

Abstract
Senile plaques, extracellular deposits of amyloid β peptide (Aβ), are one of the pathological hallmarks of Alzheimer disease (AD). As the standard immunohistochemical detection method for Aβ deposits, anti-Aβ immunohistochemistry combined with antigen retrieval (AR) by formic acid (FA) has been generally used. Here, we present a more efficient AR for Aβ antigen. On brain sections of AD and its mouse model, a double combination of either autoclave heating in EDTA buffer or digestion with proteinase K plus FA treatment reinforced Aβ immunoreactivity. A further triple combination of digestion with proteinase K (P), autoclave heating in EDTA buffer (A), and FA treatment (F), when employed in this order, gave a more enhanced immunoreactivity. Our PAF method prominently visualized various forms of Aβ deposits in AD that have not been clearly detected previously and revealed numerous minute-sized plaques both in AD and the mouse model. Quantification of Aβ loads showed that the AR effect by the PAF method was 1.86-fold (in the aged human brain) and 4.64-fold (in the mouse brain) higher than that by the FA method. Thus, the PAF method could have the potential to be the most sensitive tool so far to study Aβ pathology in AD and its mouse model.
Keywords: Alzheimer disease, amyloidβ, antigen retrieval, APP-SL mouse, autoclave heating, formic acid, immunohistochemistry, minute plaque, PAF method, proteinase K
Senile plaques (SPs) and neurofibrillary tangles (NFTs) are two pathological hallmarks that characterize brains afflicted with Alzheimer disease (AD). SPs are extracellular deposits of amyloid β peptide (Aβ) mainly consisting of 40 and 42 residues, which are cleavage products of the amyloid precursor proteins (APPs) (Masters et al. 1985; Kang et al. 1987; Iwatsubo et al. 1994). Aβ is a hydrophobic self-aggregating peptide, and the aggregation of soluble Aβ monomers leads to the composition of insoluble fibrillar polymers, Aβ fibrils. NFTs are intracellular aggregated bundles of a hyperphosphorylated form of the microtubule-associated protein tau (Lee et al. 1991; Ballatore et al. 2007).
Although it is not yet completely elucidated whether SPs and NFTs are the causes or the results of AD onset, the aggregation of Aβ is believed to be implicated in the upper stream of the cascade of AD pathogenesis as a pivotal player in the development of dementia: the amyloid hypothesis (Selkoe 1991; Hardy and Higgins 1992; Hardy and Selkoe 2002). Therefore, the detection of SPs or Aβ deposits with high specificity and sensitivity is essential for elucidating the roles of parenchymal Aβ deposition and its implication for the pathogenesis of AD as well as its pathological diagnosis. In 1987, our attempt to attain sensitive Aβ immunohistochemistry (IHC) results on formalin-fixed paraffin-embedded (FFPE) tissue sections led to the development of Aβ antigen retrieval (AR) by formic acid (FA) (Kitamoto et al. 1987). This straightforward method dramatically enhances the detection level of Aβ deposits in the AD brain, and since then, anti-AβIHC coupled with FA treatment has been the standard method in the field of Aβ pathology. There is no guarantee, however, that this method can expose all of the existing Aβ deposits without the remains, and we consider that there might be room for improvement of the AR technique. In fact, we had a chance to find irregular and larger forms of Aβ staining, known as fleecy amyloid deposits (Thal et al. 1999), which appear different from the usual SPs in the entorhinal cortex of some AD cases. These structures of Aβ aggregates stained too faintly to be recognized clearly, and we thought that the AR mediated by FA was not efficient enough to detect these Aβ structures. Thus, we tackled the development of a new AR method with a higher efficiency than the conventional FA method. We could substantially improve Aβ IHC by applying two other AR procedures prior to FA treatment. This new AR method enhanced the detection level of numerous SPs and various Aβ deposits that have not been clearly detected by the conventional method and provides a tool to uncover new aspects of Aβ pathology in AD and its mouse models.
Materials and Methods
Brain Specimens
Human brain specimens were derived from patients with AD (n=11; age range = 63–79 years), non-AD aged individuals with Aβ plaques (n=4; age range = 63–77 years), and negative controls for Aβ IHC, who have no family history of AD, including non-AD aged individuals without Aβ plaques (n=10; age range = 64–94 years) and healthy young individuals (n=6; age range = 21–38 years). For quantification of the Aβ loads, we examined a series of aged human individuals with varying degrees of the Aβ burden (n=54; age range = 69–94 years). As transgenic AD mouse models, we used the APP–Swedish/London (SL) lines 7–5 and 7–9, which overexpress human APPSwe/Lon harboring both the Swedish- and London-type mutations. The levels of APP, Aβ40, and Aβ42 in the brain tissues of the mice of line 7–5 are higher than the corresponding levels in the mice of line 7–9 (Shin et al. 2007). The outline of the ages of each of them is as follows: line 7–5 of APP-SL mice aged 6 months (n=3), 8 months (n=8), 9 months (n=2), 10 months (n=2), 11 months (n=1), 12 months (n=4), 13 months (n=6), 15 months (n=1), 16 months (n=5), and 18 months (n=1); line 7–9 of APP-SL mice aged 3 months (n=3), 6 months (n=3), 9 months (n=3), 12 months (n=2), 13 months (n=3), 15 months (n=1), 16 months (n=2), 18 months (n=2), 19 months (n=1), and 36 months (n=1). The fixation time of brains was 7–13 days with 20% buffered formalin in humans and 3–4 days with 10% buffered formalin in mice. The use of human brains for this work was approved by the Institutional Review Board of Tohoku University Graduate School of Medicine and Tokyo Metropolitan Geriatric Hospital & Institute of Gerontology, and all the animal experiments were done according to the Guidelines for Animal Care and Use at Otsuka Pharmaceutical Co. Ltd.
AR Procedures
FA pretreatment has been the standard AR method for Aβ IHC. In FA pretreatment, brain tissue sections were incubated in 98% FA (Wako Pure Chemical Industries; Osaka, Japan) for 5 min at room temperature. A challenging trial to largely improve the FA method was performed by combining and applying other AR methods prior to FA treatment. The other AR methods used in this study include heating that employs immersion of tissue sections in 10 mM EDTA (pH 3.0, pH 6.0, and pH 10.0) (Murayama et al. 1999), 0.05% citraconic anhydride (pH 3.0, pH 7.4, and pH 10.0) (Namimatsu et al. 2005), and 0.1 M sodium citrate (pH 3.0, pH 7.2, and pH 10.0) (Bataille et al. 2006) solutions, and distilled water (DW) (pH 3.0 adjusted with hydrochloric acid, pH 7.1, and pH 10.0 adjusted with sodium hydroxide), using an autoclave at 105C or 121C for 10 min (Shin et al. 1991) or using a microwave oven at 90C intermittently but for a total of about 10 min; the proteolytic digestion of tissue sections was performed at 37C for 30 min with 1.0 µg/ml of proteinase K (PK) (Wako Pure Chemical Industries) and 100.0 µg/ml trypsin (Wako Pure Chemical Industries) dissolved in 1.0 mM CaCl2/50 mM Tris buffer (pH 7.6). After each AR treatment, these sections were washed with tap water for at least 5 min and then incubated in DW for at least 5 min.
Immunostaining
With pretreatment of various combinations of the AR methods, immunostaining was performed as described (Murayama et al. 1999; Shin et al. 2007) using the polyclonal Aβ antibody 4702 (1:1500) (Shin et al. 2007) and monoclonal Aβ antibodies 6E10 (1:2000–4000; Senetek, Maryland Heights, MO) and 4G8 (1:20,000; Senetek). The concentrations of these antibodies were optimized in consideration of both the immunoreactivity and backgrounds of IHC. Brain sections were incubated with primary antibodies in 0.1% Tween-20/Tris-buffered saline (Tris, 50 mM; NaCl, 500 mM; pH 7.6) containing 5% nonfat dried milk for about 15 hr at room temperature. To exclude nonspecific staining unrelated to these polyclonal and monoclonal antibodies, immunostaining was performed with omission of the antibodies but with all other procedures unchanged in some experiments. Secondary antibodies of EnVision+ system HRP-labeled polymer (Dako; Glostrup, Denmark) were used for the detection of antigen primary mouse or rabbit antibody complexes by diaminobenzidine (DAB). The incubation of the secondary antibodies was for about 1 hr at room temperature. The immunostained brain sections were counterstained with hematoxylin.
Microscopes
The photomicrographs of human and murine samples were captured by an Axiophot2 microscope (Carl Zeiss; Oberkochen, Germany) with Axio Vision version 4.6.3.0 software (Carl Zeiss). In quantification of the Aβ load in the hippocampus of the murine samples, the same system was used in order to assemble sequential micrographs into a single larger one. For measuring the Aβ load, aged human sample photomicrographs were captured by C9600 NanoZoomer (Hamamatsu Photonics; Hamamatsu, Japan) with NDP.view software (Hamamatsu Photonics) because this system is conveniently applicable for capturing and comparing the same regions from serial sections.
Measurement of the Area of Aβ Deposits
In the human brains, we selected three microscopic fields in the fusiform gyrus, which are adequately separated from each other and contain relatively higher Aβ loads. We photographed precisely the same fields in each serial section immunostained following the FA method or PK digestion (P), EDTA autoclaving (A), and FA treatment (F) (in that order; referred to as “PAF”) method. All images were from regions of 1408 µm × 1874 µm. In the mouse brains, we selected the whole hippocampus, and its image was constructed from the photomicrographs of 872 µm × 1100 µm by using panorama module of AxioVision version 4.6.3 (Carl Zeiss). All the images were analyzed by ImageJ version 1.43 m (National Institutes of Health; Bethesda, MD) as follows: 1) each raw image was resolved into three images by the color deconvolution setting in hematoxylin and eosin and DAB; 2) the DAB color image among the three resolved images was selected for analysis; 3) the threshold value of the selected image was set to zero as the minimum value and at the optically optimum value set as the maximum value; 4) the thresholded areas of cerebral amyloid angiopathy and artifacts were excluded by selecting and filling them; 5) areas of the Aβ loads (%) to be measured were within a circle (diameter = 1408 µm), the center of which being in the middle of each image in the aged human brains, and within the circumscribed edge of the hippocampus drawn using the selection tools in the mouse brains; and 6) area fractions of the residual thresholded objects within these selections were measured.
Statistical Analyses
All the statistical analyses were performed with SPSS version 17.0 (SPSS; Chicago, IL).
Results
Development of the Enhanced AR Method for Aβ IHC
For the development of a more efficient Aβ AR method, our strategy was to modify and reinforce the retrieving effects of FA by applying other AR procedures prior to FA treatment. Such AR procedures included autoclave heating in EDTA buffer (the chelating autoclave method) (Murayama et al. 1999) and digestion with PK. With each of these AR procedures followed by FA treatment or with FA treatment alone, immunostaining using the polyclonal anti-Aβ 4702 antibody was performed on brain tissue sections derived from AD patients and APP-SL line 7–5 mice. In immunostaining using the 4702 antibody with no AR, almost no plaques were detected in the AD brains or only a few in the mouse brains (Suppl. Fig. S1). Thus, this 4702 antibody was conveniently used to easily evaluate the effectiveness of the Aβ AR methods.
Compared to the AR procedures above followed by FA treatment and FA treatment alone, both combinations of AR enhanced the Aβ immunoreactive intensity and increased the loads of Aβ plaques, albeit with low to high enhancing effects (Fig. 1A–C,E–G). However, when we applied heating to the EDTA solution, counterstaining of tissue sections with hematoxylin was remarkably thin compared with counterstaining in the FA only method. Notably, the reversed application of the two AR procedures, that is, application of FA treatment and either autoclave heating in EDTA buffer or digestion with PK in this order, showed limited and almost no enhancement, respectively, compared with the single application of FA treatment (data not shown). These results prompted us to try a triple combination of these three AR procedures. The use of the PAF method produced a remarkably stronger enhancement of Aβ immunoreactivity than the two double combinations above (Fig. 1B–D,F–H). In the triple combinations of the three AR methods in different orders other than that used in the PAF method, varying degrees of tissue damage ensued, especially in human brains. Therefore, we refrained from estimating those triple AR combinations. To confirm that this PAF method is universally applicable to Aβ IHC, we examined other Aβ antibodies including 6E10 (Fig. 2A, B, E, F) and 4G8 (Fig. 2C, D, G, H). All of these antibodies showed enhanced Aβ immunoreactivity following the PAF method compared with the FA method, albeit with varied enhancing effects. Omission of the polyclonal or monoclonal primary antibodies in Aβ IHC with the PAF method totally abolished positive immunostaining (Suppl. Fig. S2), which excludes the possibility that immunoreactivity augmented and disclosed following the PAF method was due to nonspecific staining. In addition, no artifactual immunostaining was observed in the brain sections from the normal younger individuals following Aβ IHC assisted by the PAF method (Suppl. Fig. S3).
Figure 1.
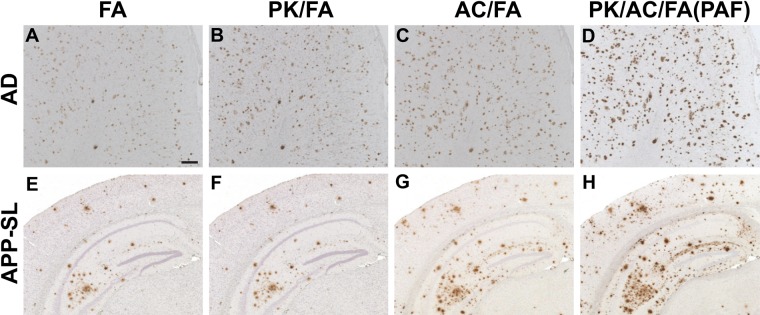
Enhancement of formic acid (FA)–mediated amyloid β peptide (Aβ) antigen retrieval. Serial brain tissue sections from a 74-year-old male patient with Alzheimer disease (A–D) and from a 13-month-old amyloid precursor protein–Swedish/London (APP-SL) mouse of line 7–5 (E–H) were immunostained with anti-Aβ antibody 4702 following pretreatment by FA alone (FA) (A, E); combination of digestion with proteinase K and FA (PK/FA) (B, F); combination of autoclave heating in EDTA buffer and FA (AC/FA) (C, G); and triple combination of digestion with proteinase K, autoclave heating in EDTA buffer, and FA (PK/AC/FA) (D, H). Pictures (A–D) are from the temporal cortex. Scale bar = 200 µm (A–H).
Figure 2.

General application of the proteinase K digestion (P), EDTA autoclaving (A), and formic acid (FA) treatment (F) (in that order; referred to as “PAF”) method in amyloid β peptide (Aβ) immunohistochemistry. Serial (A–B, C–D, E–F, G–H) brain tissue sections from a 74-year-old male patient with Alzheimer disease (the same patient as shown in Fig. 1) (A–D) and a 16-month-old (E, F) and a 15-month-old (G, H) amyloid precursor protein–Swedish/London (APP-SL) mouse of line 7–5 were immunostained with monoclonal anti-Aβ antibodies 6E10 (A, B, E, F) and 4G8 (C, D, G, H) following pretreatment by the FA (A, C, E, G) and PAF methods (B, D, F, H). Pictures are from the cingulate cortex (A, B) and the frontal cortex (C, D). Scale bar = 200 µm (A–H).
IHC Analysis of AD Brains by the PAF Method
Serial sections of the AD brains pretreated with either the PAF or FA method were immunostained with the anti-Aβ 4702 antibody, analyzed for pathological Aβ deposits, and compared between these two methods (Fig. 3). In the cerebral cortex and hippocampus of the sections pretreated with the PAF method, larger Aβ plaques (diameter > ~15 µm) showed immunoreactive enhancement with an enlarged robust contour, although there was no apparent increase in the number of these larger Aβ plaques. The prominent effect given by the PAF method was the disclosure of numerous minute-sized (diameter < ~15 µm) fine-granular plaques (hereafter referred to as “minute plaques”) in these brain regions, which were not evidently detected by the FA method (Figs. 1A, D and 3A–D). Remarkably, the number of these minute plaques increased with elevation of the total Aβ load (data not shown). Thus, all of the AD brains examined by the PAF method contained a much higher load of Aβ plaques in the cerebral cortex and hippocampus than those by the FA method. In the entorhinal cortex adjacent to the subiculum, large and irregular contours of Aβ staining appeared, which were composed of fine- to coarse-granular or diffuse Aβ deposits following the PAF method. These were distributed from near the subpial layer into the deep cortex (Fig. 3G, H), which were reported as fleecy Aβ deposits (Thal et al. 1999). These Aβ deposits were only faintly or not appreciably stained following FA treatment (Fig. 3E, F). In the cerebral white matter, small but significant amounts of Aβ deposits in the diffuse or granular form were previously shown to occur (Wisniewski et al. 1989; Behrouz et al. 1991; Uchihara et al. 1995). The PAF method gave immunoreactive enhancement and revealed larger amounts of granular Aβ deposits in the cerebral white matter (Fig. 3I–L). The PAF method also enhanced Aβ immunoreactivity of ribbon-like infiltration in the subpial layer of the cerebral cortex and that of cerebral amyloid angiopathy in the vessels of the brain (data not shown). Thus, the PAF method dramatically enhanced the detection level of a spectrum of all morphological forms of Aβ deposits.
Figure 3.

Amyloid β peptide (Aβ) pathology of Alzheimer disease (AD) brains enhanced by the proteinase K digestion (P), EDTA autoclaving (A), and formic acid (FA) treatment (F) (in that order; referred to as “PAF”) method. Serial brain tissue sections from a 72-year-old female AD patient (the identical patient as shown in Suppl. Fig. S1A,B) (A–D), a 74-year-old male AD patient (the same patient as shown in Fig. 1) (E–H), and a 74-year-old female AD patient (I–L) were immunostained with the 4702 antibody following pretreatment by the FA (A, B, E, F, I, J) and PAF methods (C, D, G, H, K, L). Pictures are from the frontal cortex (A–D), the entorhinal cortex (E–H), and the frontal white matter (I–L). B, D, F, H, J, and L are higher magnification pictures of areas outlined by squares in A, C, E, G, I, and K, respectively. Scale bars = 200µm (A, C, I, K); 50 µm (B, D, F, H); 100µm (E, G); and 20 µm (J, L).
IHC Analysis of the AD Mouse Model by the PAF Method
We examined the brains of the AD APP-SL mice in the same way as for the AD brains. In our previous (Shin et al. 2007) and present studies using conventional Aβ IHC coupled with the FA method, younger APP-SL mice (< ~9 months) showed no occurrence of Aβ deposition, and older mice (≥ ~9 months) exhibited deposition of Aβ plaques that increased its burden with age. Application of Aβ IHC assisted by the PAF method to those younger mice also failed to reveal Aβ deposition. Therefore, in mice showing no evidence of Aβ deposition as evaluated by the FA method, the PAF method did not create any occurrence of Aβ deposition. Thus, these two methods show no difference in their ability to demonstrate the absence of Aβ deposition. In the older mice showing evidence of Aβ deposition, the PAF method was more efficient than the FA method for AR. The enhanced immunoreactive profile was shown to enlarge the sizes and to increase the numbers and immunointensities of Aβ plaques (Figs. 1E,H and 2E–H). Notably, minute Aβ plaques appeared to have a similar morphology as those seen in the AD brains. These minute plaques occur in brain samples containing significant amounts of Aβ burden and prevail dominantly with severity of Aβ burden, as was shown in the AD brain. In 9-month-old APP-SL mice that show an initial appearance of Aβ deposition, Aβ deposits were indiscernible when evaluated by the FA method. These deposits were prominently visualized as distinct Aβ deposits by the PAF method (data not shown). Thus, the PAF method could have an advantage for the retrieval of antigens in Aβ IHC in comparison with the FA method in the AD mouse model as well as humans.
Efficiency of Aβ AR by the PAF Method
We measured the areas of Aβ deposit loads in the serial sections immunostained with the 4702 antibody following the PAF or FA method, and the total sums of the Aβ-loaded areas per the whole area analyzed were compared between the two methods. In the aged human brains, Aβ deposit loads measured in the fusiform cortex by the PAF method were significantly correlated with those by the FA method (p=9 × 10−69) (Fig. 4A). We compared the ratio of the Aβ deposit area from the PAF method with the ratio from the FA method. The AR effect of the PAF method (Fig. 4C) was significantly higher than that of the FA method (1.86-fold at the median) (p=2 × 10−28). In the APP-SL line 7–9 mice, Aβ deposit loads measured in the whole hippocampus by the PAF method were also significantly correlated with those by the FA method (p=0.003) (Fig. 4B). The enhancing effect of the PAF method compared with that of the FA method (the ratio as described above in aged human brains) was significantly higher and 4.64-fold at the median (p=0.01) (Fig. 4D). Thus, the PAF method produced Aβ deposit loads that were consistently and significantly larger than those produced by the FA method.
Figure 4.

Effect of amyloid β peptide (Aβ) antigen retrieval (AR) by the proteinase K digestion (P), EDTA autoclaving (A), and formic acid (FA) treatment (F) (in that order; referred to as “PAF”) method over that by the FA method. (A, B) In immunohistochemistry (IHC) with the 4702 antibody, Aβ loads (%) measured in the fusiform cortex of each case from the aged human brains (A) or in the hippocampus of each from the amyloid precursor protein–Swedish/London (APP-SL) mice (B) following the PAF method were plotted against those following the FA method. Significant correlations between the two AR methods were verified both in the aged human brains (Spearman rank correlation coefficient, r s=0.92; p=9 × 10−69) and in the mouse brains (r s=0.80; p=0.003). (C, D) The effect of Aβ AR by the PAF method was significantly higher than that by the FA method both in the aged human and in the mouse brains. In IHC with the 4702 antibody, Aβ-loaded areas measured following the PAF method compared with those following the FA method (set at 1.00; the dotted lines) were 1.59-fold at the 25th percentile, 1.86-fold at the 50th percentile, and 2.31-fold at the 75th percentile in the aged human brains (C) and 3.78-fold at the 25th percentile, 4.64-fold at the 50th percentile, and 12.32-fold at the 75th percentile in the mouse brains (D). (C) Ten (≥3.39) and (D) one (92.79) outliers are not shown. **p=2 × 10−28, *p=0.01; analyzed by Wilcoxon signed-rank test. n=162 from 54 individuals where three regions per case examined (A, C), and n=11 (B, D).
Evaluation of PK versus Trypsin in Enzymatic Digestion and Some Other Solutions in Autoclave Heating
To obtain a more effective enzymatic digestion than PK digestion, we additionally tested trypsin in the double combination of enzymatic digestion and the FA method. Trypsin digestion, when applied prior to the FA method, produced Aβ immunostaining slightly higher in its intensity than the FA method only. However, its efficacy was comparable to that of PK digestion (Suppl. Fig. S4). To obtain a more effective autoclave heating than that in the solution of 10 mM EDTA (pH 6.0) at 121C, we additionally tested solutions of EDTA (pH 3.0 and pH 10.0), DW (pH 3.0, pH 7.1, and pH 10.0), citraconic anhydride (pH 3.0, pH 7.4, and pH 10.0), and sodium citrate (pH 3.0, pH 7.2, and pH 10.0) as well as temperatures of 90C, 105C, and 121C. Among these different conditions, DW (pH 10.0 and 105C) and sodium citrate (pH 7.2 and 105C) produced high Aβ AR effects. A similar effect was observed for EDTA (pH 6.0 and 121C), although sodium citrate slightly damaged the tissue sections (Suppl. Fig. S5). The triple combinations using additional PK or trypsin digestion as the initial step produced higher Aβ AR effects than each of those double combinations, although heating in sodium citrate solution or basic water damaged the tissue sections. Further nonspecific staining was observed in the brain sections of the APP-SL mice applied by the triple combination of trypsin digestion, EDTA autoclaving, and the FA method (Suppl. Figs. S6 and S7). Thus, the triple combination of 1) PK digestion, 2) autoclave heating in 10 mM EDTA (pH 6.0 and 121C), and 3) FA treatment produced the highest Aβ AR effects without damaging tissue sections or producing nonspecific staining.
Discussion
The masking of antigens by aldehyde fixatives or by paraffin-embedding procedures is a problem for IHC studies. To overcome this problem, enzymatic digestion, FA treatment, and high-temperature heating have been developed. Among these, FA treatment is the standard method mainly used for Aβ IHC of FFPE brain tissue sections, although it was originally developed for the immunoreactive enhancement of cerebral amyloids (prion protein and Aβ) and systemic amyloids (amyloid A and prealbumin) (Kitamoto et al. 1987). The pretreatment of protein digestion with an enzyme such as trypsin had been used for IHC but only in a limited application (Battifora and Kopinski 1986; Huang et al. 1976; Mepham et al. 1979). In 1991, the advent of the heating AR method was a breakthrough in the field of IHC. Shin et al. (1991) reported that the procedure of hydrated autoclaving uncovers the masked epitopes of the microtubule-associated protein tau, showing that high-temperature heating serves as an efficient AR method. Shi et al. (1991) reported that microwave heating also shows an AR effect by testing a variety of antigens and antibodies, establishing the milestone of AR for FFPE tissue sections. Moreover, Shi et al. (1996) devised the test battery approach, which can efficiently determine the optimum protocols of AR for each antigen by comparing the immunostaining results between different kinds of solutions, temperatures, and pH (O’Leary 2001). As one good example using the test battery approach, it was demonstrated that AR procedures can also be applied to immunoelectron microscopy for amyloid deposits composed of the κ light chain or transthyretin (Rocken and Roessner 1999).
Aβ AR by FA is proposed because of the unfolding of the conformational amyloid polymers and thereby the exposing of Aβ antigens through acidic hydrolysis (Kitamoto et al. 1987). Further, FA is suggested to esterify serine residues in Aβ peptides and to alter the conformation of the amyloid polymers, as nuclear magnetic resonance imaging has revealed (Klunk et al. 1994). On the other hand, the possible mechanisms underlying AR by high-temperature heating are summarized as follows: 1) breaking of aldehyde-induced cross-linkage involving antigenic proteins, 2) extraction of diffusible blocking proteins, 3) precipitation of antigenic proteins, and 4) increased penetration of antibodies with better access to epitopes due to rehydration of the tissue sections (Suurmeijer and Boon 1993). In addition, the application of divalent/trivalent chelators in high-temperature heating removes metal ions that mask antigenic proteins (Murayama et al. 1999; Yamamoto et al. 2002; Shin et al. 2003). If autoclave high-temperature heating with EDTA chelators (Murayama et al. 1999) is then used prior to FA treatment, with the aim of affecting its activity of Aβ AR, it is likely that FA gains permeability through the tissue sections up to the unmasked Aβ fibrils. Similarly, if digestion with PK is applied prior to FA treatment, then proteolytic digestion of blocking proteins that surround Aβ fibrils might occur and unmask and thereby expose them to FA. In short, the procedures of EDTA autoclaving and PK digestion might assist the access of FA to Aβ fibrils, resulting in reinforcement of Aβ AR of FA. Indeed, we demonstrated that the combination of preceding EDTA autoclaving or PK digestion with subsequent FA treatment enhanced the AR effects of FA, and the aforementioned hypotheses might indeed be true. In support of this hypothesis, FA treatment prior to PK digestion or EDTA autoclaving gave no or only a minimally discernible enhancement in Aβ immunoreactivity compared with single FA treatment (data not shown). Our present results, together with other previous studies, show that PK digestion and EDTA autoclaving apparently differ in the mechanisms of the reinforcement of Aβ AR. If both PK digestion and EDTA autoclaving are combined with the FA method, their effects on Aβ AR by FA might be complementary rather than equivalent. Indeed, the triple combination of PK digestion and EDTA autoclaving with FA treatment provided a further stronger enhancement of Aβ AR.
Our results based on the PAF method suggest that previous IHC studies performed by the conventional FA method might have underestimated the quantitative burden of Aβ pathology and that brains with AD and its mouse models produce much more Aβ accumulation than previously assumed. Further, the presence of minute plaques revealed by the PAF method remains to be clarified for its implication for Aβ pathology of AD and its mouse models. Notably, the molecular layer of the hippocampal dentate gyrus is the brain region that produces larger amounts of the minute plaques in the APP-SL mice than in the aged humans. This observation might explain partly, albeit not totally, why the retrieving effects differ between the human and mouse brains.
This powerful PAF method could reveal numerous SPs and various Aβ deposits that have not been detected so far. Therefore, the PAF method could serve as a sensitive IHC tool to give new insights into Aβ pathology of AD and its mouse models. We speculate that the PAF method may be the Aβ AR method with the highest efficiency so far and could be used in place of the conventional FA method.
Supplementary Material
Acknowledgments
We thank H. Kudo and H. Murayama for technical assistance and Daniel Berrar (Tokyo Institute of Technology) for the proofreading of this article and helpful comments.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by the Starter Research Subvention of Tohoku University Graduate School of Medicine and Grant-in-Aid for Scientific Research on Innovative Areas (Comprehensive Brain Science Network) from the Ministry of Education, Science, Sports and Culture of Japan.
Supplementary material for this article is available on the Journal of Histochemistry & Cytochemistry Web site at http://jhc.sagepub.com/supplemental.
References
- Ballatore C, Lee VM, Trojanowski JQ. 2007. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 8:663–672 [DOI] [PubMed] [Google Scholar]
- Bataille F, Troppmann S, Klebl F, Rogler G, Stoelcker B, Hofstadter F, Bosserhoff AK, Rummele P. 2006. Multiparameter immunofluorescence on paraffin-embedded tissue sections. Appl Immunohistochem Mol Morphol. 14:225–228 [DOI] [PubMed] [Google Scholar]
- Battifora H, Kopinski M. 1986. The influence of protease digestion and duration of fixation on the immunostaining of keratins: a comparison of formalin and ethanol fixation. J Histochem Cytochem. 34:1095–1100 [DOI] [PubMed] [Google Scholar]
- Behrouz N, Defossez A, Delacourte A, Mazzuca M. 1991. The immunohistochemical evidence of amyloid diffuse deposits as a pathological hallmark in Alzheimer’s disease. J Gerontol. 46:B209–B212 [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 297:353–356 [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. 1992. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 256:184–185 [DOI] [PubMed] [Google Scholar]
- Huang SN, Minassian H, More JD. 1976. Application of immunofluorescent staining on paraffin sections improved by trypsin digestion. Lab Invest. 35:383–390 [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. 1994. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 13:45–53 [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. 1987. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 325:733–736 [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Ogomori K, Tateishi J, Prusiner SB. 1987. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 57:230–236 [PubMed] [Google Scholar]
- Klunk WE, Xu CJ, Pettegrew JW. 1994. NMR identification of the formic acid-modified residue in Alzheimer’s amyloid protein. J Neurochem. 62:349–354 [DOI] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos L, Jr., Trojanowski JQ. 1991. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 251:675–678 [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. 1985. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 82:4245–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mepham BL, Frater W, Mitchell BS. 1979. The use of proteolytic enzymes to improve immunoglobulin staining by the PAP technique. Histochem J. 11:345–357 [DOI] [PubMed] [Google Scholar]
- Murayama H, Shin RW, Higuchi J, Shibuya S, Muramoto T, Kitamoto T. 1999. Interaction of aluminum with PHFtau in Alzheimer’s disease neurofibrillary degeneration evidenced by desferrioxamine-assisted chelating autoclave method. Am J Pathol. 155:877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namimatsu S, Ghazizadeh M, Sugisaki Y. 2005. Reversing the effects of formalin fixation with citraconic anhydride and heat: a universal antigen retrieval method. J Histochem Cytochem. 53:3–11 [DOI] [PubMed] [Google Scholar]
- O’Leary TJ. 2001. Standardization in immunohistochemistry. Appl Immunohistochem Mol Morphol. 9:3–8 [PubMed] [Google Scholar]
- Rocken C, Roessner A. 1999. An evaluation of antigen retrieval procedures for immunoelectron microscopic classification of amyloid deposits. J Histochem Cytochem. 47:1385–1394 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. 1991. The molecular pathology of Alzheimer’s disease. Neuron. 6:487–498 [DOI] [PubMed] [Google Scholar]
- Shi SR, Cote RJ, Yang C, Chen C, Xu HJ, Benedict WF, Taylor CR. 1996. Development of an optimal protocol for antigen retrieval: a ‘test battery’ approach exemplified with reference to the staining of retinoblastoma protein (pRB) in formalin-fixed paraffin sections. J Pathol. 179:347–352 [DOI] [PubMed] [Google Scholar]
- Shi SR, Key ME, Kalra KL. 1991. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 39:741–748 [DOI] [PubMed] [Google Scholar]
- Shin RW, Iwaki T, Kitamoto T, Tateishi J. 1991. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab Invest. 64:693–702 [PubMed] [Google Scholar]
- Shin RW, Kruck TP, Murayama H, Kitamoto T. 2003. A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer’s disease. Brain Res. 961:139–146 [DOI] [PubMed] [Google Scholar]
- Shin RW, Ogino K, Shimabuku A, Taki T, Nakashima H, Ishihara T, Kitamoto T. 2007. Amyloid precursor protein cytoplasmic domain with phospho-Thr668 accumulates in Alzheimer’s disease and its transgenic models: a role to mediate interaction of Abeta and tau. Acta Neuropathol. 113:627–636 [DOI] [PubMed] [Google Scholar]
- Suurmeijer AJ, Boon ME. 1993. Notes on the application of microwaves for antigen retrieval in paraffin and plastic tissue sections. Eur J Morphol. 31:144–150 [PubMed] [Google Scholar]
- Thal DR, Sassin I, Schultz C, Haass C, Braak E, Braak H. 1999. Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N-terminal truncated fragments of Abeta. J Neuropathol Exp Neurol. 58:210–216 [DOI] [PubMed] [Google Scholar]
- Uchihara T, Kondo H, Akiyama H, Ikeda K. 1995. White matter amyloid in Alzheimer’s disease brain. Acta Neuropathol. 90:51–56 [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J. 1989. Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol. 78:337–347 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Shin RW, Hasegawa K, Naiki H, Sato H, Yoshimasu F, Kitamoto T. 2002. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J Neurochem. 82:1137–1147 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.