uniformity of ligand binding.
Acknowledgments
We are grateful for the use and assistance provided by the Flow Cytometry and Advanced Tissue and Cellular Microscopy Cores at TMHRI. We acknowledge use of the TMHRI Scanning Electron Microscopy core, which is directed and managed by authors of this manuscript. We also thank James P. Barrish, Technical Specialist in the Department of Pathology Electron Microscopy Facility at Texas Children’s Hospital, for TE microscopy sample preparation and imaging. This research was supported by funding provided by TMHRI, and ST Wong is supported by NIH U54CA149169.
Footnotes
Supplemental Information Available
Data regarding the influence of cytokines on APC phenotype, LPS quantitation, LAL analysis, AFM images and surface measurements, additional IL-1β secretion data, and cytochalasin blockade of phagocytosis data are included in online supplemental data. This information is available free of charge via the internet at http://pubs.acs.org/.
References
- 1.Sanarico N, Ciaramella A, Sacchi A, Bernasconi D, Bossu P, Mariani F, Colizzi V, Vendetti S. Human monocyte-derived dendritic cells differentiated in the presence of IL-2 produce proinflammatory cytokines and prime Th1 immune response. J Leukoc Biol. 2006;80(3):555–62. doi: 10.1189/jlb.1105690. [DOI] [PubMed] [Google Scholar]
- 2.Ueno H, Klechevsky E, Schmitt N, Ni L, Flamar AL, Zurawski S, Zurawski G, Palucka K, Banchereau J, Oh S. Targeting human dendritic cell subsets for improved vaccines. Semin Immunol. 2011;23(1):21–7. doi: 10.1016/j.smim.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Maldonado-Lopez R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, Thielemans K, Leo O, Urbain J, Moser M. CD8alpha+ and CD8alpha- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. 1999;189(3):587–92. doi: 10.1084/jem.189.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci U S A. 1999;96(3):1036–41. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315(5808):107–11. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]; (d) Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L, Shortman K. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med. 1992;176(1):47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Maldonado-Lopez R, De Smedt T, Pajak B, Heirman C, Thielemans K, Leo O, Urbain J, Maliszewski CR, Moser M. Role of CD8alpha+ and CD8alpha- dendritic cells in the induction of primary immune responses in vivo. J Leukoc Biol. 1999;66(2):242–6. doi: 10.1002/jlb.66.2.242. [DOI] [PubMed] [Google Scholar]
- 4.(a) Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106(3):255–8. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]; (b) Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 5.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 6.Manicassamy S, Pulendran B. Modulation of adaptive immunity with Toll-like receptors. Semin Immunol. 2009;21(4):185–93. doi: 10.1016/j.smim.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Watts C, Zaru R, Prescott AR, Wallin RP, West MA. Proximal effects of Toll-like receptor activation in dendritic cells. Curr Opin Immunol. 2007;19(1):73–8. doi: 10.1016/j.coi.2006.11.014. [DOI] [PubMed] [Google Scholar]; (b) Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440(7085):808–12. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 8.Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, Elhasi S, Samuel J, Lavasanifar A. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26(39):5046–57. doi: 10.1016/j.vaccine.2008.07.035. [DOI] [PubMed] [Google Scholar]
- 9.Baldrick P, Richardson D, Elliott G, Wheeler AW. Safety evaluation of monophosphoryl lipid A (MPL): an immunostimulatory adjuvant. Regul Toxicol Pharmacol. 2002;35(3):398–413. doi: 10.1006/rtph.2002.1541. [DOI] [PubMed] [Google Scholar]
- 10.(a) Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]; (b) Martin M, Michalek SM, Katz J. Role of innate immune factors in the adjuvant activity of monophosphoryl lipid A. Infect Immun. 2003;71(5):2498–507. doi: 10.1128/IAI.71.5.2498-2507.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ismaili J, Rennesson J, Aksoy E, Vekemans J, Vincart B, Amraoui Z, Van Laethem F, Goldman M, Dubois PM. Monophosphoryl lipid A activates both human dendritic cells and T cells. J Immunol. 2002;168(2):926–32. doi: 10.4049/jimmunol.168.2.926. [DOI] [PubMed] [Google Scholar]
- 11.Marciani DJ. Vaccine adjuvants: role and mechanisms of action in vaccine immunogenicity. Drug Discov Today. 2003;8(20):934–43. doi: 10.1016/s1359-6446(03)02864-2. [DOI] [PubMed] [Google Scholar]
- 12.Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, Mellman I, Ledizet M, Fikrig E, Flavell RA, Fahmy TM. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27(23):3013–21. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24(3):317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 14.(a) Serda RE, Ferrati S, Godin B, Tasciotti E, Liu X, Ferrari M. Mitotic trafficking of silicon microparticles. Nanoscale. 2009;1(2):250–9. doi: 10.1039/b9nr00138g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Serda RE, Mack A, Pulikkathara M, Zaske AM, Chiappini C, Fakhoury J, Webb D, Godin B, Conyers JL, Liu XW, Bankson JA, Ferrari M. Cellular Association and Assembly of a Multistage Delivery System. Small. 2010 doi: 10.1002/smll.201000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han HD, Shahzad MM, Liu X, Bhavane R, Gu J, Fakhoury JR, Chiappini C, Lu C, Matsuo K, Godin B, Stone RL, Nick AM, Lopez-Berestein G, Sood AK, Ferrari M. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70(9):3687–96. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serda RE, Gu J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M. The association of silicon microparticles with endothelial cells in drug delivery to the vasculature. Biomaterials. 2009;30(13):2440–8. doi: 10.1016/j.biomaterials.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 17.Serda RE, Mack A, van de Ven AL, Ferrati S, Dunner K, Jr, Godin B, Chiappini C, Landry M, Brousseau L, Liu X, Bean AJ, Ferrari M. Logic-Embedded Vectors for Intracellular Partitioning, Endosomal Escape, and Exocytosis of Nanoparticles. Small. 2010 doi: 10.1002/smll.201000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adriani G, de Tullio MD, Ferrari M, Hussain F, Pascazio G, Liu X, Decuzzi P. The preferential targeting of the diseased microvasculature by disk-like particles. Biomaterials. 2012 doi: 10.1016/j.biomaterials.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamala T. Hock immunization: a humane alternative to mouse footpad injections. J Immunol Methods. 2007;328(1–2):204–14. doi: 10.1016/j.jim.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiappini C, Tasciotti E, Fakhoury JR, Fine D, Pullan L, Wang YC, Fu L, Liu X, Ferrari M. Tailored porous silicon microparticles: fabrication and properties. Chemphyschem. 2010;11(5):1029–35. doi: 10.1002/cphc.200900914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss J, Burnham NA, Camesano TA. Atomic force microscopy study of the role of LPS O-antigen on adhesion of E. coli. J Mol Recognit. 2009;22(5):347–55. doi: 10.1002/jmr.955. [DOI] [PubMed] [Google Scholar]
- 22.Ulrich J. MPL immunostimlant: Adjuvant formulations. Vaccine Adjuvants. 2000;42:273–282. [Google Scholar]
- 23.Morishige T, Yoshioka Y, Inakura H, Tanabe A, Yao X, Narimatsu S, Monobe Y, Imazawa T, Tsunoda S, Tsutsumi Y, Mukai Y, Okada N, Nakagawa S. The effect of surface modification of amorphous silica particles on NLRP3 inflammasome mediated IL-1beta production, ROS production and endosomal rupture. Biomaterials. 2010;31(26):6833–42. doi: 10.1016/j.biomaterials.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 24.(a) Gerdes HH, Bukoreshtliev NV, Barroso JF. Tunneling nanotubes: a new route for the exchange of components between animal cells. FEBS Lett. 2007;581(11):2194–201. doi: 10.1016/j.febslet.2007.03.071. [DOI] [PubMed] [Google Scholar]; (b) Gurke S, Barroso JF, Gerdes HH. The art of cellular communication: tunneling nanotubes bridge the divide. Histochem Cell Biol. 2008;129(5):539–50. doi: 10.1007/s00418-008-0412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrati S, Shamsudeen S, Summers H, Rees P, Abbey JVA, Schmulen J, Liu X, Wong STC, Bean AJ, Ferrari M, Serda RE. Inter-Endothelial Transport of Microvectors using Cellular Shuttles and Tunneling NanoTubes. Small. 2012 doi: 10.1002/smll.201200472. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falo LD, Jr, Kovacsovics-Bankowski M, Thompson K, Rock KL. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat Med. 1995;1(7):649–53. doi: 10.1038/nm0795-649. [DOI] [PubMed] [Google Scholar]
- 27.Kanchan V, Panda AK. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials. 2007;28(35):5344–57. doi: 10.1016/j.biomaterials.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi H, Miura S, Nagata H, Tsuzuki Y, Hokari R, Ogino T, Watanabe C, Azuma T, Ishii H. In situ demonstration of dendritic cell migration from rat intestine to mesenteric lymph nodes: relationships to maturation and role of chemokines. J Leukoc Biol. 2004;75(3):434–42. doi: 10.1189/jlb.0603250. [DOI] [PubMed] [Google Scholar]
- 29.Li GB, Lu GX. Adherent cells in granulocyte-macrophage colony-stimulating factor-induced bone marrow-derived dendritic cell culture system are qualified dendritic cells. Cellular immunology. 2010;264(1):4–6. doi: 10.1016/j.cellimm.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304(5673):1014–8. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 31.Tong J, McIntosh TJ. Structure of supported bilayers composed of lipopolysaccharides and bacterial phospholipids: raft formation and implications for bacterial resistance. Biophysical journal. 2004;86(6):3759–71. doi: 10.1529/biophysj.103.037507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deleu M, Nott K, Brasseur R, Jacques P, Thonart P, Dufrene YF. Imaging mixed lipid monolayers by dynamic atomic force microscopy. Biochim Biophys Acta. 2001;1513(1):55–62. doi: 10.1016/s0005-2736(01)00337-6. [DOI] [PubMed] [Google Scholar]
- 33.Lee AG. Lipid-protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta. 2003;1612(1):1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 34.Chitta S, Santambrogio L, Stern LJ. GMCSF in the absence of other cytokines sustains human dendritic cell precursors with T cell regulatory activity and capacity to differentiate into functional dendritic cells. Immunol Lett. 2008;116(1):41–54. doi: 10.1016/j.imlet.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 35.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305(5687):1153–7. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 36.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388(6644):782–7. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 37.Wilson NS, El-Sukkari D, Villadangos JA. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood. 2004;103(6):2187–95. doi: 10.1182/blood-2003-08-2729. [DOI] [PubMed] [Google Scholar]
- 38.Arend WP, Massoni RJ. Characteristics of bacterial lipopolysaccharide induction of interleukin 1 synthesis and secretion by human monocytes. Clin Exp Immunol. 1986;64(3):656–64. [PMC free article] [PubMed] [Google Scholar]
- 39.Bocker U, Sirenko OI, Morris JS, Sartor RB, Singer MV, Haskill JS, Watson JM. Expression and localization of IL-1beta mRNA is interrelated with cytoskeletal rearrangement in monocytes stimulated by adherence: a light microscopy in situ hybridization study. Immunology and cell biology. 2001;79(5):444–53. doi: 10.1046/j.1440-1711.2001.01031.x. [DOI] [PubMed] [Google Scholar]
- 40.Tilney NL. Patterns of lymphatic drainage in the adult laboratory rat. J Anat. 1971;109(Pt 3):369–83. [PMC free article] [PubMed] [Google Scholar]
- 41.Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332(1–2):170–4. doi: 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Willimann K, Legler DF, Loetscher M, Roos RS, Delgado MB, Clark-Lewis I, Baggiolini M, Moser B. The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur J Immunol. 1998;28(6):2025–34. doi: 10.1002/(SICI)1521-4141(199806)28:06<2025::AID-IMMU2025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (b) Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95(1):258–63. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumen C, Mempel TR, Mazo IB, von Andrian UH. Intravital microscopy: visualizing immunity in context. Immunity. 2004;21(3):315–29. doi: 10.1016/j.immuni.2004.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.