

Abstract
Pleural and peritoneal mesotheliomas (MMs) are chemoresistant tumors with no effective therapeutic strategies. The authors first injected multifunctional, acid-prepared mesoporous spheres (APMS), microparticles functionalized with tetraethylene glycol oligomers, intraperitoneally into rodents. Biodistribution of APMS was observed in major organs, peritoneal lavage fluid (PLF), and urine of normal mice and rats. After verification of increased mesothelin in human mesotheliomas injected into severe combined immunodeficient (SCID) mice, APMS were then functionalized with an antibody to mesothelin (APMS-MB) or bovine serum albumin (BSA), a nonspecific protein control, and tumor targeting was evaluated by inductively coupled plasma mass spectrometry and multifluorescence confocal microscopy. Some APMS were initially cleared via the urine over a 24 hr period, and small amounts were observed in liver, spleen, and kidneys at 24 hr and 6 days. Targeting with APMS-MB increased APMS uptake in mesenteric tumors at 6 days. Approximately 10% to 12% of the initially injected amount was observed in both spheroid and mesenteric MM at this time point. The data suggest that localized delivery of APMS-MB into the peritoneal cavity after encapsulation of drugs, DNA, or macromolecules is a novel therapeutic approach for MM and other tumors (ovarian and pancreatic) that overexpress mesothelin.
Keywords: malignant mesothelioma, targeted microparticle, mesothelin, amorphous silica
Occupational exposure to airborne asbestos is associated with the development of numerous diseases, including pulmonary and pleural fibrosis, lung cancer, and malignant mesotheliomas (MMs) (Mossman et al. 1990). MM is an aggressive tumor arising from mesothelial cells lining the pleura, peritoneum, and pericardium. In advanced stage MM (stages III and IV), current treatment options are limited to draining of retained fluid from the chest cavity to relieve associated pain, debulking of the tumor mass by surgical resection, and a combination of systemic administration of cisplatin drugs alone or in combination with one or more of several approved alkylating agents or folate agonists such as pemetrexed (Grosso and Scagliotti 2012). The use of radiation therapy is of debate. In the case of diffuse malignant peritoneal mesothelioma, radical debulking surgery is followed by heated intraperitoneal (IP) chemotherapy (Sugarbaker et al. 2006). Local administration allows for a higher concentration of certain drugs such as cisplatin and doxorubicin (DOX) (Adriamycin) with lower systemic side effects, whereas the addition of heat increases the cytotoxic effect of chemotherapeutic agents (Mirarabshahii et al. 2012).
Treatment of chemoresistant tumors, such as MM, is usually hindered by the systemic toxicity of intravenously and intraperitoneally administered chemotherapeutics, such as DOX. Intracavitary chemotherapy (Hesdorffer, Chabot, DeRosa, et al. 2008; Hesdorffer, Chabot, Keohan, et al. 2008; Van der Speeten, Stuart, Mahteme, et al. 2009; Van der Speeten, Stuart, and Sugarbaker 2009), including the administration of hyperthermic drugs (van Ruth, Baas, et al. 2003; van Ruth, van Tellingen, et al. 2003; Richards et al. 2006), has increased patient survival time by an average of only 6 months. Thus, there is a need for more effective treatment strategies (Zauderer and Krug 2011). Toxic, systemic administration of chemotherapeutics or radiation in combination with the low therapeutic efficacy of current treatment regimens suggests that localized delivery methods are needed to target MMs (Albelda 1997). To help meet this need, we hypothesized that acid-prepared mesoporous spheres (APMS) are an effective drug delivery device with tissue targeting potential.
APMS microparticles are amorphous silica particles (1–2 µm diameter) with a disordered pore structure, a large specific surface area, and a large pore volume. APMS particle diameter and pore size are easily tunable for optimal delivery of specific agents (Gallis and Landry 2002; Nassivera et al. 2002). As previously published, these characteristics make APMS an ideal vehicle for carrying chemotherapeutic agents, DNA plasmids, small interfering RNA (siRNA), or other macromolecules (Blumen et al. 2007; Cheng et al. 2010). Most important, amorphous silicas produce no chronic adverse biological responses in contrast to crystalline silicas, presumably because of their lack of crystalline structure and increased solubility over time (Warheit 2001). In addition, the external surfaces of APMS are easily modified with tetraethylene glycol or antibodies to facilitate targeting and uptake of the particles by cells (Cheng et al. 2010; Cheng et al. 2012). Finally, we have shown that APMS can be targeted with an anti-mesothelin antibody in vitro, suggesting their use in vivo to target MM tumors overexpressing mesothelin (Cheng et al. 2010).
Mesothelin is a differentiation antigen, with expression normally limited to the mesothelial cells lining the pleura, pericardium, and peritoneum (Chang and Pastan 1996; Ho et al. 2007). However, mesothelin is overexpressed in several human cancers, including virtually all MMs, ovarian cancers (70% of cases), lung cancers (50% of cases), and pancreatic/biliary adenocarcinomas (100% of cases) (Miettinen and Sarlomo-Rikala 2003; Rump et al. 2004; Ho et al. 2005; Hassan, Broaddus, et al. 2007; Hassan and Ho 2008). The expression frequency and characterization of mesothelin in many other cancers, including squamous cell carcinomas of the cervix, lung, head, and neck, as well as endometrial adenocarcinomas, have been thoroughly reviewed (Hassan et al. 2004; Hassan and Ho 2008). The 71-kD protein encoded by the mesothelin gene is further processed to a 31-kD protein, megakaryocyte potentiating factor, which is released into the serum. The remaining 40-kD fragment (mesothelin) remains bound to the cell membrane by a glycosylphosphatidylinositol (GPI) anchor (Rump et al. 2004; Hassan, Bullock, et al. 2007). The expression of mesothelin in the serum of patients with MM results in the production of mesothelin-specific IgG antibodies which enables a protective, host humoral immune response (Ho et al. 2005).
Here we hypothesized that surface functionalization of APMS with a mouse monoclonal antibody recognizing the extracellular domain of human mesothelin (MB) with high affinity would be advantageous in targeting MMs. The MB antibody contains only mouse sequences that recognize human mesothelin, making it is less immunogenic than a human-mouse chimeric antibody. For example, previous studies with the mesothelin antibody MORAb-009 showed binding inhibition of mesothelin that resulted in cell cytotoxicity, mediated by a humoral immune response in both in vitro and in vivo models of ovarian cancer (Hassan and Ho 2008; Hassan et al. 2010). In studies here, Alexa Fluor and gadolinium (Gd)–functionalized APMS were used to study the targeting potential of MB. Using several imaging approaches, we also show that mesothelin is expressed by human MM cells growing subcutaneously (SQ) or IP in a mouse xenograft model. Last, we show that the highest fraction of APMS-MB injected IP in a severe combined immunodeficient (SCID) mouse model occurs in both MM spheroids and mesenteric tumors over time (1–6 days), with uptake by both tumor cells and tumor-associated macrophages (TAMs).
Materials and Methods
Synthesis of APMS and Subsequent Modifications
Amorphous silica microparticles premodified with surface tetraethylene glycol (hereafter simply referred to as APMS) were used for subsequent modification in experiments described here (Cheng et al. 2010). APMS microparticles were further modified with Gd chelate, an antibody to MB, bovine serum albumin (BSA), and/or Alexa Fluor dyes as previously described (Cheng et al. 2010; Steinbacher et al. 2010).
Cell Cultures
Human mesothelial LP9/TERT-1 (LP9) cells are an hTERT-immortalized human mesothelial cell line (Dickson et al. 2000) obtained from Dr. James Rheinwald (Dana Farber Cancer Research Institute; Boston, MA). Human MM cell lines PPM Mill (H2373), PPM Gar (H2461), PPM Gat (HP-1), PPM Rob (H2595), PPM Ada (H2596), and PPM Gord (H2818) were previously isolated by Dr. Harvey I. Pass (NYU Langone Medical Center; New York, NY) (Pass et al. 1995). The HMESO MM line, previously described (Reale et al. 1987), and the Gard MM cell line were both obtained from Dr. Joseph R. Testa (Fox Chase Cancer Center, Philadelphia, PA). LP9 cells and MM cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 50/50 (Mediatech; Manassas, VA) supplemented with 10% fetal bovine serum (FBS), 0.1 µg/ml hydrocortisone (Sigma, St. Louis, MO), 2.5 µg/ml insulin, 2.5 µg/ml transferrin, 2.5 ng/ml sodium selenite (Sigma), and penicillin-streptomycin (50 U/ml penicillin G, 50 µg/ml streptomycin sulfate) (Invitrogen, Carlsbad, CA) (Hillegass et al. 2011). Normal human pleural mesothelial cells (NYU474) were isolated surgically from cancer-free patients by Dr. Pass. NYU474 cells were maintained in DMEM (Invitrogen) containing 10% FBS, 50 µM penicillin, and 100 mg/ml streptomycin (Shukla et al. 2009).
Administration of APMS-Gd Particles
Rats were injected with APMS-Gd as previously reported in imaging studies (Steinbacher et al. 2010). In brief, following anesthesia using 3% isoflurane, rats were cannulated through the lower left wall of the peritoneal cavity. APMS-Gd particles suspended in saline (0.9%) at a dose of 500 mg/kg were injected through the cannula into the peritoneal cavity followed by a flush of 100 to 200 µl saline to ensure that no particles remained within the cannula.
Tissue and Urine Sample Collection after MRI
Expelled urine was collected from rats as they recovered from anesthesia at 2, 4, 24, and 144 hr (6 days). Animals were euthanized by cervical dislocation. Necropsies were performed on each animal 6 days after APMS-Gd administration. Urine samples were collected from the bladder immediately following euthanasia. Samples were stored at 4C until further processing. Urine samples (50 µl) were processed as later described for peritoneal lavage fluid (PLF) samples. Tissue samples of heart, lungs, liver, kidneys, spleen, bladder, and bone marrow were isolated for scanning electron microscopy (SEM)/energy dispersive spectroscopy (EDS) analysis. Approximately 1-mm3 tissue samples were removed and processed as described below. Remaining tissues were preserved in 4% paraformaldehyde, paraffin embedded, and stained with hematoxylin and eosin (H&E) for evaluation by histopathology.
Scanning Electron Microscopy and Energy Dispersive Spectroscopy
Tissue samples were fixed in Karnovsky’s fixative (2.5% glutaraldehyde, 1% paraformaldehyde in 0.1 M Millonig’s phosphate buffer) at 4C for 24 hr. Samples were then washed with Millonig’s phosphate buffer (pH 7.2) and postfixed in osmium tetroxide. Fixed samples were then critical point-dried using CO2 as the transition fluid in a Samdri PVT-3B critical point dryer (Tousimis Research Corporation; Rockville, MD). Specimens were mounted on copper specimen mounts using conductive graphite tape, followed by sputter coating for 3 to 4 min with gold and palladium in a Polaron sputter coater (Model 5100; Quorum Technologies, Guelph, ON, Canada). SEM images and EDS spectra of samples were obtained using a JEOL 1210 scanning transmission electron microscope (STEM) (JEOL Ltd.; Peabody, MA) and a PGT IMIX SiLi prism detector (Bruker; Madison, WI), located in the Microscopy Imaging Center at the University of Vermont. All SEM images were acquired at an accelerating voltage of 20 kV.
SCID Mouse Xenograft Model of Human Malignant Mesothelioma
Tissue and tumor distribution, urinary concentrations, and PLF concentrations of APMS were evaluated in a SCID mouse xenograft model of MM after IP injection of human PPM Mill cells (Hillegass et al. 2010). In brief, 5 × 106 cells (in 50 µl sterile 0.9% NaCl [pH 7.4]) were injected into male, 6-week-old Fox Chase SCID mice from Charles River Laboratories (Wilmington, MA). IP injections were administered to the lower left anterior quadrant of the mouse (1 injection site/mouse). An extra aliquot of PPM Mill cells was retained in a 25-cm2 tissue culture flask after all injections into mice to verify cell viability. Ear punches were administered as needed to distinguish between animals. All animal procedures were approved by the University of Vermont College of Medicine (UVM) Institutional Animal Care and Use Committee (IACUC). Both free-floating spheroid and mesenteric tumors lining the diaphragm were observed at 4 weeks in all mice (Hillegass et al. 2010; Hillegass et al. 2011) at which time saline, APMS-MB, or APMS-BSA in 500 µl sterile 0.9% NaCl (pH 7.4) was injected IP.
Collection and Digestion of PLF Samples from SCID Mice
Mice were euthanized with 0.1 ml of Sleep Away (26% sodium pentobarbital; Webster Veterinary, Devens, MA) before 5 ml of cold phosphate-buffered saline (PBS) (Ca/Mg-free) was instilled into the peritoneal cavity of each mouse using an 18-gauge needle. The abdomen was then lightly massaged and the saline removed. Then, 1 ml of each PLF sample (prior to centrifugation) in a 1.5-ml Eppendorf tube was digested in 500 µl proteinase K/sodium dodecyl sulfate (SDS) digestion fluid. The digestion fluid contained NaCl (292.2 mg), Tris-Cl (500 µ1), SDS (2.5 ml, 10%), and proteinase K (0.5 µl, 10 mg/ml) in a total volume of 50 ml double distilled (dd)H2O. Remaining PLF was centrifuged and cell-free supernatants were stored at −20C. Samples were placed in a 50C shaking water bath overnight to remove any proteinacious material from the samples. Digested samples were passed through a 0.4-µm cellulose filter (Whatman; Piscataway, NJ), followed by five rinses of ddH2O and 100% ethanol to prevent any crystallization of particulate matter. Filters were dried at room temperature overnight in a desiccator. A representative section of the dried filter was removed with a sterile razor blade. Filter sections were mounted onto prepared copper specimen mounts using Parlodion (2.0%) amyl acetate and were sputter-coated and imaged as described under SEM methods. Three representative images of APMS particles from PLF fluid were taken for analysis from each sample.
Characterization of APMS Particles and PLF Using Cytospin Analysis
Total white blood cell counts in PLF were assessed using an ADVIA Hematology Analyzer (Siemens Diagnostics; Tarrytown, NY). Cytospins were prepared from 50,000 cells following standard protocols (Blumen et al. 2007; Hillegass et al. 2011). Cytospins were stained using a HEMA 3 kit (Fisher Scientific; Middletown, VA) as per the manufacturer’s directions. Images were captured using an Olympus BX50 upright light microscope (Olympus America; Lake Success, NY) with an attached Q Imaging Retiga 2000R digital CCD camera (Advanced Imaging Concepts, Inc.; Princeton, NJ). Five hundred cells per slide were counted for each animal in each treatment group (n=5/group/time point).
Histopathology
Tissue samples of heart, lungs, liver, kidneys, spleen, bladder, and bone marrow were resected from Wistar rats, preserved in 4% paraformaldehyde, paraffin embedded, and processed for H&E staining. Sectioning (4 µm) and staining using H&E on tissues were performed in the Department of Pathology (Fletcher Allen Health Care; Burlington, VT). Tissue sections were examined by a board-certified pathologist (Kelly J. Butnor, MD).
Immunofluorescence Confocal Microscopy
To determine if mesothelin was expressed on human MMs used in these studies and on MM cells in vitro, immunofluorescence visualization of MB was performed on sections of paraffin-embedded MM tumors derived from four different human cell lines (PPM Mill, PPM Gat, PPM Gar, and HMESO) grown in SCID mice SQ (Hillegass et al. 2010). Sections were deparaffinized in xylenes followed by hydration in ddH2O, via decreasing alcohol concentrations. Autofluorescence was blocked with 0.3 M glycine (Bio-Rad; Hercules, CA) for 10 min. After a 5 min wash in 1× PBS, antigen retrieval was performed by placing slides in 1× Dako target retrieval solution at 95–99C for 40 min (Dako; Carpinteria, CA). Slides were then cooled to room temperature before addition of 50 µl of 10% normal goat serum and 3.8% mouse IgG blocking reagent (Vector; Burlingame, CA) in 1× PBS for 1 hr in a humidified chamber. Mouse anti-mesothelin (clone MB) antibody (Rockland; Gilbertsville, PA) was diluted 1:50 in 1% BSA in 1× PBS. Tumor sections were treated with diluted primary antibody overnight in a humidified chamber at 4C. Goat anti-mouse Alexa Fluor 568 secondary antibody (Molecular Probes; Carlsbad, CA) was diluted 1:400 in PBS, and 50 µl was applied to each section for 1 hr in a dark chamber. A 1:10,000 dilution of SYTOX Green nucleic acid stain (Molecular Probes) in PBS was then applied to each section for 10 min. Slides were washed with ddH2O, then air-dried before adherence of coverslips (Aqua-Poly/Mount; Polysciences Inc., Warrington, PA) and stored at 4C. Confocal images of two to three fields per tumor were acquired using a 40× objective lens on a Bio-Rad MRC 1024ES confocal scanning laser microscope running Bio-Rad Lasersharp 2000 imaging software (Advanced Imaging Concepts, Inc.). A dual fluorescence mode was used to visualize cell nuclei (green) and mesothelin (red) in tumors. Images were scanned in sequential mode to avoid bleed-through between channels.
Western Blot Analysis
NYU isolates, LP9, and MM cell lines were processed following standard protocols after lysing in 4× sample buffer (Shukla et al. 2009). In comparative studies, MMs were removed from mice, total protein was recovered using Lysing Matrix A Tubes (MP Biomedicals, Inc.; Solon, OH), and PLF (500 µl) was concentrated using Amicon Ultra 10 K centrifugal filter devices, as per the manufacturer’s instructions (Millipore; Cork, Ireland). Protein content in each sample was assessed using the RC DC protein assay (Bio-Rad), then resolved by SDS-PAGE, and transferred to nitrocellulose membranes. Equal loading of protein was verified by probing for β-actin (1:2000 dilution; Abcam, Cambridge, MA). Membranes were incubated with MB antibody at a dilution of 2 µg/ml (Rockland Immunochemicals; Gilbertsville, PA). Antibody binding was visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech; Piscataway, NJ) according to the manufacturer’s protocol (Hillegass et al. 2011).
Inductively Coupled Plasma Mass Spectrometry for Detection of Silicon from APMS
Inductively coupled plasma mass spectrometry (ICP-MS) analysis for silicon to detect APMS was performed on heart, liver, lungs, spleen, kidneys, bladder, and tumor tissues as well as urine samples from each mouse in saline, APMS-MB, and APMS-BSA treatment groups (n=5 mice/group) at the Trace Element Analysis Core Facility at Dartmouth College (Hanover, NH). Tissue samples were digested for analysis in 5% (w/v) teramethylammonium hydroxide for 18 hr at 80C and diluted with ddH2O water to a final base concentration of 1%.
Immunofluorescence Staining for TAM and APMS Microparticles in MM Tissue
Localization of APMS-MB antibody or APMS-BSA in free-floating spheroids and mesenteric tumors was assessed from frozen sections (10 µm thick). Tissue sections were fixed in Z-Fix (Anatech Ltd.; Battle Creek, MI) for 15 min. Slides were then rinsed in 1× PBS/1.0% BSA (3× for 5 min), incubated in PBS/1.0% BSA/0.1% Triton X-100 for 10 min, rinsed in 1× PBS/1.0% BSA (2× for 5 min), and blocked in 10% goat serum for 30 min. Slides were incubated with monoclonal rat anti-mouse Mac-2 (Mac-2) (Cedar Lane; Burlington, NC) at 1:200 dilution in PBS/1.0% BSA overnight at 4C. Slides were rinsed in 1× PBS/1.0% BSA (2× for 5 min) and incubated with a fluorophore-conjugated secondary antibody (Alexa Fluor 488 goat anti-rat; Invitrogen) for 60 min. They then were rinsed in 1× PBS/1.0% BSA (2× for 5 min) and incubated with 4,′6-diamidino-2-phenylindole (DAPI) (1:200) dilution in PBS/1.0% BSA (10 µg/ml final concentration) (Roche Applied Science; Indianapolis, IN) for 15 min. Slides were rinsed in 1× PBS/1.0% BSA (2× for 5 min), air dried, mounted with Aqua Poly/Mount, coverslipped, and imaged using a Zeiss LSM 510 META confocal laser scanning microscope (CLSM) (Zeiss Microimaging; Thornwood, NY).
Quantification of Percentage Mac-2+ (Macrophage-containing) and Alexa Fluor 647 (A647) (Particle-containing) Tumor Areas at 24 hr and 6 Days Postinjection of APMS-MB or APMS-BSA Microparticles
Because fluorescence microscopy suggested that the two particle preparations were associated with both TAMs and MM cells, we attempted to determine the approximate area of TAM-containing tumor tissue and whether injection of microparticles altered these profiles over time. Two tiled CLSM images ([4 images × 4 images] [2048 × 2048 pixels]) of three slides (prepared as described above) per treatment group (n=3) were acquired using a Zeiss LSM 510 META confocal laser scanning microscope using a plan-neofluar 25×/0.81 neofluar objective lens. Three channel images were captured at excitation wavelengths of 450 nm (DAPI, blue, to image nuclei), 543 nm (Mac-2, green, to image macrophages), and 633 nm (A647, to image APMS particles) and then opened in MetaMorph image and analysis software (Universal Imaging Corporation, a subsidiary of Molecular Devices, Sunnyvale, CA) as a set of three planes representing separate fluorophores. A random number generator (www.random.org) was used to select five tiles from each image. Graphic outlines highlighting the representative tissue in each of the five selected tiles were traced on each image for quantification. The fluorescence intensity from antibody binding in each representative area was assessed by assigning a minimum pixel intensity threshold value (based on control tissue specimens of mouse aorta stained positively for Mac-2) for the Mac-2 antibody (55) and A647 (57). The mean intensity and integrated intensity of these two planes were then determined automatically in each of the thresholded areas by measuring all pixels with intensity values greater than the set minimum threshold. The intensity values were logged into an Excel spreadsheet (Microsoft Corp.; Redmond, WA) for analysis (Brooks et al. 2009).
Statistical Analysis
All data, excluding the quantification of the percent Mac-2+ and A647-positive MM tissue region of interest area, were evaluated by analysis of variance using the Student Neuman-Keul’s procedure for adjustment of multiple pairwise comparisons between treatment groups. Statistical significance was determined as p≤0.05. The percent Mac-2+ and A647-positive area of percent threshold areas was analyzed with a repeated-measures analysis of variance using the linear mixed models procedure of the SAS System for Windows, version 9.3 (SAS; Cary, NC). Time and treatment means were compared within each of the fluorescence types. Mesenteric and spheroid tumors were each analyzed separately. Percent threshold areas by Mac-2 and A647 fluorescence were correlated using Pearson’s product moment correlation. Correlations were obtained separately for mesenteric and spheroid tumors.
Results
Tissue Distribution of APMS and Subsequent Clearance from the Bladder in Wistar Rats over Time
Previous studies from our group used APMS microparticles administered SQ or IP to tumor-bearing SCID mice as a novel method of chemotherapeutic drug delivery (Hillegass et al. 2011). These studies produced exciting data showing an equal reduction in MM burden at lower doses of DOX (0.33 mg/kg) delivered by APMS as opposed to injection of DOX alone (1.0 mg/kg) (Hillegass et al. 2011) but did not address the biodistribution or excretion of these microparticles over time. We hypothesized that APMS injected into the peritoneal fluid and not binding to or taken up by human MMs in the peritoneal fluid would be filtered by the kidneys and cleared via the urine. To address the question of biodistribution and clearance of APMS over time, we also previously developed a novel preparation of APMS-Gd that enabled in vivo MRI analysis after injection of APMS into the peritoneal cavities of healthy Wistar rats (500 mg/kg) (Steinbacher et al. 2010). These results showed that APMS-Gd is a novel microparticle-based imaging agent that can be visualized in vivo, produces no adverse pathological alterations, and is cleared within 144 hr (6 days).
SEM/EDS Analysis Confirms APMS Microparticles in Tissues and Urine from Wistar Rats
SEM micrographs and corresponding EDS spectra show the presence of APMS-Gd in the organs of Wistar rats 6 days after IP injection (Fig. 1A–D). SEM micrographs show APMS-Gd on the exterior surface of liver tissue, on the interior tissue of the spleen, within renal tubules in the kidneys, and on the interior surface of the bladder. EDS analysis of the particles on or in tissues (black arrows) in the SEM images corresponds to prominent silicon signals, whereas a representative off-particle spectrum (Fig. 1E, white arrow) shows only a strong osmium signal (line arrow; Os is a component of the tissue fixation process) but no silicon signal. Analysis of urine collected at 0.5, 2, 4, and 24 hr after injection of APMS also corroborated with previously reported MRI imaging results (Steinbacher et al. 2010). APMS-Gd microparticles were located in urine using SEM and EDS at 0.5 to 24 hr (Fig. 2A–D). An off-particle spectrum (Fig. 2E, white arrow) showed only an intense peak for gold (Au, a component of sputter coating, line arrow) but an absence of silicon. No APMS-Gd could be found at 144 hr in urine. These data demonstrate that APMS-Gd initially appeared in the kidneys and urine and were cleared from the bladder within 6 days.
Figure 1.

Acid-prepared mesoporous spheres (APMS)–gadolinium (Gd) microparticles occur initially in major clearance organs as verified by scanning electron microscopy (SEM)/energy dispersive spectroscopy (EDS). Representative tissue samples (1 mm3) were removed from organs of Wistar rats (n=3) at 144 hr (6 days). In panels A to D, APMS-Gd microparticles (indicated by black arrows) are present in SEM micrographs (left) of the liver (A) (8000×, scale bar = 2 µm), spleen (B) (2500×, scale bar = 5 µm), renal tubule of kidney (C) (4000×, scale bar = 5 µm), and the internal surface of the bladder (D) (4000×, scale bar = 5 µm). Corresponding EDS spectra (right) indicate the presence of silicon (black arrows) in APMS-Gd microparticles. Panel E shows an off-target (white arrow) control micrograph of bladder tissue (4000×, scale bar = 5 µm). The corresponding spectra reveal an absence of silicon (line arrow).
Figure 2.

Acid-prepared mesoporous spheres (APMS)–gadolinium (Gd) microparticles are excreted in the urine. Urine samples were passively collected from Wistar rats (n=3) following each MRI time point (0.5, 2, 4, 24 hr). Urine samples were digested, filtered, and prepared for scanning electron microscopy (SEM)/energy dispersive spectroscopy (EDS) analysis with gold and palladium coating. Panels A to D present SEM micrographs (left) of APMS-Gd microparticles (indicated by black arrows). In panels A and E, magnification is 12,000×, scale bar = 1 µm. B and C are at 8000× magnification; scale bar = 2 µm. D is at 5000× magnification; scale bar = 2 µm. The corresponding EDS spectra (right) display prominent silicon peaks (black arrows), the main component of APMS microparticles. Panel E represents an off-target (white arrow) SEM micrograph control. The corresponding EDS spectrum reveals an absence of silicon (line arrow). No particles could be found for analysis in the 144-hr (6-day) sample.
APMS-MB and APMS-BSA Microparticles Are Retained in the Peritoneal Fluid and Do Not Alter Inflammatory Cell Profiles in PLF
We next used targeted approaches with APMS-MB versus APMS-BSA comparatively. To assess the retention of APMS microparticles in the peritoneal cavity and possible inflammatory effects, PLF was isolated at 24 hr or 6 days after a single IP APMS injection. SEM micrographs of representative samples showed that APMS microparticles were present in PLF at 6 days (Fig. 3A, B). Having confirmed the presence of APMS in PLF, we then examined changes in inflammatory cell profiles in PLF from MM tumor-bearing SCID mice. No significant changes in inflammatory cell differential counts between groups were observed at 24 hr after injection. However, in both the APMS-MB and APMS-BSA groups (Fig. 3C), the number of macrophages increased significantly from 24 hr to 6 days, whereas the number of neutrophils and eosinophils in these groups decreased significantly. Although experimental problems prevented statistical significance from being calculated for the saline groups at 24 hr (n=1), the similar trends in all three groups led us to conclude that changes in the inflammatory cell profiles between 24 hr and 6 days is likely caused by increased tumor burden rather than an effect of the particles.
Figure 3.

Acid-prepared mesoporous spheres (APMS) are retained in the peritoneal cavity of mice (n=5 mice/group) up to 144 hr (6 days) postinjection and do not alter inflammatory cell profiles in peritoneal lavage fluid (PLF) at 24 hr or 6 days. PLF was filtered and prepared for scanning electron microscopy (SEM)/energy dispersive spectroscopy (EDS) analysis. SEM micrographs (A, B) show both on-target (black arrow) and off-target images (white arrow) of APMS microparticles (10,000× magnification, scale bar = 2 µm). Corresponding EDS spectra to the right indicate the presence (black arrow) or absence (lined arrowhead) of silicon. Differential cell counts of the 24-hr and 6-day time points (mean ± standard error of the mean) (n=3/group/time point). *Significantly different from the 24-hr time point (p<0.05) (C). Abbreviations: MAC, macrophages; NEU, neutrophils; EOS, eosinophils; BASO/MAST, basophils/mast cells; LYM, lymphocytes.
Mesothelin Is Expressed by MM Cell Lines In Vitro and in a Xenograft Model of MM
Literature precedent showed that mesothelin protein is overexpressed in human MMs in vivo, making this an ideal target for APMS modified with an anti-mesothelin antibody. Indeed, PPM Mill, PPM Gat, PPM Gar, and HMESO paraffin-embedded MM tumor sections all stained positively for expression of mesothelin (Fig. 4A). However, Western blots of only four of seven MM cell lines in vitro exhibited visible bands for the 40-kD portion of mesothelin protein (PPM Mill, PPM Gar, PPM Gord, and PPM Rob). Immortalized human peritoneal mesothelial cells (LP9) and human primary pleural mesothelial cells (NYU474) did not express the mesothelin protein (Fig. 4B). MM tumors derived from the HMESO and PPM Mill, PPM Gat, PPM Gar, and PPM Gard cell lines were then assessed for mesothelin protein after growing either SQ or IP in SCID mice (Fig. 4C). These experiments provided evidence of mesothelin protein produced from MM cell lines both in vitro and in vivo. The PPM Mill cell line produced mesothelin consistently in vivo and in vitro and has been characterized in the SCID xenograft model (Hillegass et al. 2010). Therefore, we chose the PPM Mill cell line for our targeting studies. In these studies, PLF taken from IP injected animals contained both normal and malignant mesothelial cells comprising either APMS functionalized with the MB antibody or BSA at 24 hr and 6 days (Fig. 4D). In both the APMS-MB and APMS-BSA treatment groups, particles could be seen surrounding cell nuclei showing that APMS persist after localized administration up to 1 week. No particles or abnormalities were seen in saline control PLF samples.
Figure 4.

Mesothelin is expressed in mesothelioma (MM) tumors. Paraffin sections of MM tumors derived from four different MM cell lines (PPM Mill, PPM Gat, PPM Gar, and HMESO) grown in severe combined immunodeficient (SCID) mice were stained with mouse anti-mesothelin antibody (clone MB) (A). Alexa Fluor 568 secondary antibody was used to visualize mesothelin (red), and SYTOX Green nucleic acid stain was used to visualize cell nuclei (green). Micrographs were taken at 400× magnification, scale bar = 50 µm. Mesothelin protein was assessed by Western blot analysis on human MM cell lines (B) and in human MM tumors grown either intraperitoneally (IP) or subcutaneously (SQ) (C). β-Actin was used as a loading control. Micrographs of Hema 3 differential stained cytospins from peritoneal lavage fluid (PLF) show uptake of acid-prepared mesoporous spheres (APMS)–MB and APMS-BSA microparticles (n=5 mice/group). Particles surround the cell nuclei at 24 hr and 6 days (black arrows) (D). Micrographs were taken at 400× magnification, scale bar = 50 µm.
Functionalization of APMS with an Anti-mesothelin Antibody (APMS-MB) Significantly Increases the Fraction of Administered Dose to MM Tissues Using ICP-MS
To test the hypothesis that APMS-MB would be taken up by MM tumors more avidly than APMS-BSA control particles, we examined tumors (pooled spheroid and mesenteric) and major organs of treated mice by ICP-MS at 24 hr and 6 days. At 24 hr, less than 2% of the administered dose of APMS was observed in the lungs, liver, spleen, and kidneys, whereas in both APMS-MB and APMS-BSA groups, greater than 6% of the administered dose was present in the pooled tumors (Fig. 5A). At 6 days, approximately 3.5% of the total administered amounts were observed in the liver. However, greater than 10% of the delivered doses of APMS-MB and APMS-BSA were observed in tumors, and the fraction of the dose in the APMS-MB tumors was significantly increased compared with the fraction of dose observed in APMS-BSA tumors (Fig. 5B). We then examined the delivery of particles as a function of concentration of SiO2 (ppm) (i.e., APMS) in both spheroid and mesenteric tumors at 24 hr and 6 days (Fig. 5C). APMS-MB-treated mesenteric tumors contained a significantly higher concentration of SiO2 than mesenteric tumors treated with APMS-BSA at 6 days, indicating that APMS-MB is retained in tumors over time to a greater extent than APMS-BSA.
Figure 5.
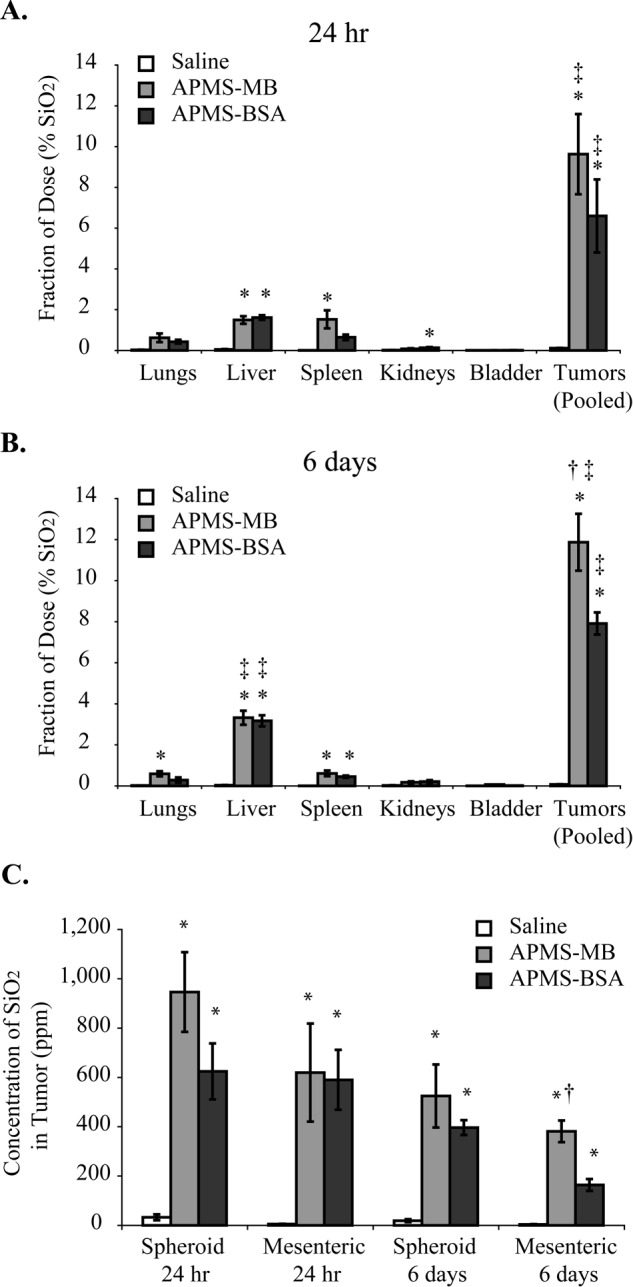
Acid-prepared mesoporous spheres (APMS) microparticles functionalized with a mesothelin antibody (MB) significantly increased the fraction of administered dose taken up by mesotheliomas (MMs) at 6 days (n=5 mice/group). The fraction of the administered dose of APMS in major organs and tumor tissue was analyzed by inductively coupled plasma mass spectrometry (ICP-MS) (A, B). The concentration of SiO2 in tumor tissue in parts per million (ppm) is significantly greater in tumors treated with APMS-MB as compared with the APMS-BSA group at 6 days (C). *Significantly different from saline control of the same organ/tumor group. †Significantly different from APMS-BSA group. ‡Significantly different from all other organ/tumor groups in the same treatment group (p<0.05). For saline and APMS-MB treatment groups, five individual mice were assessed per group per time point. For APMS-BSA treatment, four mice were assessed at each time point.
Uptake of APMS by MM Cells and TAMs In Vivo Is Enhanced by Multifunctionalization with an Anti-mesothelin Antibody
To complement results of ICP-MS studies and determine the cell types associated with APMS-MB and APMS-BSA in tumors, we assessed the association of APMS with TAMs and MM cells visually using CLSM. Cryo-sections from in vivo MM mesenteric tumor tissues were examined using A647-labeled particles at 24 hr and 6 days postinjection of APMS. As expected, no particles were observed in tumor tissue injected with saline alone (Fig. 6A, B). APMS-BSA and APMS-MB microparticles, at both 24 hr and 6 days, could be visualized in association with MM tumor cells as punctate areas of fluorescence (indicated by white arrows) (Fig. 6C–F). In both treatment groups, APMS microparticles were also engulfed by TAMs (shown in green in Fig. 6) and surrounded the cell nuclei. Larger, tiled images of MM tumors from the APMS-MB and APMS-BSA treatment groups show APMS-MB microparticles are translocated from the outer surface of the tumor (24 hr postinjection) to the center (by 6 days) (Suppl. Fig. S1). The APMS-BSA microparticles remain in the tumor periphery at 6 days (Suppl. Fig. S2).
Figure 6.

Acid-prepared mesoporous spheres (APMS) microparticles were taken up by mesenteric mesotheliomas (MMs), retained for up to 6 days, and possibly trafficked by tumor-associated macrophages (TAMs) (n=5 mice/group). Confocal microscopy analysis of frozen sections of MM tumor tissue at 24 hr and 6 days shows no particles in saline-injected control tumors (scale bar = 50 µm) (A, B). APMS-BSA and APMS-MB were taken up by tumor tissue at 24 hr and remained in the tumor tissue at 6 days following intraperitoneal (IP) injection (C–F) (scale bar = 50 µm). In all panels, APMS-MB or APMS-BSA are indicated with A647 (red) (white arrows), cell nuclei are stained with DAPI (blue), and TAMs are stained with Mac-2 (green). Panels B, C, E and F are of the tumor exterior (tumor edge) vs. panels A and D of the tumor interior.
Quantification of Percent Positive Area in Mesenteric and Spheroid MM Tissue Analyzed for Mac-2 and A647 (APMS) Fluorescence Intensity
To show that the particles do not increase the TAM burden in established tumors, we performed quantitative evaluation of the percentage of mesenteric and spheroid MM tissue area staining positively for TAMs (Mac-2). The percentage of MM tissue area containing APMS-MB or APMS-BSA (both A647-tagged) particles was assessed by CLSM in cryo-sections (as described above) from tiled images. Randomly selected regions of interest from each sample (n=18/tumor) at each time point were analyzed for the percentage of the region of interest meeting the minimum fluorescence intensity threshold set by control sample tissues (Fig. 7). In accordance with results from tiled images, TAM+ areas comprised less than 5% of the spheroid or mesenteric tumor area examined. At 24 hr, significant decreases were seen in the percentage of Mac-2+ area in the APMS-MB treatment group (p<0.0001) and the 24-hr APMS-BSA treatment group (p<0.001) compared with the 24-hr saline control (Fig. 7A). There were no significant differences in either Mac-2+ or A647+ areas at 6 days in the spheroid or areas of mesenteric MMs at either 24 hr or 6 days.
Figure 7.
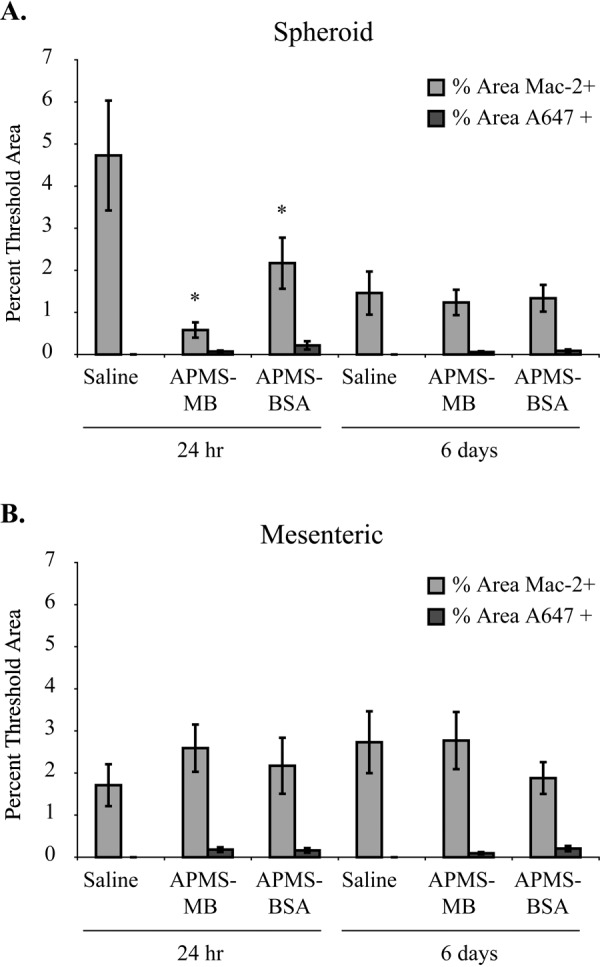
The percentage of Mac-2+ area within regions of interest analyzed in spheroid mesothelioma (MM) tumor tissue was significantly decreased in acid-prepared mesoporous spheres (APMS)–MB and APMS-BSA treated animals at 24 hr as compared with saline controls (*p<0.05) (n=1 tumor per mouse/3 mice per group) (A). Five randomly chosen regions of interest from tiled (4 × 4 tiles) confocal fluorescent microscopy images were opened in MetaMorph image analysis software to determine differences in the percent of threshold areas positive for the minimum fluorescence intensity of Mac-2 and A647 (APMS preparations). Minimum intensity thresholds were set by Mac-2+ control samples of mouse aorta and visual assessment of multiple A647+ areas. No significant differences were seen in Mac-2+ percent threshold areas by particles in mesenteric MM tissues (B).
Discussion
Here we demonstrate that APMS, a non-toxic, amorphous silica microparticle, associates with human MM tumor cells and TAMs in a mouse xenograft model of MM. A significantly greater fraction of the delivered dose of APMS-MB was taken up by tumors compared with a non-targeting control microparticle (APMS-BSA). Mesoporous silica nanoparticles and other nanoparticles have recently been highlighted as advantageous platforms for the delivery of drugs and biomolecules due to their large internal surface area and ability to be chemically modified for enhanced uptake and targeting (Hudson et al. 2008). However, nanoparticles are readily taken up by cellular organelles, cause cytotoxicity alone, and are distributed systemically throughout the body, whereby they can cross the blood-brain barrier (Borm and Kreyling 2004; Pantarotto et al. 2004; Oberdorster, Maynard, et al. 2005; Oberdorster, Oberdorster, et al. 2005; Xia et al. 2006; Handy et al. 2008). These properties of nanoparticles raise concern for their acute and long-term effects as drug delivery vehicles. APMS microparticles circumvent these issues as their diameter (1–2 µm) prevents them from entering cellular organelles and prevents them from persisting in the systemic circulation after localized delivery (Cheng et al. 2010; Steinbacher et al. 2010). Previously we have shown that APMS modified with tetraethylene glycol on the external surface are readily taken up by mesothelial cells in vitro and remain in the cytosol, avoiding endosomal degradation of cargo (Blumen et al. 2007). We recently reported that APMS-Gd, a novel porous silica microparticle containing a Gd chelate, can be used as a contrast agent that is able to be tracked in real time using MRI (Steinbacher et al. 2010). The use of this approach in studies described here allowed us to verify that APMS microparticles, once administered IP, are excreted with no obvious adverse effects on organ pathology or inflammation. In previous published studies by Steinbacher et al. (2010) and Hillegass et al. (2011), the toxicity of APMS (both unloaded and loaded with DOX) was evaluated. Studies by Steinbacher et al. (2010) showed that doses up to 500 mg/kg were well tolerated in Wistar rats (non–tumor bearing) without histological evidence of toxicity to the major organs as evaluated by a board-certified pathologist. In studies by Hillegass et al. (2011), SCID mice bearing peritoneal MMs were administered APMS loaded with DOX. These mice were treated with 7.8 × 108 APMS/mouse (equivalent to 0.33 mg/kg DOX) 3× weekly for 1 week. Adverse health effects were not seen, nor was there evidence of toxicity present in histological samples evaluated by a board-certified pathologist. However, diffuse hepatocytic cell swelling was observed in animals receiving injections of DOX alone (1 mg/kg). These studies led us to believe that APMS particles do not pose a severe risk to the major clearance/filtration organs and that the low levels of APMS (4% of administered dose) have similar retention patterns, as reported in other studies of microparticle accumulation (Martin et al. 2005).
One of the challenges with chemotherapeutic approaches in MM and other chemoresistant tumors is maintaining the longevity of the given agent in the desired locale. APMS microparticles remained in the peritoneal cavity up to 6 days after a single IP injection without altering the inflammatory cell profile, making them advantageous for the delivery of chemotherapeutic agents to peritoneal tumors. We have previously published that APMS preloaded with DOX and administered either SQ or IP to established MM tumors in SCID mice reduced tumor burden and volume at smaller doses (0.33 mg/kg) in comparison to administration of DOX alone at a dose of 1.0 mg/kg (Hillegass et al. 2011). Here we show that functionalization of APMS with a targeting antibody for mesothelin (MB) results in enhanced uptake of APMS by MM tumors in vivo and that APMS-MB have potential as a more efficacious chemotherapeutic delivery vehicle than particles modified with a nonspecific protein. However, it is also important to note that APMS, even without functionalization with MB, are localized in tumor tissue and TAMs. Inherent properties of APMS make it an appropriate delivery device for an even broader range of malignant diseases.
Recent studies have demonstrated that albumin can bind to surface glycoproteins such as gp60 (albondin) on capillary endothelial cells and mesothelial cells and activates transcytosis through an as yet unknown signaling mechanism (Gotloib and Shostak 1995; Minshall et al. 2000; Vogel et al. 2001; John et al. 2003). This association between albumin and endothelial cells allows for active transport across the cell membrane, reinforcing the validity of the data presented here showing some uptake of APMS-BSA particles by tumor cells. Due to the observed mechanism of active transport of serum albumin across the endothelial and mesothelial cell membranes, albumin microparticles and nanoparticles have become an area of interest in drug delivery research. Studies have shown that serum albumin micro/nanospheres are readily taken up by macrophages and degraded within a week following administration (Schafer et al. 1994), unlike the persistence of APMS microparticles observed here.
When we examined MM tumors by immunofluorescence for TAMs, a high proportion was observed. Given these findings, it is not surprising that we see a high level of APMS-BSA microparticles in MM tumors or that we see the greatest difference in silica concentration between the targeted and non-targeted groups at 6 days (Fig. 5C). A review of the literature reveals that localized administration of antibody-targeted, drug-loaded microparticles to tumors using similar approaches has not been published. Moreover, studies using IP delivery of microparticles of various compositions for drug delivery show less success with regard to microparticle retention by tumor tissue. For example, one study (Li and Howell 2009) examining CD44-targeted Hyplat microparticles (produced by cross-linking hyaluronan via its carboxylate groups with cisplatin) reported tumor retention values between 5 and 10 ppb as assessed by inductively coupled plasma-optical emission spectrometry. We demonstrated that APMS-MB particles had tumor tissue retention values between 800 and 1000 ppm SiO2 over the same time frame.
Regardless of macrophage involvement or active transport of albumin-modified particles into cells, statistical analysis of our data indicated that the retained dose (12%) of administered APMS-MB targeted microparticles is significantly higher than the non-targeted APMS-BSA microparticles (8%) showing that APMS-MB microparticles are more effective at targeting MM tumor tissue. Furthermore, mesothelin is highly expressed not only in MM but also in more than 70% of ovarian cancers, 100% of pancreatic tumors, and 50% of lung cancers, making this approach relevant to a broad range of malignant diseases.
Mesothelin is shed from the cell surface mediated by the “sheddase” activity of ADAM17/TACE. TACE is a transmembrane glycoprotein known for its role in releasing epidermal growth factor receptor (EGFR) ligands from the cell’s surface, which in turn regulates the activation of the EGFR pathway and can contribute to drug resistance (Ho et al. 2005; Ho et al. 2006; Hassan and Ho 2008; Zhang et al. 2011; Pastan and Zhang 2012). Mesothelin has been found to bind to the cell surface antigen MUC16/CA125. MUC16/CA125 is elevated in the serum of women with ovarian carcinoma and is overexpressed in 70% of human ovarian cancers (Bast et al. 1983). Thus, it has been implicated in the spread of ovarian cancer throughout the peritoneal cavity (Rump et al. 2004; Gubbels et al. 2006). Mesothelin has also been examined as a biomarker for the progression of ovarian cancer and mesothelioma (Scholler et al. 1999; Robinson et al. 2003; Robinson et al. 2005; Scherpereel et al. 2006). Several phase I and II clinical trials focusing on mesothelin as a therapeutic target for mesothelioma and ovarian and pancreatic cancers have been conducted or are currently ongoing. These studies have used anti-mesothelin recombinant immunotoxins (SS1 [dsFv] PE38 [Fv portion of antibody] SS1 and truncated Pseudomonas exotoxin) alone and in combination with chemotherapeutics (Hassan, Bullock, et al. 2007; Kreitman et al. 2009) or the anti-mesothelin antibody MORAb-009 (a chimeric IgG/k/SS1 [dsFv] fusion antibody), leading to an antibody-dependent cell-mediated cytotoxic response (Hassan et al. 2007; Hassan et al. 2010). The mesothelin tumor vaccine (CRS-207) is currently in a phase II trial in combination with the pancreatic cancer vaccine GVAX (Le et al. 2012), and adoptive T-cell immunotherapy using mesothelin and antibody drug conjugate therapies are currently being developed for preclinical trials. The shedding of mesothelin into the interstitial space limits the interaction of targeted therapies and may prevent tumor penetration (Pastan and Zhang 2012).
We have shown here that APMS-MB are successfully internalized by tumor tissues. APMS-MB may have a dual advantage in that the MB antibody can effect immune changes such as antibody-dependent cell-mediated cytotoxic response and can direct the drug/biomolecule payloads to mesothelin-overexpressing tumor cells. In addition, the potential to bind free mesothelin may hinder the spread of cancerous cells throughout the peritoneal cavity. A further advantage of our treatment modality is highlighted by the identification of macrophages in MM tumors, implicating the involvement of these cells and their potential to traffic APMS to MM tumors in vivo.
MMs exhibit notably high resistance to apoptosis in vivo, which is thought to contribute to heightened chemoresistance of this tumor type (Fennell and Rudd 2004). Focus has recently shifted to the interactions between tumor cells, non-malignant cells, the extracellular matrix, and their combined effects on tumor cell growth, invasion, migration, and resistance to apoptosis (Kobayashi et al. 1993). The end effect of interactions between malignant and non-malignant cells such as TAMs has been termed multicellular resistance (Kobayashi et al. 1993). In more than 80% of epithelial cancers, tumor stage and disease prognosis correlate with tumor macrophage burden. TAMs are known to be present throughout solid MM tumors in both human and murine MMs and are thought to contribute to tumor development (Bielefeldt-Ohmann et al. 1994; Hegmans et al. 2006). Multicellular resistance of TAMs may cause changes in the tumor microenvironment, including immunosuppression that can aid in the establishment of tumors or avoidance of immunodetection (Mantovani et al. 2006). TAMs have also been shown to increase angiogenic and other growth factors that, when removed by ablation of TAMs via liposome-encapsulated clodronate (CLIP) in a murine model of diffuse peritoneal MM, leads to loss of cell/tumor survival, invasion, and metastatic properties (Miselis et al. 2008).
Although the TAMs in our study were present in large numbers and appeared to phagocytose both APMS-MB and APMS-BSA, their role in MM and particle trafficking is unclear. Macrophages can be divided into two phenotypes: the M1 “classically activated” and the M2 “alternatively activated” subtypes. Recent studies indicate that TAMs displaying an M2 phenotype are characterized by increased expression of interleukin (IL)–10 that enhances alternatively activated macrophage differentiation and production of CCL17 and CCL22 chemokines (Mantovani et al. 2004; Miselis et al. 2008). In addition, the production of CCL17 and CCL22 recruits TH2 lymphocytes, thereby inhibiting TH1 cell function and creating an immunosuppressive microenvironment (Mantovani et al. 2006).
We attempted here to quantify changes in tumor proportions of TAMs that may be in response to APMS-MB or APMS-BSA treatment and to evaluate if targeted microparticles were accumulating in tumors with an increased TAM burden. We did not discover any significant changes in TAM burden in established mesenteric tumors between targeted, non-targeted, or saline-treated MMs. However, we did demonstrate a significant reduction in TAM burden in the APMS-MB- and APMS-BSA-treated MMs in spheroid MMs at 24 hr, indicating our microparticles may have an effect on TAM recruitment to developing tumors. We hypothesize that if TAMs are contributing to the trafficking of microparticles to MM tissue, the contributions are not significantly changing the distribution of microparticles within the tissue between treatment groups, nor are APMS significantly altering the TAM component of the tumor microenvironment.
In summary, we have shown that APMS modified with an antibody to mesothelin provide a unique therapeutic delivery device that can be both imaged and targeted in an in vivo model of IP MM in SCID mice. We have previously shown that APMS preloaded with a therapeutic agent such as DOX can reduce MM tumor growth (Hillegass et al. 2011), thus suggesting a novel platform for localized, targeted, multimodality treatments for MM. This approach may increase the efficacy of current chemotherapeutic treatments for MM and other intracavitary tumors while lowering systemic toxicity of drugs in patients.
Supplementary Material
Acknowledgments
We thank Dr. Harvey I. Pass and Dr. Joseph R. Testa for providing isolates of human mesothelial cells and MM cell lines; Marilyn Wadsworth for assistance with confocal microscopy; Michele Von Turkovich for assistance with SEM; Neil P. Jerome for assistance with MRI experiments conducted at Dartmouth College; Dr. Raffit Hassan and Dr. Mitchell Ho for helpful editorial advice and comments during the preparation of this article; Drs. Ira Pastan, Mitchell Ho, and Raffit Hassan for contribution of one MB antibody; and Alan Howard and the Statistical Software Support & Consulting Services at the University of Vermont for assistance with data analysis. We also thank Jennifer L. Díaz, Department of Pathology at the University of Vermont, for editorial assistance with this manuscript.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Mesothelioma Applied Research Foundation (BTM), an STTR grant R41 CA126155 from the National Cancer Institute (CCL), and T32 ES007122 from the National Institute of Environmental Health Sciences (BTM for SLM, JMH, JLS); 1S10 RR08173–01A1 from the National Center for Research Resources (NCRR) for purchase of the Bio-Rad 1024 confocal laser scanning microscope (DJT); and 1S10 RR01924B from the NCRR for the purchase of the Zeiss 510 META confocal laser scanning microscope (DJT).
Supplementary material for this article is available on the Journal of Histochemistry & Cytochemistry Web site at http://jhc.sagepub.com/supplemental.
References
- Albelda SM. 1997. Gene therapy for lung cancer and mesothelioma. Chest. 111:144S–149S [DOI] [PubMed] [Google Scholar]
- Bast RC, Jr, Klug TL, St John E, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, Leavitt T, Griffiths CT, Parker L, et al. 1983. A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med. 309:883–887 [DOI] [PubMed] [Google Scholar]
- Bielefeldt-Ohmann H, Fitzpatrick DR, Marzo AL, Jarnicki AG, Himbeck RP, Davis MR, Manning LS, Robinson BW. 1994. Patho- and immunobiology of malignant mesothelioma: characterisation of tumour infiltrating leucocytes and cytokine production in a murine model. Cancer Immunol Immunother. 39:347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumen SR, Cheng K, Ramos-Nino ME, Taatjes DJ, Weiss DJ, Landry CC, Mossman BT. 2007. Unique uptake of acid-prepared mesoporous spheres by lung epithelial and mesothelioma cells. Am J Respir Cell Mol Biol. 36:333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borm PJ, Kreyling W. 2004. Toxicological hazards of inhaled nanoparticles—potential implications for drug delivery. J Nanosci Nanotechnol. 4:521–531 [DOI] [PubMed] [Google Scholar]
- Brooks EG, Trotman W, Wadsworth MP, Taatjes DJ, Evans MF, Ittleman FP, Callas PW, Esmon CT, Bovill EG. 2009. Valves of the deep venous system: an overlooked risk factor. Blood. 114:1276–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Pastan I. 1996. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A. 93:136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Blumen SR, MacPherson MB, Steinbacher JL, Mossman BT, Landry CC. 2010. Enhanced uptake of porous silica microparticles by bifunctional surface modification with a targeting antibody and a biocompatible polymer. ACS Appl Mater Interfaces. 2:2489–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, El-Boubbou K, Landry CC. 2012. Binding of HIV-1 gp120 glycoprotein to silica nanoparticles modified with CD4 glycoprotein and CD4 peptide fragments. ACS Appl Mater Interfaces. 4:235–243 [DOI] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 20:1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell DA, Rudd RM. 2004. Defective core-apoptosis signalling in diffuse malignant pleural mesothelioma: opportunities for effective drug development. Lancet Oncol. 5:354–362 [DOI] [PubMed] [Google Scholar]
- Gallis KW, Landry CC. 2002. Mesoporous silicates and method of making same. US Patent No. 6,334,988 [Google Scholar]
- Gotloib L, Shostak A. 1995. Endocytosis and transcytosis of albumin gold through mice peritoneal mesothelium. Kidney Int. 47:1274–1284 [DOI] [PubMed] [Google Scholar]
- Grosso F, Scagliotti GV. 2012. Systemic treatment of malignant pleural mesothelioma. Future Oncol. 8:293–305 [DOI] [PubMed] [Google Scholar]
- Gubbels JAA, Belisle J, Onda M, Rancourt C, Migneault M, Ho M, Bera TK, Connor J, Sathyanarayana BK, Lee B. 2006. Mesothelin-MUC 16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy RD, Henry TB, Scown TM, Johnston BD, Tyler CR. 2008. Manufactured nanoparticles: their uptake and effects on fish—a mechanistic analysis. Ecotoxicology. 17:396–409 [DOI] [PubMed] [Google Scholar]
- Hassan R, Bera T, Pastan I. 2004. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 10:3937–3942 [DOI] [PubMed] [Google Scholar]
- Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. 2007. Anti-mesothelin immunotoxin SS1P in combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 13:7166–7171 [DOI] [PubMed] [Google Scholar]
- Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, Pastan I. 2007. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 13:5144–5149 [DOI] [PubMed] [Google Scholar]
- Hassan R, Cohen SJ, Phillips M, Pastan I, Sharon E, Kelly RJ, Schweizer C, Weil S, Laheru D. 2010. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 16:6132–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan R, Ebel W, Routhier EL, Patel R, Kline JB, Zhang J, Chao Q, Jacob S, Turchin H, Gibbs L, et al. 2007. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 7:20. [PMC free article] [PubMed] [Google Scholar]
- Hassan R, Ho M. 2008. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 44:46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegmans JP, Hemmes A, Hammad H, Boon L, Hoogsteden HC, Lambrecht BN. 2006. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur Respir J. 27:1086–1095 [DOI] [PubMed] [Google Scholar]
- Hesdorffer ME, Chabot J, DeRosa C, Taub R. 2008. Peritoneal mesothelioma. Curr Treat Options Oncol. 9:180–190 [DOI] [PubMed] [Google Scholar]
- Hesdorffer ME, Chabot JA, Keohan ML, Fountain K, Talbot S, Gabay M, Valentin C, Lee SM, Taub RN. 2008. Combined resection, intraperitoneal chemotherapy, and whole abdominal radiation for the treatment of malignant peritoneal mesothelioma. Am J Clin Oncol. 31:49–54 [DOI] [PubMed] [Google Scholar]
- Hillegass JM, Blumen SR, Cheng K, MacPherson MB, Alexeeva V, Lathrop SA, Beuschel SL, Steinbacher JL, Butnor KJ, Ramos-Niño ME. 2011. Increased efficacy of doxorubicin delivered in multifunctional microparticles for mesothelioma therapy. Int J Cancer. 129:233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillegass JM, Shukla A, Lathrop SA, MacPherson MB, Beuschel SL, Butnor KJ, Testa JR, Pass HI, Carbone M, Steele C, et al. 2010. Inflammation precedes the development of human malignant mesotheliomas in a SCID mouse xenograft model. Ann N Y Acad Sci. 1203:7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M, Bera TK, Willingham MC, Onda M, Hassan R, FitzGerald D, Pastan I. 2007. Mesothelin expression in human lung cancer. Clin Cancer Res. 13:1571–1575 [DOI] [PubMed] [Google Scholar]
- Ho M, Hassan R, Zhang J, Wang QC, Onda M, Bera T, Pastan I. 2005. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 11:3814–3820 [DOI] [PubMed] [Google Scholar]
- Ho M, Onda M, Wang QC, Hassan R, Pastan I, Lively MO. 2006. Mesothelin is shed from tumor cells. Cancer Epidemiol Biomarkers Prev. 15:1751. [DOI] [PubMed] [Google Scholar]
- Hudson SP, Padera RF, Langer R, Kohane DS. 2008. The biocompatibility of mesoporous silicates. Biomaterials. 29:4045–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John TA, Vogel SM, Tiruppathi C, Malik AB, Minshall RD. 2003. Quantitative analysis of albumin uptake and transport in the rat microvessel endothelial monolayer. Am J Physiol Lung Cell Mol Physiol. 284:L187–L196 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Man S, Graham CH, Kapitain SJ, Teicher BA, Kerbel RS. 1993. Acquired multicellular-mediated resistance to alkylating agents in cancer. Proc Natl Acad Sci U S A. 90:3294–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. 2009. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 15:5274–5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, Sterman DH, Hassan R, Lutz E, Moyer B, et al. 2012. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 18:858–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SD, Howell SB. 2009. CD44-targeted microparticles for delivery of cisplatin to peritoneal metastases. Mol Pharm. 7:280–290 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A. 2004. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. 40:1660–1667 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. 2006. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 25:315–322 [DOI] [PubMed] [Google Scholar]
- Martin FJ, Melnik K, West T, Shapiro J, Cohen M, Boiarski AA, Ferrari M. 2005. Acute toxicity of intravenously administered microfabricated silicon dioxide drug delivery particles in mice: preliminary findings. Drugs R D. 6:71–81 [DOI] [PubMed] [Google Scholar]
- Miettinen M, Sarlomo-Rikala M. 2003. Expression of calretinin, thrombomodulin, keratin 5, and mesothelin in lung carcinomas of different types: an immunohistochemical analysis of 596 tumors in comparison with epithelioid mesotheliomas of the pleura. Am J Surg Pathol. 27:150–158 [DOI] [PubMed] [Google Scholar]
- Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. 2000. Endothelial cell-surface gp60 activates vesicle formation and trafficking via Gi-coupled Src kinase signaling pathway. J Cell Biol. 150:1057–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarabshahii P, Pillai K, Chua TC, Pourgholami MH, Morris DL. 2012. Diffuse malignant peritoneal mesothelioma—an update on treatment. Cancer Treat Rev. 38:605–612 [DOI] [PubMed] [Google Scholar]
- Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. 2008. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther. 7:788–799 [DOI] [PubMed] [Google Scholar]
- Mossman BT, Bignon J, Corn M, Seaton A, Gee JB. 1990. Asbestos: scientific developments and implications for public policy. Science. 247:294–301 [DOI] [PubMed] [Google Scholar]
- Nassivera T, Eklund AG, Landry CC. 2002. Size-exclusion chromatography of low-molecular-mass polymers using mesoporous silica. J Chromatogr A. 973:97–101 [DOI] [PubMed] [Google Scholar]
- Oberdorster G, Maynard A, Donaldson K, Castranova V, Fitzpatrick J, Ausman K, Carter J, Karn B, Kreyling W, Lai D, et al. 2005. Principles for characterizing the potential human health effects from exposure to nanomaterials: elements of a screening strategy. Part Fibre Toxicol. 2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdorster G, Oberdorster E, Oberdorster J. 2005. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 113:823–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantarotto D, Briand JP, Prato M, Bianco A. 2004. Translocation of bioactive peptides across cell membranes by carbon nanotubes. Chem Commun (Camb). 2004:16–17 [DOI] [PubMed] [Google Scholar]
- Pass HI, Stevens EJ, Oie H, Tsokos MG, Abati AD, Fetsch PA, Mew DJ, Pogrebniak HW, Matthews WJ. 1995. Characteristics of nine newly derived mesothelioma cell lines. Ann Thorac Surg. 59:835–844 [DOI] [PubMed] [Google Scholar]
- Pastan I, Zhang Y. 2012. Modulating mesothelin shedding to improve therapy. Oncotarget. 3:114–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reale FR, Griffin TW, Compton JM, Graham S, Townes PL, Bogden A. 1987. Characterization of a human malignant mesothelioma cell line (H-MESO-1): a biphasic solid and ascitic tumor model. Cancer Res. 47:3199–3205 [PubMed] [Google Scholar]
- Richards WG, Zellos L, Bueno R, Jaklitsch MT, Janne PA, Chirieac LR, Yeap BY, Dekkers RJ, Hartigan PM, Capalbo L, et al. 2006. Phase I to II study of pleurectomy/decortication and intraoperative intracavitary hyperthermic cisplatin lavage for mesothelioma. J Clin Oncol. 24:1561–1567 [DOI] [PubMed] [Google Scholar]
- Robinson BW, Creaney J, Lake R, Nowak A, Musk AW, de Klerk N, Winzell P, Hellstrom KE, Hellstrom I. 2003. Mesothelin-family proteins and diagnosis of mesothelioma. Lancet. 362:1612–1616 [DOI] [PubMed] [Google Scholar]
- Robinson BW, Creaney J, Lake R, Nowak A, Musk AW, de Klerk N, Winzell P, Hellstrom KE, Hellstrom I. 2005. Soluble mesothelin-related protein—a blood test for mesothelioma. Lung Cancer. 49:S109–S111 [DOI] [PubMed] [Google Scholar]
- Rump A, Morikawa Y, Tanaka M, Minami S, Umesaki N, Takeuchi M, Miyajima A. 2004. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 279:9190–9198 [DOI] [PubMed] [Google Scholar]
- Schafer V, von Briesen H, Rubsamen-Waigmann H, Steffan AM, Royer C, Kreuter J. 1994. Phagocytosis and degradation of human serum albumin microspheres and nanoparticles in human macrophages. J Microencapsul. 11:261–269 [DOI] [PubMed] [Google Scholar]
- Scherpereel A, Grigoriu B, Conti M, Gey T, Gregoire M, Copin MC, Devos P, Chahine B, Porte H, Lassalle P. 2006. Soluble mesothelin-related peptides in the diagnosis of malignant pleural mesothelioma. Am J Respir Crit Care Med. 173:1155–1160 [DOI] [PubMed] [Google Scholar]
- Scholler N, Fu N, Yang Y, Ye Z, Goodman GE, Hellstrom KE, Hellstrom I. 1999. Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc Natl Acad Sci U S A. 96:11531–11536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Bosenberg MW, MacPherson MB, Butnor KJ, Heintz NH, Pass HI, Carbone M, Testa JR, Mossman BT. 2009. Activated cAMP response element binding protein is overexpressed in human mesotheliomas and inhibits apoptosis. Am J Pathol. 175:2197–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher JL, Lathrop SA, Cheng K, Hillegass JM, Butnor KJ, Kauppinen RA, Mossman BT, Landry CC. 2010. Gd-labeled microparticles in MRI: in vivo imaging of microparticles after intraperitoneal injection. Small. 6:2678–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarbaker P, Yan T, Stuart O, Yoo D. 2006. Comprehensive management of diffuse malignant peritoneal mesothelioma. Eur J Surg Oncol. 32:686–691 [DOI] [PubMed] [Google Scholar]
- Van der Speeten K, Stuart OA, Mahteme H, Sugarbaker PH. 2009. A pharmacologic analysis of intraoperative intracavitary cancer chemotherapy with doxorubicin. Cancer Chemother Pharmacol. 63:799–805 [DOI] [PubMed] [Google Scholar]
- Van der Speeten K, Stuart OA, Sugarbaker PH. 2009. Pharmacokinetics and pharmacodynamics of perioperative cancer chemotherapy in peritoneal surface malignancy. Cancer J. 15:216–224 [DOI] [PubMed] [Google Scholar]
- van Ruth S, Baas P, Haas RL, Rutgers EJ, Verwaal VJ, Zoetmulder FA. 2003. Cytoreductive surgery combined with intraoperative hyperthermic intrathoracic chemotherapy for stage I malignant pleural mesothelioma. Ann Surg Oncol. 10:176–182 [DOI] [PubMed] [Google Scholar]
- van Ruth S, van Tellingen O, Korse CM, Verwaal VJ, Zoetmulder FA. 2003. Pharmacokinetics of doxorubicin and cisplatin used in intraoperative hyperthermic intrathoracic chemotherapy after cytoreductive surgery for malignant pleural mesothelioma and pleural thymoma. Anticancer Drugs. 14:57–65 [DOI] [PubMed] [Google Scholar]
- Vogel SM, Minshall RD, Pilipovic M, Tiruppathi C, Malik AB. 2001. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am J Physiol Lung Cell Mol Physiol. 281:L1512–L1522 [DOI] [PubMed] [Google Scholar]
- Warheit DB. 2001. Inhaled amorphous silica particulates: what do we know about their toxicological profiles? J Environ Pathol Toxicol Oncol. 20:133–141 [PubMed] [Google Scholar]
- Xia T, Kovochich M, Brant J, Hotze M, Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR, Nel AE. 2006. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 6:1794–1807 [DOI] [PubMed] [Google Scholar]
- Zauderer MG, Krug LM. 2011. The evolution of multimodality therapy for malignant pleural mesothelioma. Curr Treat Options Oncol. 12:163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chertov O, Zhang J, Hassan R, Pastan I. 2011. Cytotoxic activity of immunotoxin SS1P is modulated by TACE-dependent mesothelin shedding. Cancer Res. 71:5915–5922 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.