

Abstract
Glutamate transporters (GLT-1, GLAST, EAAC1) limit the actions of excitatory amino acids. Because a disturbed transporter operation can cause or aggravate neurological diseases, transporters are of considerable neuropathological interest. Human samples, however, are seldom obtained fresh. Here, we used mice brains to study how fast glutamate transporters are degraded after death. Immunoblots showed that terminal GLT-1 epitopes (within residues 1–26 and 518–573) had mostly disappeared after 24 hr. GLAST termini (1–25 and 522–543) degraded slightly slower. In contrast, epitopes within central parts of GLT-1 (493–508) and the EAAC1 C-terminus (510–523) were readily detectable after 72 hr. The decline in immunoreactivity of the GLT-1 and GLAST termini was also seen in tissue sections, but proteolysis did not happen synchronously in all cells. At 24 hr, scattered cells remained strongly immunopositive, while the majority of cells were completely immunonegative. GLAST and GLT-1 co-localized in neocortical tissue, but at 12 hr, many GLAST-positive cells had lost the GLT-1 termini. The uneven disappearance of labeling was not observed with the antibodies to GLT-1 residues 493–508. The immunoreactivity to this epitope correlated better with the reported glutamate uptake activity. Thus, postmortem delay may affect epitopes differently, possibly causing erroneous conclusions about relative expression levels.
Keywords: postmortem proteolysis, glutamate transporter, knockout mice, Slc1a1, Slc1a2, Slc1a3
Glutamate is the major excitatory neurotransmitter in the mammalian brain and is inactivated by cellular uptake catalyzed by glutamate transporter proteins: GLAST (EAAT1; slc1a3), GLT-1 (EAAT2; slc1a2), EAAC1 (EAAT3; slc1a1), EAAT4 (slc1a6), and EAAT5 (slc1a7). GLAST and EAAC1 are, respectively, selectively expressed in astrocytes and neurons (Lehre et al. 1995; Schmitt et al. 1997; Holmseth et al. 2012a). In contrast, GLT-1 is mostly in astrocytes (Danbolt et al. 1992; Levy et al. 1993; Lehre et al. 1995), and only a few percentage are in axon terminals (Chen et al. 2004; Furness et al. 2008). Perturbations in glutamate uptake have been described in several neurodegenerative disorders, and it is important to obtain reliable data on the distribution and expression of glutamate transporters in humans (Bergles et al. 1999; Conti and Weinberg 1999; Danbolt 2001; Beart and O’Shea 2007; Jiang and Amara 2011).
Most, if not all, mature astrocytes in the rat cerebral cortex express both GLT-1 and GLAST and target the proteins to all of their ramifications (e.g., Lehre et al. 1995; Haugeto et al. 1996; Danbolt 2001). This creates a fine mesh where tissue prisms devoid of transporters are tiny (For virtual microscopy, see Holmseth et al. [2009]: http://www.rbwb.org. Choose “Neurotransporter Atlas” and then “Access Repository and Virtual Microscope.”).
Some investigators have reported that GLT-1 and GLAST distributions in humans are similar to those seen in rodents (e.g., Bjørnsen et al. 2007; Melone et al. 2011), while other investigators have observed less co-localization of GLAST and GLT-1 in humans and depicted large patches of tissue lacking glutamate transporters (e.g., Fray et al. 1998; Banner et al. 2002). We asked if the different results could be due to the time from death to tissue preservation. This is a legitimate question considering reported glutamate transporter labeling redistribution in human samples (Tessler et al. 1999; Melone et al. 2011) and the demonstration by Western blotting that the C-termini of glial glutamate transporters are proteolyzed quickly after death (Beckstrøm et al. 1999). In the case of GLT-1, the C-terminal cleavage site was between an epitope within residues 493–508 and an epitope within residues 518–525 (Beckstrøm et al. 1999). Subsequent studies identified a caspase-3 cleavage site between residues 505 and 506 (G. Gegelashvili et al. 2002; M. Gegelashvili et al. 2002; Boston-Howes et al. 2006). The immunoreactivity to the 493–508 epitope correlated better with the transport activity, suggesting that truncated transporters were still active (Beckstrøm et al. 1999), which is in agreement with a recent report (Leinenweber et al. 2011).
Here, we compare immunoblotting and immunocytochemistry at several different postmortem intervals using both N-terminal and C-terminal antibodies to both GLT-1 and GLAST as well as antibodies to the C-terminus of EAAC1 and the central parts of GLT-1. We show that the degradation is likely to involve several different enzymes and that the various epitopes are degraded with different rates. Further, the degradation of GLT-1 and GLAST varies greatly between cells. This may explain the different observations on human tissue described above.
Materials and Methods
Materials
Chemicals, reagents, and equipment for electrophoresis were the same described previously (Zhou et al. 2012). Paraformaldehyde and glutaraldehyde were from TAAB (Reading, UK). The primary antibodies to glutamate transporters used in the present study are summarized in Table 1. The antigenic peptides representing parts of GLAST, GLT-1, and EAAC1 are referred to by capital letters “A,” “B,” and “C,” respectively, followed by numbers indicating the corresponding amino acid residues in the respective sequences (numbering from the rat sequences). The antibodies’ names are thereby informative. Because antibody batches may differ from each other, antibody batches are further identified by the unique identification number (“Ab#”); they are given by our electronic laboratory information system (software provided by Science Linker AS; Oslo, Norway). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Table 1.
Primary Antibodies to Transporter Proteins
| Ab# | Code | Antibody Name | Animal No. | HS | T.Prot | Peptide Name | Peptide (Antigen) Sequence | Reference |
|---|---|---|---|---|---|---|---|---|
| 22 | 19950129 | Anti-A1 | 20492 | Rb | GLAST | A1–25 | MTKSNGEEPRMGSRME RFQQGVRKRC-(amide) | Dehnes et al. 1998 |
| 286 | 19980716 | Anti-A1 | 5383 | Sh | GLAST | A1–25 | MTKSNGEEPRMGSR MERFQQGVRKRC-(amide) | Unpublished |
| 314 | 19980729 | Anti-A522 | 8D0161 | Rb | GLAST | A522–541 | PYQLIAQDNEPEKPVADSET-(amide) | Holmseth et al. 2009 |
| 210 | 19970621 | Anti-B2 | 3261 | Sh | GLT-1 | B2–26 MAP | ASTEGANNMPKQVEV RMHDSHLSSE-(MAP) | Unpublished |
| 360 | 20020710 | Anti-B12 | 26970 | Rb | GLT-1 | B12–26 | KQVEVRMHDSHLSSE-(amide) | Furness et al. 2008 |
| 8 | 19950128 | Anti-B493 | 3024 | Sh | GLT-1 | B493–508 | YHLSKSELDTIDSQHR-(amide) | Unpublished |
| 95 | 19940529 | Anti-B493 | 84946 | Rb | GLT-1 | B493–508 | YHLSKSELDTIDSQHR-(amide) | Ullensvang et al. 1997 |
| 1125 | 19981009 | Anti-B518 | M | GLT-1 | B518–525 | TQSVYDDT | Levy et al. 1993 | |
| 9C4 clone | ||||||||
| 355 | 20020905 | Anti-B563 | 1B0707 | Rb | GLT-1 | B563–573 | SVEEEPWKREK-(free acid) | Holmseth et al. 2009 |
| 371 | 20030103 | Anti-C491 | 1B0683 | Rb | EAAC1 | C491–523 | CLDNEDSDTKKSYVNGGFSVDKSDTISFTQTSQF-(free acid) | Holmseth et al. 2005 |
| 565 | 20051031 | Anti-C510 | 4131 | Sh | EAAC1 | C510–524 | VDKSDTISFTQTSQF- (free acid) | Holmseth et al. 2012b |
Antibodies were produced as described before (Danbolt et al. 1998). Briefly, peptides representing parts of the sequences of the rat GLT-1 (EAAT2; slc1a2) (Pines et al. 1992), rat GLAST (EAAT1; slc1a3) (Storck et al. 1992; Tanaka 1993), and rat EAAC1 (EAAT3; slc1a1) (Kanai and Hediger 1992; Bjørås et al. 1996) were synthesized as C-terminal amides, free acids, or multiple antigenic peptides (MAPs) as indicated. With the exception of the MAP peptide, the other peptides were coupled to carrier proteins (keyhole limpet hemocyanin) with glutaraldehyde before being used to immunize hosts (HS: Rb = rabbits; Sh = sheep). The antipeptide antibodies were isolated by affinity chromatography using columns with immobilized peptide (coupled to N-hydroxysuccinimide–activated agarose) and tested as described (Holmseth et al. 2006; Holmseth et al. 2012b). The monoclonal antibody (anti-B518; 9C4 clone) was made by immunizing mice with a purified glutamate transporter (Danbolt et al. 1990) and afterwards identifying the exact epitope (Levy et al. 1993). Ab# = antibody number; M = mouse; T.Prot. = target protein; HS = host species.
Animals, Immunizations, and Collection of Tissue
All animal experimentation was carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 80–23), revised 1996, and the European Communities Council Directive of November 24, 1986 (86/609/EEC). Formal approval to conduct the experiments described was obtained from the animal subjects review board of our institutions. Mice in the C57Black/6 background deficient in GLT-1 (EAAT2; slc1a2) (Tanaka et al. 1997) or GLAST (EAAT1; slc1a3) (Watase et al. 1998), hereafter referred to as GLT1-KO and GLAST-KO, respectively, were kept at the animal facility at the Governmental Institute of Public Health (Oslo, Norway). The mice were bred as heterozygotes so that knockout mice and wild-type mice could be found in the same litters. Other animals were kept in the animal facility at the Institute of Basic Medical Sciences. Sheep were immunized with synthetic peptides (Table 1) and bled as described (Danbolt et al. 1998) but using subcutaneous rather than intracutaneous injections.
Tissue Preparation
Mice (34–71 days old) were killed by cervical dislocation and decapitated. The heads were stored in plastic bags at room temperature from 1–72 hr before the brains were dissected. Control brains were dissected immediately (0 hr). The room temperature was not monitored, but day-night fluctuations were small and probably in the range of 18–25C. The tissue was either homogenized in 32 volumes of 1% sodium dodecyl sulfate with 10 mM sodium phosphate buffer (pH 7.4), 5 mM EDTA, and 1 mM PMSF for immunoblotting (see below) or immersion fixed (6–10 hr, 4C; 4% formaldehyde in 0.1 M sodium phosphate buffer, pH 7.4) for immunocytochemistry. The immersion-fixed tissue was cryoprotected in sucrose (10, 20, and 30% w/v), cut using a Cryotome Microm HM450 (thickness = 40 µm; Thermo Fisher Scientific, Waltham, MA), and developed with antibodies as described below. Sections from wild-type and knockout mice were developed together.
Electrophoresis and Immunoblotting
Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto nitrocellulose membranes as described (Lehre et al. 1995). The blots were washed in Tris-HCl–buffered saline, incubated in blocking solution consisting of 1% (w/v) bovine serum albumin and 0.05% (v/v) Tween 20 in Tris-HCl–buffered saline, and then incubated (overnight, room temperature) with primary antibodies (Table 1) as indicated and developed with alkaline phosphatase–conjugated secondary antibodies as described before (Danbolt et al. 1992).
Immunocytochemistry
This was performed exactly as described previously (Holmseth et al. 2009). We validated the immunolabeling by processing tissues from wild-type, mice and from GLT-1 (Tanaka et al. 1997) and GLAST (Watase et al. 1998) of knockout mice in parallel (for discussion of the importance of this, see Holmseth et al. [2012b]). Briefly, for immunoperoxidase labeling, free-floating microtome sections (40 μm thick) were treated with hydrogen peroxide to quench endogenous peroxidase, and with 1 M ethanolamine-HCl (pH 7.4) in sodium phosphate buffer (30 min) to block free aldehyde groups. Sections were then washed in Tris-buffered saline with Tween 20 (TBST), blocked with 10% newborn calf serum (NCS) in TBST (300 mM NaCl, 0.5% Triton X-100, and 100 mM Tris-HCl, pH 7.4) for 1 hr to saturate unspecific protein binding sites, and incubated overnight with primary antibodies diluted in 10% NCS in TBST as above, followed by secondary antibodies. Triton X-100 was included to maximize penetration of the immunoreagents (for discussion, see Danbolt et al. [1998]). The sections were observed in an Axioskop 2 plus equipped with an AxioCam MRc r1.2 camera (Zeiss; Jena, Germany) and Zeiss camera software. Contrast and brightness were not adjusted after acquisition. When stated (“Montage”), several overlapping images were merged using Adobe Photoshop (version 7; Adobe Systems, San Jose, CA).
For immunofluorescent labeling, the sections were rinsed (3 × 5 min) in TBST, treated with 1 M ethanolamine, washed as above, and incubated (1 hr) in TBST containing 10% NCS and 3% bovine serum albumin. This was followed by incubation with primary antibodies and finally with secondary antibodies (Alexa Fluor 488 donkey anti-rabbit IgG and Alex Fluor 555 donkey anti-sheep IgG; Molecular Probes, Eugene, OR) in 1:1000 dilution. The sections were observed in a Zeiss Axioplan 2 microscope equipped with a Zeiss LSM 5 Pascal confocal scanner head with Zeiss LSM software (version 3.2 SP2). Pinhole size was around 1 area unit, optimized for each wavelength to ensure confocality. Excitation wavelengths were 488 and 555 nm, with corresponding emission wavelengths at 520 and 568 nm. Contrast and brightness were not adjusted after acquisition.
Results
Western Blots Reveal Different Rates of Glutamate Transporter Proteolysis
Immunoblots (Fig. 1) showed that both the N-terminus and the C-terminus of mouse GLT-1 were quickly proteolyzed, being mostly absent after 24 hr, while the central parts of the GLT-1 protein were readily detectable (using the anti-B493 antibodies) even after 72 hr. Further, the immunoreactivity of the anti-B518 (9C4) monoclonal antibody to residues 518–525 disappeared as fast as that of the anti-B563 antibodies, suggesting that the cleavage site must be somewhere between the epitope recognized by the anti-B493 and anti-B518 (9C4) antibodies. This is in agreement with our previous data from rats (Beckstrøm et al. 1999). We add here that the fastest proteolysis appeared to be on the C-terminal side because the band recognized by anti-B563 antibodies disappeared without broadening, while the band recognized by the anti-B12 antibodies (to residues 12–26) broaden before disappearing. Also, the termini of GLAST were prone to proteolysis as the immunoreactivity with both the anti-A1 antibodies (not shown) and the anti-A522 antibodies disappeared, although not quite as fast as that of the anti-B12 and the anti-B563 antibodies. In contrast, the immunoreactivity detected by the anti-C491 antibody to EAAC1 was only slowly degrading, being readily detectable even at 72 hr. Because antibodies to the central parts of GLAST are not available, we have not tested if the central part of GLAST is similarly resistant to proteolysis.
Figure 1.

Postmortem proteolysis of GLT-1. Mouse forebrains were stored 0–72 hr at room temperature before being solubilized and immunoblotted with glutamate transporter antibodies as indicated. Note that the epitopes recognized by the anti-B12 antibodies to the N-terminus (Ab#360, 0.2 μg/ml) and the anti-B563 (Ab#355, 0.2 μg/ml) and the anti-B518 (Ab#1125, anti-B518, 1:150) antibodies to the C-terminus of GLT-1 had mostly disappeared after 24 hr, while the main part of the GLT-1 protein was still intact as indicated by the reactivity of the anti-B493 antibody (Ab#95, 0.2 μg/ml). Also note that the C-terminal antibodies gave images without band broadening. The weak lower band observed with the N-terminal antibodies likely represents GLT-1 without the C-terminus. Similarly, the labeling obtained with the anti-B493 antibodies comprised the band representing the intact GLT-1 and comprised two lower bands probably representing various combinations of partly proteolyzed variants. Immunoblots were also labeled with anti-C491 antibodies to EAAC1 (Ab#371, 0.7 μg/ml) and anti-A522 antibodies to GLAST (Ab#314, 0.2 μg/ml). Note that GLAST and EAAC1 were not proteolyzed as quickly as GLT-1, and immunoreactivity was still present after 48 hr. Each lane on the blots developed with anti–GLT-1 antibodies contained 10 μg of protein, while each lane on the two other blots contained 30 μg of protein.
Immunocytochemistry Reveals Nonsynchronous Proteolysis
When a protein is fragmented, this will be apparent on immunoblots. However, membrane proteins may still be held together as long as they are sitting in the membrane. The possibility therefore existed that the termini would be visible in tissue sections even though they were no longer detectable on the Western blots. We probed mouse forebrain sections with glutamate transporter antibodies. The anti-B12 (Fig. 2A) and anti-B2 (Fig. 2B1) antibodies to the N-terminus gave strong labeling with control sections (0 hr) from wild-type mice. Most of this immunoreactivity, however, had disappeared by 24 hr and was completely gone after 48 hr. The same pattern was observed with anti-B563 antibodies to the C-terminus (Fig. 2A). In contrast, the two different antibodies to residues 493–508 (anti-B493) gave rise to strong labeling even after 48 hr (Fig. 2A, B1). Thus, the overall labeling intensity of sections appeared to parallel the labeling intensity of immunoblots.
Figure 2.

Postmortem changes in GLT-1 immunoreactivity in mouse brain tissue sections. (A) The sections were developed with rabbit GLT-1 antibodies: anti-B12, anti-B493, and anti-B563. (B1) The sections were developed with sheep GLT-1 antibodies: anti-B2 and anti-B493. Sections from GLT-1 knockout (KO) mice (3 weeks old) were used as negative control. (B2) Immunoblots showing the specificity of the sheep anti-B2 and anti-B493 antibodies. The antibodies were used in concentrations as indicated. Scale bars = 2 mm.
However, when the sections were examined at higher magnification (Fig. 3), it was evident that the loss of labeling observed with the antibodies to the termini (anti-B12 and anti-B563) did not occur evenly in all cells. There was a patchy loss of labeling. At 12 hr (Fig. 3), some patches had unchanged labeling, some had increased labeling, and some were completely devoid of labeling. The staining pattern looked like a mosaic of tiles with different colors and with diameters at around 50 µm. This corresponds to the reported sizes of astrocyte domains in mice (Oberheim et al. 2009). (An astrocyte domain is the volume of tissue defined by the branches extending from one astrocyte. There is limited overlap between neighboring astrocyte domains [Oberheim et al. 2009].)
Figure 3.

Some of the sections from Figure 2 are shown at higher magnification. (A) Note the uneven patchy labeling in the neocortex (N) observed with antibodies to the termini of GLT-1. This is in contrast to the labeling observed with the anti-B493 antibodies. Also note that the labeling in the striatum (S) was more even than that in the neocortex. However, at 24 hr, patchy labeling appeared also in the striatum with the anti-B12 and anti-B563 antibodies. At this time, there was hardly any labeling at all with these antibodies in the neocortex, whereas the labeling with the anti-B493 antibodies was almost unchanged. (B) Higher magnification images from the neocortex at 12 hr postmortem. Scale bars = 100 μm (A) and 50 μm (B).
As the labeling with the anti-B493 antibodies was mostly preserved, it followed that some cells contained truncated GLT-1 (Fig. 3: compare the images of the neocortex at 12 and 24 hr). This was confirmed by double labeling. At 0 hr, cells were positive for all the GLT-1 antibodies used, but at 24 hr, they were almost exclusively positive for the anti-B493 antibodies (data not shown).
The labeling of sections did not change dramatically over the first 6 hr (Fig. 4). In contrast, there were major changes during the next 6-hr period. Hippocampus CA1–3 was one of the regions with the earliest changes.
Figure 4.
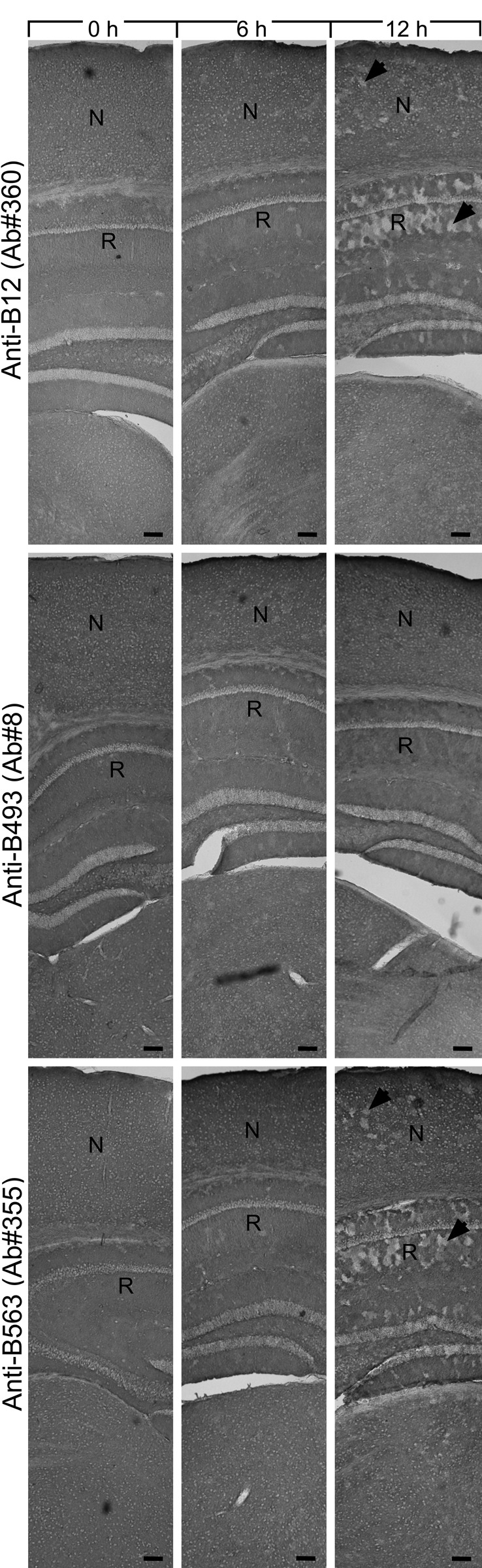
Changes in GLT-1 labeling during the first 12 hr. Mice brains were kept for 0, 6, and 12 hr as indicated and developed with GLT-1 antibodies as indicated (same concentrations as in Fig. 2). Note that the labeling was fairly stable the first few hours with all antibodies, but at 12 hr, there were unlabeled patches (arrowheads) in the hippocampus (R, stratum radiatum, CA1) and the neocortex (N). Montage. Scale bars = 100 μm.
Different Rates of Proteolysis Resulted in Cells Only Containing GLAST
A similar pattern was seen with the anti-A522 antibodies to the C-terminus of GLAST (Fig. 5), but the proteolysis was somewhat slower than the C-terminus of GLT-1 (Figs. 1 and 5A). The slower degradation of GLAST was confirmed by double labeling using rabbit anti-A522 and sheep anti-B2 (Fig. 6). At 0 hr, the two proteins co-localized, but at 12 hr, some cells were positive with anti-A522 and negative with anti-B2.
Figure 5.
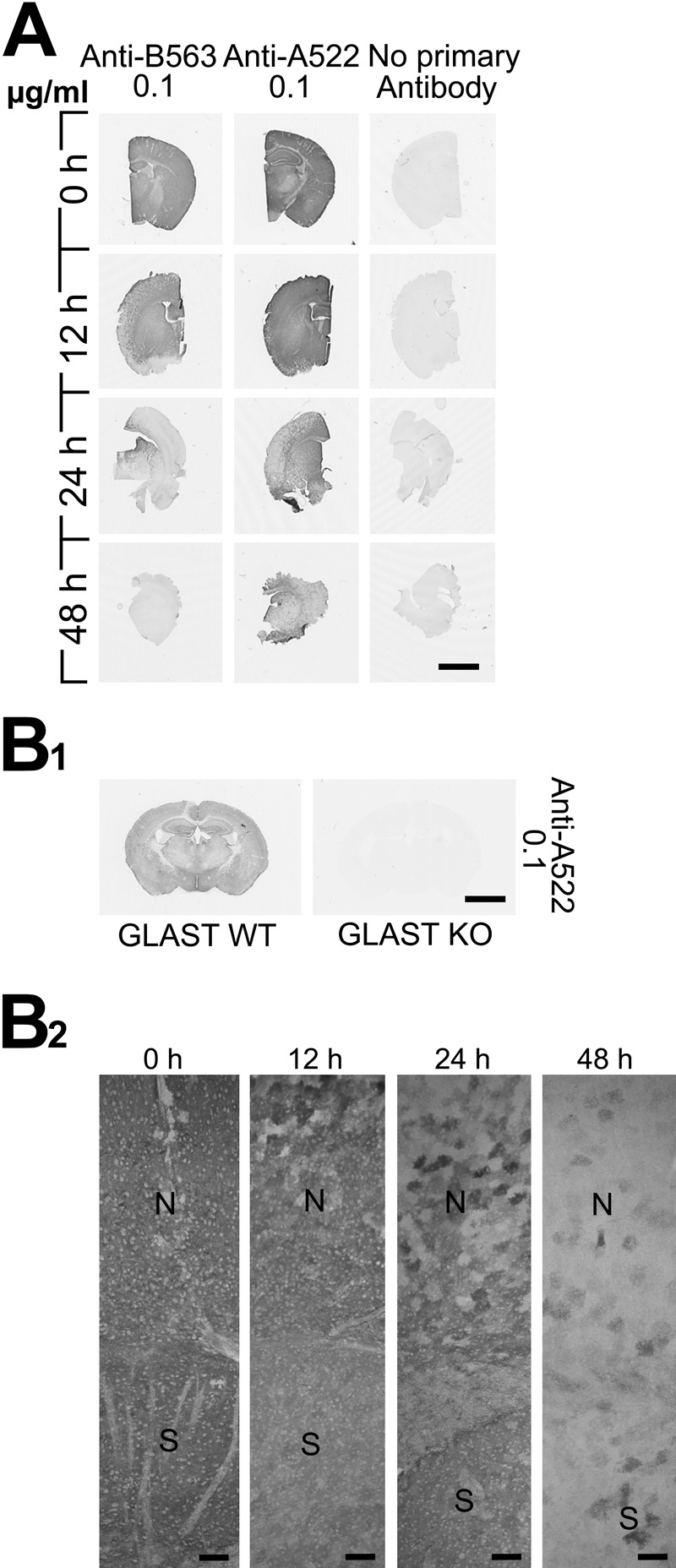
Postmortem changes in GLAST immunoreactivity in the mouse brain compared to GLT-1. (A) The sections were labeled with GLT-1 antibody anti-B563 (Ab#355) and GLAST antibody anti-522 (Ab#314). (B1) Brain section from a GLAST knockout mouse was labeled with 0.1 ug/ml anti-A522 (Ab#314) as a specificity control. (B2) Light micrographs at higher magnification showing GLAST (Ab#314). Scale bars = 2 mm (A, B1) and 100 μm (B2).
Figure 6.

The GLT-1 termini are proteolyzed faster than those of GLAST. Sections of the mouse neocortex were double labeled with rabbit anti-A522 antibodies (Ab#314, 0.06 μg/ml) to GLAST and sheep anti-B2 antibodies (Ab#210, 1 μg/ml) to GLT-1. GLAST and GLT-1 co-localized at 0 hr (A1–3). At 12 hr (B1–3), however, there was only a partial co-localization. Several cells no longer contained GLT-1. This gave a misleading impression that these cells were only expressing GLAST. Scale bars = 20 μm.
Discussion
The present study addresses the stability of glutamate transporter proteins after death, but the methodological issues described here are potentially also relevant for a large number of other clinically significant proteins.
Postmortem Proteolysis Is Likely to Involve Several Enzymes
We reproduced in mice our previous immunoblot observations from rats (Beckstrøm et al. 1999) that the degradation of the termini of GLT-1 is much faster than the degradation of the central parts of the protein. Further, the C-terminal cleavage site is between the epitope recognized by the anti-B493 antibodies and the anti-B518 antibodies. This is in agreement with the fact that there is a caspase-3 cleavage site between residues 505 and 506 (G. Gegelashvili et al. 2002; M. Gegelashvili et al. 2002; Boston-Howes et al. 2006). This site, however, is unique for GLT-1 and therefore cannot explain the degradation of GLAST, nor can it explain the degradation of the N-termini. Thus, it seems likely that more enzymes are involved. It is also notable that EAAC1 was not affected to the same degree.
Truncated GLT-1 Is Still Active
It is important to note (Leinenweber et al. 2011) that GLT-1 is neither inactivated by cleaving at the caspase-3 site (causing removal of the last 68 amino acid residues) nor by the elimination of the first 26 residues at the N-terminus. These findings are in agreement with the findings that about 80% of the transport activity could be reconstituted in liposomes even after 48 hr (Beckstrøm et al. 1999) and that there is functional glutamate uptake in synaptosomes prepared from human autopsy samples for more than 2 days after death (e.g., Schwarcz 1981).
Consequences of the Different Rates of Proteolysis
As explained above, the observed proteolysis is likely to involve several different enzymes operating at different rates. One implication of this is that the proportions of the various epitopes change as a function of the postmortem interval. In particular, the altered ratio between the C-terminus of GLAST and C-terminus of GLT-1 is particularly evident between 12 and 24 hr postmortem. At this time, there are many cells that are still GLAST positive but negative when probed with antibodies to the termini of GLT-1. The patchy disappearance of GLT-1 termini starts already after a few hours in the hippocampus. In human samples (see Fig. 5E in Mathern et al. [1999]), this is apparent already in the hippocampus fixed 7 hr after death. Importantly, this patchy labeling is not seen with the other transporters studied at this time point.
Because expression and regulation of enzymes may be altered in disease, it follows that the postmortem degradation may occur at different rates in control brains and in diseased brains. In principle, it is also possible to hypothesize that proteolysis may be initiated in vivo. Differences in proteolytic activity have, in fact, been suggested. For instance, Beckstrøm et al. (1999) found that the relationship between anti-B12 immunoreactivity and anti-B493 immunoreactivity was different in samples from controls and from patients with Alzheimer disease. Thus, the lengths of acceptable postmortem intervals need to be determined in each case.
The Cellular Distributions of GLT-1 and GLAST are Probably Similar in Humans and Rats
A large number of articles describing glutamate transporter distribution in human brain samples have been published. Here, we randomly looked through some of these articles and selected some that contain both good images and good descriptions of key parameters (Milton et al. 1997; Fray et al. 1998; Tessler et al. 1999; Mathern et al. 1999; Proper et al. 2002; Banner et al. 2002; Matute et al. 2005; Bjørnsen et al. 2007; Woltjer et al. 2010; Melone et al. 2011; Desilva et al. 2012). When viewing the data presented in these articles together, a clear picture emerges. The labeling pattern seen in samples obtained during surgery and fixed immediately resembles those shown here from mouse brains fixed immediately after death. Also, with increasing postmortem delay, the labeling becomes increasingly patchy like that shown here for mice.
The result of this meta-analysis does not rule out species differences in expression levels and in proportions of the different transporter subtypes and splice variants as suggested (Williams et al. 2005), but it clearly demonstrates, in agreement with Melone et al. (2011), that the distributions in rats and humans are so similar that well-controlled quantitative measurements are required in order to identify differences.
Transporter-Deficient Patches In Vivo
It should be noted that small GLT-1–deficient patches have sometimes been observed in rodent brain tissue fixed in vivo by perfusion. For instance, patchy expression of GLT-1 in vivo has been reported to be increased after treatment with the antipsychotic drug clozapine (Melone et al. 2001) and after ingestion of cycad flour (Wilson et al. 2003). Mechanisms that can be expected to lead to true glutamate transporter deficiency within small patches of tissue include internalization of transporters (e.g., Susarla and Robinson 2008), altered expression (e.g., Figiel et al. 2007), or retraction of astrocyte branches (e.g., Reichenbach et al. 2010). However, the possibility that transporters are truly absent from small volumes of tissue has not been much studied. Here, we show the opposite, namely, failure to detect the protein that is actually present. Postmortem proteolysis (as shown here) and variable splicing (e.g., Utsunomiya-Tate et al. 1997; Münch et al. 2003; Rauen et al. 2004; Holmseth et al. 2009; Scott et al. 2011) can lead to loss of the epitopes recognized by the antibodies. In these cases, the transporters are there but go undetected with the antibodies used. In this respect, most of the reports in the literature describing patchy expression are ambiguous either because only C-terminal antibodies were used, because of long postmortem delays, or because information on key parameters is missing.
Conclusions
The present study demonstrates that the postmortem interval has a major impact on the labeling pattern and that epitopes on the central parts of GLT-1 are more robust markers of in vivo GLT-1 protein levels than those on the termini. Further, the proteolysis described here is probably catalyzed by several different enzymes operating at different rates. Finally, postmortem proteolysis of the termini does not occur synchronously in all cells but at very different rates in different cells. When one astrocyte loses all of its immunoreactivity, then the entire astrocyte domain becomes immunonegative. This explains the patchy labeling pattern observed in tissue that has been stored for several hours before fixation and suggests that the reported differences between rodents and humans with respect to glutamate transporter distribution may, in part, be a postmortem artifact.
However, not all published studies can be adequately interpreted because important information is lacking. In addition to describing postmortem interval, labeling specificity (Lorincz and Nusser 2008; Holmseth et al. 2012b), and fixation (e.g., Melone et al. 2009; Holmseth et al. 2012b), it is important to precisely describe the epitope recognized by the antibodies.
The exact rates of proteolysis, and thereby acceptable postmortem intervals, are likely to depend on several factors, including temperature. Further, it should be taken into consideration that the expression levels of the enzymes involved in the postmortem degradation may change in disease. It is therefore possible that there are situations where the rates of degradation differ between control brains and diseased brains. Consequently, postmortem proteolysis rates should be investigated whenever autopsy material is used.
Acknowledgments
We thank Henriette Danbolt for technical assistance and Kohichi Tanaka for the GLT-1 (Tanaka et al. 1997) and GLAST (Watase et al. 1998) knockout mice.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Norwegian Research Council (FUGE II 183727-S10).
References
- Banner SJ, Fray AE, Ince PG, Steward M, Cookson MR, Shaw PJ. 2002. The expression of the glutamate re-uptake transporter excitatory amino acid transporter 1 (EAAT1) in the normal human CNS and in motor neurone disease: an immunohistochemical study. Neuroscience. 109:27–44 [DOI] [PubMed] [Google Scholar]
- Beart PM, O’Shea RD. 2007. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 150:5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstrøm H, Julsrud L, Haugeto Ø, Dewar D, Graham DI, Lehre KP, Storm-Mathisen J, Danbolt NC. 1999. Interindividual differences in the levels of the glutamate transporters GLAST and GLT, but no clear correlation with Alzheimer’s disease. J Neurosci Res. 55:218–229 [DOI] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE. 1999. Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol. 9:293–298 [DOI] [PubMed] [Google Scholar]
- Bjørås M, Gjesdal O, Erickson JD, Torp R, Levy LM, Ottersen OP, Degree M, Storm-Mathisen J, Seeberg E, Danbolt NC. 1996. Cloning and expression of a neuronal rat brain glutamate transporter. Brain Res Mol Brain Res. 36:163–168 [DOI] [PubMed] [Google Scholar]
- Bjørnsen LP, Eid T, Holmseth S, Danbolt NC, Spencer DD, de Lanerolle NC. 2007. Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures. Neurobiol Dis. 25:319–330 [DOI] [PubMed] [Google Scholar]
- Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH, Trotti D. 2006. Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem. 281:14076–14084 [DOI] [PubMed] [Google Scholar]
- Chen WZ, Mahadomrongkul V, Berger UV, Bassan M, Desilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. 2004. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 24:1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Weinberg RJ. 1999. Shaping excitation at glutamatergic synapses. Trends Neurosci. 22:451–458 [DOI] [PubMed] [Google Scholar]
- Danbolt NC. 2001. Glutamate uptake. Prog Neurobiol. 65:1–105 [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Lehre KP, Dehnes Y, Chaudhry FA, Levy LM. 1998. Localization of transporters using transporter-specific antibodies. Methods Enzymol. 296:388–407 [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Pines G, Kanner BI. 1990. Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry. 29:6734–6740 [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, Kanner BI. 1992. An [Na++ K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 51:295–310 [DOI] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. 1998. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci. 18:3606–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desilva T, Borenstein N, Volpe J, Kinney H, Rosenberg P. 2012. Expression of EAAT2 in neurons and protoplasmic astrocytes during human cortical development. J Comp Neurol. Epub Apr 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figiel M, Allritz C, Lehmann C, Engele J. 2007. Gap junctional control of glial glutamate transporter expression. Mol Cell Neurosci. 35:130–137 [DOI] [PubMed] [Google Scholar]
- Fray AE, Ince PG, Banner SJ, Milton LD, Usher PA, Cookson MR, Shaw PJ. 1998. The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur J Neurosci. 10:2481–2489 [DOI] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, et al. 2008. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience. 157:80–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Dehnes Y, Balazs R, Cribbs DH, Cotman CW, Rodriguez-Kern A, Gegelashvili M. 2002. Caspase-dependent regulation and receptor-like properties of glutamate transporters. Society for Neuroscience Abstracts. 28:441.16 [Google Scholar]
- Gegelashvili M, Rodriguez-Kern A, Dehnes Y, Danbolt NC, Balazs R, Cribbs DH, Cotman CW, Gegelashvili G. 2002. Glutamate transporters: caspase-dependent regulation and receptor-like properties. Neuropharmacology. 43:287 [Google Scholar]
- Haugeto Ø, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, Nielsen M, Lehre KP, Danbolt NC. 1996. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 271:27715–27722 [DOI] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Bjørnsen LP, Boulland JL, Furness DN, Bergles D, Danbolt NC. 2005. Specificity of antibodies: unexpected cross reactivity of antibodies directed against the EAAT3 (EAAC) glutamate transporter. Neuroscience. 136:649–660 [DOI] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, et al. 2012a. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci. 32:6000–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Lehre KP, Danbolt NC. 2006. Specificity controls for immunocytochemistry. Anat Embryol (Berl). 211:257–266 [DOI] [PubMed] [Google Scholar]
- Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, Danbolt NC. 2009. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience. 162:1055–1071 [DOI] [PubMed] [Google Scholar]
- Holmseth S, Zhou Y, Follin-Arbelet V, Lehre K, Bergles D, Danbolt N. 2012b. Specificity controls for immunocytochemistry: the antigen pre-adsorption test can lead to inaccurate assessment of antibody specificity. J Histochem Cytochem. 60:174–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Amara SG. 2011. New views of glutamate transporter structure and function: advances and challenges. Neuropharmacology. 60:172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. 1992. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 360:467–471 [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. 1995. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 15:1835–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinenweber A, Machtens J-P, Begemann B, Fahlke C. 2011. Regulation of glial glutamate transporters by C-terminal domains. J Biol Chem. 286:1927–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Rolstad B, Danbolt NC. 1993. A monoclonal antibody raised against an [Na+- K+] coupled L-glutamate transporter purified from rat brain confirms glial cell localization. FEBS Lett. 317:79–84 [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. 2008. Specificity of immunoreactions: the importance of testing specificity in each method. J Neurosci. 28:9083–9086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L. 1999. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 52:453–472 [DOI] [PubMed] [Google Scholar]
- Matute C, Melone M, Vallejo-Illarramendi A, Conti F. 2005. Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia. 49:451–455 [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Conti F. 2009. Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia. 57:108–117 [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Ducati A, Iacoangeli M, Conti F. 2011. Cellular and synaptic localization of EAAT2a in human cerebral cortex. Front Neuroanat. 4:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melone M, Vitellaro-Zuccarello L, Vallejo-Illarramendi A, Pérez-Samartin A, Matute C, Cozzi A, Pellegrini-Giampietro DE, Rothstein JD, Conti F. 2001. The expression of glutamate transporter GLT-1 in the rat cerebral cortex is down-regulated by the antipsychotic drug clozapine. Mol Psychiatry. 6:380–386 [DOI] [PubMed] [Google Scholar]
- Milton ID, Banner SJ, Ince PG, Piggott NH, Fray AE, Thatcher N, Horne CH, Shaw PJ. 1997. Expression of the glial glutamate transporter EAAT2 in the human CNS: an immunohistochemical study. Brain Res Mol Brain Res. 52:17–31 [DOI] [PubMed] [Google Scholar]
- Münch C, Zhu B-G, Leven A, Stamm S, Einkorn H, Schwalenstocker B, Ludolph AC, Riepe MW, Meyer T. 2003. Differential regulation of 5’ splice variants of the glutamate transporter EAAT2 in an in vivo model of chemical hypoxia induced by 3-nitropropionic acid. J Neurosci Res. 71:819–825 [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, et al. 2009. Uniquely hominid features of adult human astrocytes. J Neurosci. 29:3276–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjørås M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. 1992. Cloning and expression of a rat brain L-glutamate transporter. Nature. 360:464–467 [DOI] [PubMed] [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN. 2002. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 125:32–43 [DOI] [PubMed] [Google Scholar]
- Rauen T, Wiessner M, Sullivan R, Lee A, Pow DV. 2004. A new GLT1 splice variant: cloning and immunolocalization of GLT1c in the mammalian retina and brain. Neurochem Int. 45:1095–1106 [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Derouiche A, Kirchhoff F. 2010. Morphology and dynamics of perisynaptic glia. Brain Res Rev. 63:11–25 [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Puschel B, Kugler P. 1997. Cellular and regional distribution of the glutamate transporter GLAST in the CNS of rats: nonradioactive in situ hybridization and comparative immunocytochemistry. J Neurosci. 17:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R. 1981. Effects of tissue storage and freezing on brain glutamate uptake. Life Sci. 28:1147–1154 [DOI] [PubMed] [Google Scholar]
- Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR. 2011. Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging. 32:553.e1–553.e11 [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. 1992. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A. 89:10955–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susarla BT, Robinson MB. 2008. Internalization and degradation of the glutamate transporter GLT-1 in response to phorbol ester. Neurochem Int. 52:709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K. 1993. Expression cloning of a rat glutamate transporter. Neurosci Res. 16:149–153 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Hori S, et al. 1997. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT- 1. Science. 276:1699–1702 [DOI] [PubMed] [Google Scholar]
- Tessler S, Danbolt NC, Faull RLM, Storm-Mathisen J, Emson PC. 1999. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience. 88:1083–1091 [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. 1997. Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci. 9:1646–1655 [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y. 1997. Tissue specific variants of glutamate transporter GLT-1. FEBS Lett. 416:312–316 [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, et al. 1998. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 10:976–988 [DOI] [PubMed] [Google Scholar]
- Williams SM, Sullivan RK, Scott HL, Finkelstein DI, Colditz PB, Lingwood BE, Dodd PR, Pow DV. 2005. Glial glutamate transporter expression patterns in brains from multiple mammalian species. Glia. 49:520–541 [DOI] [PubMed] [Google Scholar]
- Wilson JM, Khabazian I, Pow DV, Craig UK, Shaw CA. 2003. Decrease in glial glutamate transporter variants and excitatory amino acid receptor down-regulation in a murine model of ALS-PDC. Neuromolecular Med. 3:105–117 [DOI] [PubMed] [Google Scholar]
- Woltjer RL, Duerson K, Fullmer JM, Mookherjee P, Ryan AM, Montine TJ, Kaye JA, Quinn JF, Silbert L, Erten-Lyons D, et al. 2010. Aberrant detergent-insoluble excitatory amino acid transporter 2 accumulates in Alzheimer disease. J Neuropathol Exp Neurol. 69:667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Holmseth S, Hua R, Lehre AC, Olofsson AM, Poblete-Naredo I, Kempson SA, Danbolt NC. 2012. The betaine-GABA transporter (BGT1, slc6a12) is predominantly expressed in the liver and at lower levels in the kidneys and at the brain surface. Am J Physiol Renal Physiol. 302:F316–F328 [DOI] [PubMed] [Google Scholar]