

Abstract
Efficient and highly diastereo- and enantioselective conjugate additions of phenyldimethylsilyl units to acyclic and cyclic dienones and dienoates are disclosed. The C–Si bond forming reactions are catalyzed by 2.0–2.5 mol % of a copper complex of a chiral monodentate N-heterocyclic carbene; the requisite reagent, PhMe2Si–B(pin), is commercially available or can be easily prepared. Transformations generate allylsilanes in up to 98% yield and >99:1 enantiomeric ratio, and proceed with complete 1,4-selectivity, unless the dienone or dienoate carries a trisubstituted alkene conjugated to the carbonyl group; in the latter cases, 1,6-addition products are obtained exclusively and in up to >98% Z selectivity.
Silicon-containing organic molecules are of substantial utility in chemical synthesis1 and allylsilanes are one of the most useful among them.2 A valuable subset of chiral allylsilanes consists of those containing a carbonyl unit β to the silyl or the alkene unit (cf. A and B, Scheme 1);3 such entities have served in enantioselective preparation of several structurally complex natural products.3,4 One direct approach to accessing the carbonylcontaining allylsilanes in the enantiomerically enriched form5,6,7,8,9,10 involves site- and enantioselective silyl conjugate additions to acyclic or cyclic α,β,γ,δ-unsaturated dienones or dienoates. We envisioned that monodentate chiral N-heterocyclic carbenes11 (NHCs) and their derived Cu-based complexes, developed recently in these laboratories and used for catalytic enantioselective silyl conjugate additions,12,13 might be utilized to access allylsilanes represented by A, B and C (Scheme 1). If 1,6-addition were to be predominant (i.e., B and C are formed as major products), we would have to establish whether the alkene, likely to be generated as part of a β,γ-unsaturated carbonyl due to kinetic protonation of dienolate at the α position, is formed with high stereoselectivity.
Scheme 1. Enantioselective Synthesis of Carbonyl-Containing Allylsilanes by NHC–Cu-Catalyzed Silyl Conjugate Addition.
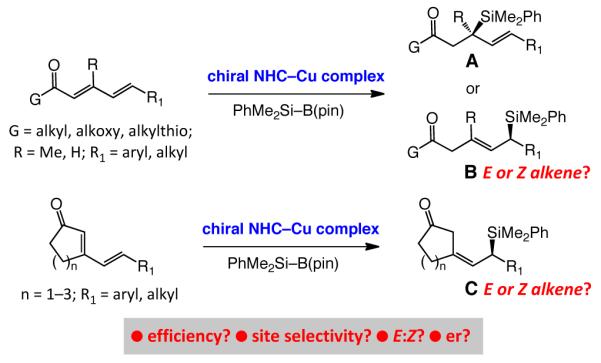
Herein, we report the results of our investigations regarding the development of an efficient catalytic enantioselective method for silyl conjugate additions to dienones and dienoates. The resulting protocols allow access to an assortment of allylsilanes in 69–98% yield and 94.5:5.5 to more than 99:1 enantiomeric ratio (er). The requisite chiral ligands are prepared in three–four steps in ~50% overall yield;11 the silylboron reagent [(dimethylphenylsilyl)pinacolatoboron, (1)] is commercially available or can be prepared easily on multigram scale.14 Acylic unsaturated ketones, esters and thioesters as well as five-, six- and seven-membered ring cyclic dienones can be used as substrates. Depending on the substrate structure, 1,4 or 1,6 modes of addition predominate. In the case of 1,6-addition products,15 trisubstituted alkenes are obtained with 78% to >98% Z selectivity.
We began by probing the ability of a selection of NHC–Cu complexes, derived from imidazolinium salts 4–6, to promote conjugate addition of the versatile phenyldimethylsilyl moiety to dienone 2a through reaction performed in the presence of silylboron reagent 1. Although reasonable efficiency and enantioselectivity is observed in every case (Table 1), with C2-symmetric 6b serving as catalyst precursor, allylsilane 3a is isolated in 96% yield and 99:1 er, and formation of the 1,4-addition product is strongly favored (<2% δ C–Si bond formation). We then determined that enantioselective synthesis of 3a can be performed with 2.2 mol % 6b (2.0 mol % CuCl, 4.4 mol % NaOt-Bu, −78 °C, 2.0 h) with identical efficiency and enantioselectivity (96% yield, 99:1 er). Similar to the NHC–Cu-catalyzed transformations involving B2(pin)2 to generate tertiary C–B bonds,16 silyl conjugate additions proceed readily in the absence of MeOH, the presence of which is required in some instances.17 Thus, the NHC–Cu-enolate appears to be sufficiently reactive, likely due to the strong donor ability of the heterocyclic ligand,16 to react with the sizable silylboron reagent in order to regenerate the catalytically active NHC–Cu–SiMe2Ph species.
Table 1.
Silyl Addition to an Acyclic Dienone: NHC–Cu Screeninga
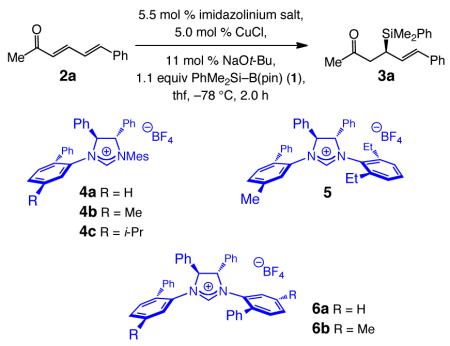
| entry | imidazolinium salt | conv (%)b | yield (%)c | erd |
|---|---|---|---|---|
| 1 | 4a | 80 | 77 | 86:14 |
| 2 | 4b | >98 | 92 | 93:7 |
| 3 | 4c | 88 | 87 | 91:9 |
| 4 | 5 | 94 | 91 | 96:4 |
| 5 | 6a | >98 | 94 | 95:5 |
| 6 | 6b | >98 | 96 | 99:1 |
Reactions performed under N2 atm.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Yields of purified products (±5%).
Determined by HPLC analysis. See the Supporting Information (SI) for details. Mes = 2,4,6-Me3C6H2.
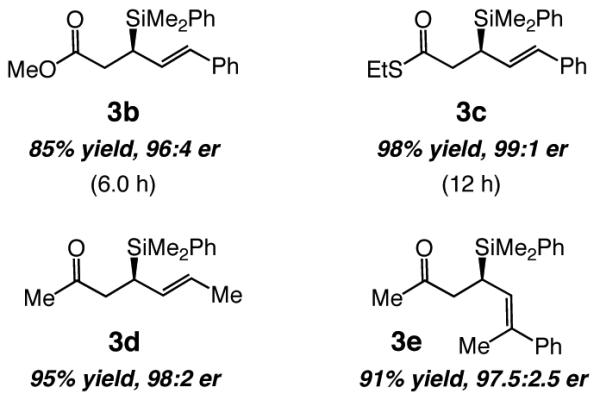
The Cu-catalyzed additions have significant scope, as the data presented in Scheme 2 indicate. With 2.7 mol % 6b (2.5 mol % CuCl, 5.5 mol % NaOt-Bu, −78 °C), β-silyl-substituted carboxylic ester 3b is formed in 85% yield and 96:4 er, although the transformation is less facile and requires six hours to proceed to completion. Conjugate addition to the corresponding thioester (cf. 3c) is still slower (>98% conv in 12 h at −78 °C), but the desired product is isolated in exceptionally high yield and enantiomeric purity (98% yield, 99:1 er). Cu-catalyzed enantioselective syntheses of β-silylketones 3d and 3e illustrate that substrates bearing an alkyl-substituted dienes (vs Ph in 3a-c), or those that contain a sterically demanding trisubstituted olefin, serve as equally effective starting points.
Scheme 2. Enantiomerically Enriched Acyclic Carbonyl-Containing Allylsilanesa.
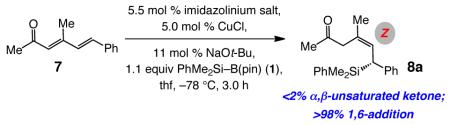
Examining the reactions of acyclic dienones that contain amethyl substituent at their β carbon came next (cf. 7, Table 2). The question here was whether the NHC–Cu-catalyzed additions would occur at the C4 site to generate a Si-substituted quaternary carbon stereogenic center, or if C–Si bond formation occurs at the δ site. In both cases, there is only 1,6-addition and the β,γ-unsaturated ketone is formed exclusively. Unlike transformations with the less substituted dienone 2a, the Cu complex derived from imidazolinium salt 6a promotes a more efficient reaction and delivers the desired product (8a) with higher enantiomeric purity (entries 1–2, Table 1: 95:5 vs 83:17 er). Although both processes furnish the Z trisubstituted olefin predominantly, the reaction with 6a is more stereoselective (91% vs 80% Z). As mentioned above, a hallmark of chiral monodentate NHC ligands is the ease with which their structure can be modified.11,12 Accordingly, further screening led us to establish that use of C2-symmetric imidazolinium salt 9 (entry 3, Table 2) leads to the formation of 8a with superior Z- and enantioselectivity (93% Z, >99:1 er).
Table 2.
Addition to a β-Substituted Acyclic Dienone: NHC–Cu Screeninga
| entry | imidazolinium salt | conv (%);b | Z:E b | erc |
|---|---|---|---|---|
| 1 | 6a | >98 | 91:9 | 95:5 |
| 2 | 6b | 77 | 80:20 | 83:17 |
| 3 | 9 | 81 | 93:7 | >99:1 |
Reactions performed under N2 atm.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Determined by HPLC analysis; er values correspond to Z alkene isomers. See the SI for details.
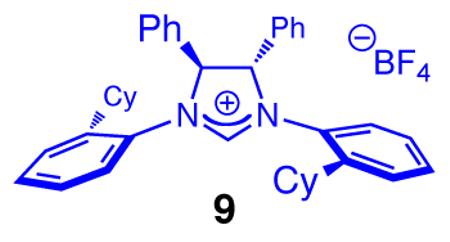
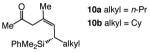
Enantioselective synthesis of 8a proceeds to >98% conv with 2.5 mol % 9 within six hours, under the conditions shown in Table 3; the allylsilane is generated in 93% Z selectivity and >99:1 er (entry 1). As the findings in entries 2–3 of Table 3 indicate, variations in the electronic attributes of the aryl substituent do not strongly affect Z or enantioselectivity. Formation of alkyl-substituted allylsilanes 10a-b (entries 4–5, Table 3) is equally efficient and exceptionally enantioselective (>99:1 er) but the degree of alkene stereoisomeric purity is somewhat diminished (89% Z). Conjugate additions to the corresponding carboxylic and alkylthioesters, as shown in entries 6–7 of Table 3, generate the expected allylsilanes with complete Z- and enantioselectivity (<2% E and ≥99:1 er); the conjugate addition with the S-containing substrate, however, demands higher temperatures (−50 vs −78 °C) and longer reaction times (48 vs 12 h) to proceed to completion.
Table 3.
Additions to β-Substituted Acyclic Dienones: Scopea
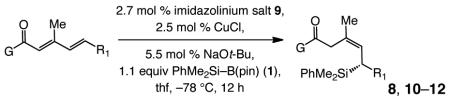
| entry | allylsilane product | yield (%);b | Z:E c | erd |
|---|---|---|---|---|
| 1e |
|
81 | 93:7 | >99:1 |
| 2 | 81 | 95:5 | 99:1 | |
| 3 | 75 | 91:9 | >99:1 | |
| 4 |
|
85 | 89:11 | >99:1 |
| 5 | 72 | 89:11 | >99:1 | |
| 6 |
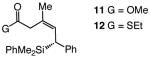
|
73 | >98:2 | >99:1 |
| 7f | 69 | >98:2 | 99:1 |
Reactions performed under N2 atm; >98% conv, <2% α,β-unsaturated carbonyl product, and >98% 1,6-addition in all cases.
Yields of purified products (±5%).
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Determined by HPLC analysis; er values correspond to the Z alkene product.
Reaction time = 6.0 h.
Performed at −50°C for 48 h. See the SI for details.
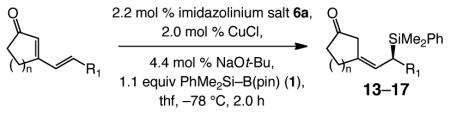
The final segment of our investigation entailed a more extensive study of additions to cyclic dienones, two examples of which, involving six-membered ring substrates in entries 2 and 7 of Table 4, were reported to occur with high Z- and enantioselectivity in our initial disclosure.12a We were particularly keen on establishing whether the reactions are sensitive to different ring sizes and variations in the steric or electronic attributes of the alkene substituent.
Table 4.
Additions to β-Substituted Cyclic Dienonesa
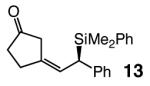
| entry | allylsilane product | yield (%)b | Z:E c | erd |
|---|---|---|---|---|
| 1e |
|
85 | 78:12 | 95:5 |
| 2 |

|
91 | 95:5 | 98:2 |
| 3 | 95 | >98:2 | 95:5 | |
| 4 | 91 | >98:2 | 97:3 | |
| 5 | 88 | >98:2 | 94.5:5.5 | |
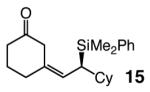
| 6 |
|
92 | 95:5 | 98:2 |
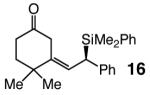
| 7 |
|
95 | <2:>98 | 96:4 |
| 8 |
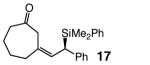
|
98 | 96:4 | 96:4 |
See Table 3.
See Table 3.
See Table 3.
See Table 3.
Product mixture contained 15–20% of the isomerized α,β-unsaturated ketone. See the SI for details.
Five- as well as seven-membered ring dienones serve as effective substrates, delivering the desired allylsilanes in 85% and 98% yield, respectively (entries 1 and 8, Table 4).18 Both products (13 and 17, Table 4) are obtained with high enantioselectivity (95:5 and 96:4 er, respectively), but the degree of Z selectivity with the cyclopentenyl system is diminished (78% vs 96% Z). Additionally, for the transformation in entry 1 of Table 4, the product mixture contains 15–20% of the derived α,β-unsaturated cyclopentenone (inseparable from 13), formed presumably as a result of adventitious isomerization. Electronic alterations within the aryl substituent do not cause any significant variations in the Z- or enantioselectivity of the silyl conjugate additions, as demonstrated by reactions shown in entries 2–5 of Table 4. Moreover, conjugate addition to the cyclic dienone in entry 6, carrying an alkyl substituent is equally efficient and stereoselective, affording 15 in 92% yield, 95% Z selectivity and 98:2 er.19
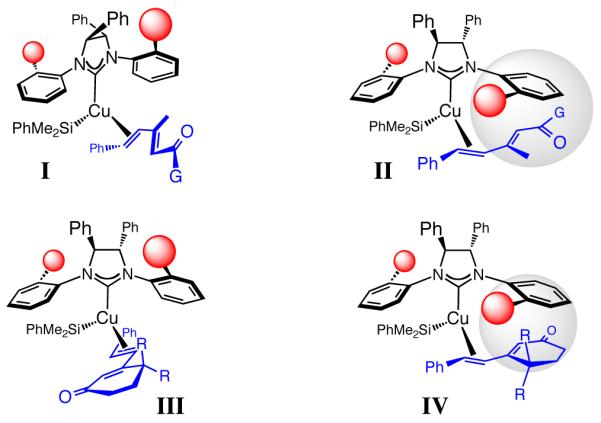
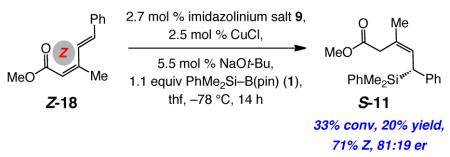
The NHC–Cu•dienone ensembles in Figure 1 provide a stereochemical model regarding the outcome of the 1,6-silyl conjugate additions. The NHC–Cu–SiMe2Ph complex is generated in situ by the pathways described before.12a Such a complex can coordinate with the γ,δ-alkene of the substrate as illustrated by I-IV, with the high Z selectivity indicating the involvement of the s-cis conformers. Preliminary DFT calculations indicate that, although, as expected, the s-trans dienes are energetically favored (by ≥2.0 kcal/mol), reaction through the intermediacy of an NHC–Cu-s-cis complex is favored.20 Elucidation of the precise reason for such a preference requires additional studies but might partly be attribute to the η4 nature of the Cu–diene coordination.20 The observed enantioselectivity can arise from the minimization of steric repulsion between the substrate and the catalyst, forcing the more sterically demandingenone moiety to be oriented as depicted in I and III (Figure 1). The relatively low Z selectivity in observed for the addition to cyclopentenone 13 (entry 1, Table 4) is difficult to explain. However, the proposed scheme is consistent with the lower Z selectivity delivered for the more sluggish and relatively nonselective transformations with to dienone Z-18 (eq 1). In the latter instance, the resulting allylic strain likely raises substantially the energy of the substrate’s s-cis conformer, rendering the derived NHC–Cu–diene complex less accessible. As a result, alternative complexes, such as the corresponding s-trans dienone or that derived from coordination through the other enantiotopic face of the olefin become competitive, causing diminution in Z- and enantioselectivity.
Figure 1. Proposed model for NHC-Cu-catalyzed enantioselective silyl conjugate additions.
 |
(1) |
The protocols described here offer a convenient and direct route to a variety of enantiomerically enriched allylsilanes. The high Z selectivity in instances where 1,6-addition is dominant is a noteworthy aspect of the NHC–Cu-catalyzed transformations; in the corresponding processes with C-based nucleophiles, only moderate stereoselectivity is observed (≤85% of one alkene isomer favored).15 Furthermore, attempts to perform NHCcatalyzed (Cu-free) silyl conjugate additions12b to representative substrates mentioned above (e.g., 2a) resulted in <10% yield of the desired allylsilanes and a complex mixture of unidentifiable entities. The above attributes, together with the high efficiency and stereoselectivity observed in the aforementioned processes should render the present class of Cu-catalyzed protocols of utility in chemical synthesis. The example shown in eq 2, involving synthesis of β-hydroxy ketone 19, the product of an enantioselective ketone aldol addition21 to an α,β-unsaturated ketone, is illustrative.
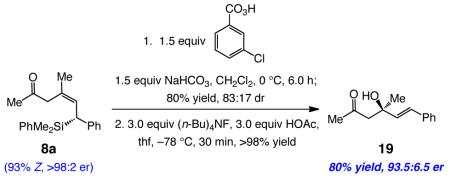 |
(2) |
Design and development of additional Cu-based chiral catalysts and methods for enantioselective synthesis of C–Si as well as C–B and C–C bonds are in progress.
Supplementary Material
Acknowledgment
Financial support was provided by the NIH (GM-57212) and the NSF (CHE-1111074). We thank Dr. Adil R. Zhugralin for his assistance with the initial DFT calculations and Boston College Research Services for access to computational facilities.
Footnotes
Supporting Information Available:
Experimental procedures and spectral, analytical data for all products (PDF). This material is available on the web: http://www.pubs.acs.org.
References
- (1).For reviews regarding the use of organosilicon species in organic synthesis, see: Chan TH, Wang D. Chem. Rev. 1992;92:995. Jones GR, Landais Y. Tetrahedron. 1996;52:7599. Fleming I, Barbero A, Walter D. Chem. Rev. 1997;97:2063. doi: 10.1021/cr941074u. Suginome M, Ito Y. Chem. Rev. 2000;100:3221. doi: 10.1021/cr9902805.
- (2).For reviews on the utility of allylsilanes in organic synthesis, see: Masse CE, Panek JS. Chem. Rev. 1995;95:1293. Barbero A, Pulido FJ. Acc. Chem. Res. 2004;37:817. doi: 10.1021/ar0400490. Chabaud L, James P, Landais Y. Eur. J. Org. Chem. 2004;3173
- (3).For a review on the utility of β-silylcarbonyls in synthesis, see: Fleming I. Science of Synthesis. Vol. 4. Thieme: Stuttgart; Germany: 2002. p. 927.
- (4).For specific examples involving the use of enantiomerically enriched allylsilanes in natural product synthesis, see: Hale MR, Hoveyda AH. J. Org. Chem. 1992;57:1643. Hu T, Takenaka N, Panek JS. J. Am. Chem. Soc. 1999;121:9229. Liu P, Panek JS. J. Am. Chem. Soc. 2000;122:1235. Arefolov A, Panek JS. J. Am. Chem. Soc. 2005;127:5596. doi: 10.1021/ja043168j.
- (5).For synthesis of enantiomerically enriched allylsilanes by catalytic cross-coupling reactions, see: Hayashi T, Konishi M, Ito H, Kumada M. J. Am. Chem. Soc. 1982;104:4962. Hayashi T, Konishi M, Okamoto Y, Kabeta K, Kumada M. J. Org. Chem. 1986;51:3772.
- (6).For synthesis of enantiomerically enriched allylsilanes by catalytic hydrosilylation, see: Hayashi T, Kabeta K, Yamamoto T, Tamao K, Kumada M. Tetrahedron Lett. 1983;24:5661. Hayashi T, Han JW, Takeda A, Tang J, Nohmi K, Mukaide K, Tsuji H, Uozumi Y. Adv. Synth. Catal. 2001;343:279.
- (7).For synthesis of enantiomerically enriched allylsilanes by catalytic allylic substitution, see: Hayashi T, Ohno A, Lu S.-j., Matsumoto Y, Fukuyo E, Yanagi K. J. Am. Chem. Soc. 1994;116:4221. Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:4554. doi: 10.1002/anie.200700841.
- (8).For synthesis of enantiomerically enriched allylsilanes by catalytic reduction of silyl-substituted allyl carbonates, see: Hayashi T, Iwamura H, Uozumi Y. Tetrahedron Lett. 1994;35:4813.
- (9).For synthesis of enantiomerically enriched allylsilanes by catalytic Si–B addition to allenes, see: Ohmura T, Taniguchi H, Suginome M. J. Am. Chem. Soc. 2006;128:13682. doi: 10.1021/ja063934h.
- (10).For synthesis of enantiomerically enriched allylsilanes by catalytic olefin metathesis reactions, see: Kiely AF, Jernelius JA, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2002;124:2868. doi: 10.1021/ja012679s. Adam J-M, de Fays L, Laguerre M, Ghosez L. Tetrahedron. 2004;60:7325.
- (11).(a) Lee K.-s., Hoveyda AH. J. Org. Chem. 2009;74:4455. doi: 10.1021/jo900589x. [DOI] [PubMed] [Google Scholar]; (b) Wu H, Radomkit S, O’Brien JM. J. Am. Chem. Soc. 2012;134:8277. doi: 10.1021/ja302929d. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For NHC–Cu-catalyzed enantioselective silyl conjugate additions, see: Lee K-s., Hoveyda AH. J. Am. Chem. Soc. 2010;132:2898. doi: 10.1021/ja910989n. For the corresponding metal-free processes, catalyzed by chiral NHCs, see: O’Brien JM, Hoveyda AH. J. Am. Chem. Soc. 2011;133:7712. doi: 10.1021/ja203031a.
- (13).For Pd- and Ru-catalyzed enantioselective silyl conjugate additions, see: Matsumoto Y, Hayashi T. Tetrahedron. 1994;50:335. Hayashi T, Matsumoto Y, Ito Y. J. Am. Chem. Soc. 1988;110:5579. Walter C, Auer G, Oestreich M. Angew. Chem., Int. Ed. 2006;45:5675. doi: 10.1002/anie.200601747. Walter C, Oestreich M. Angew. Chem., Int. Ed. 2008;47:3818. doi: 10.1002/anie.200800361. Walter C, FrÖhlich R, Oestreich M. Tetrahedron. 2009;65:5513.
- (14).Suginome M, Matsuda T, Ito Y. Organometallics. 2000;19:4647. [Google Scholar]
- (15).To the best of our knowledge, the examples reported herein, are the first cases of 1,6-conjugate additions involving Si-based nucleophiles. For enantioselective 1,6-conjugate additions of carbon-based nucleophiles, see: Hayashi T, Yamamoto S, Tokunaga N. Angew. Chem., Int. Ed. 2005;44:4224. doi: 10.1002/anie.200500678. Fillion E, Wilsily A, Liao E. Tetrahedron Asymmetry. 2006;17:2957. den Hartog T, Harutyunyan SR, Font D, Minnaard AJ, Feringa BL. Angew. Chem., Int. Ed. 2008;47:398. doi: 10.1002/anie.200703702. Hénon H, Mauduit M, Alexakis A. Angew. Chem., Int. Ed. 2008;47:9122. doi: 10.1002/anie.200803735. Nishimura T, Yasuhara Y, Sawano T, Hayashi T. J. Am. Chem. Soc. 2010;132:7872. doi: 10.1021/ja1034842. For a recent case of catalytic enantioselective 1,6-addition of S-based nucleophiles, see: Tian X, Liu Y, Melciorre P. Angew. Chem., Int. Ed. 2012;51:6439. doi: 10.1002/anie.201202392.
- (16).Lee K-s., Zhugralin AR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7253. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).For example, see: Lillo V, Prieto A, Bonet A, Díaz-Requejo MM, Ramírez J, Pérez PJ, Fernández E. Organometallics. 2009;28:659. O’Brien JM, Lee K-s., Hoveyda AH. J. Am. Chem. Soc. 2010;132:10630. doi: 10.1021/ja104777u.
- (18).Similar yields and enantioselectivities are obtained with the Cu complex derived from 9. For instance, 16 is obtained in 79% yield (>98% conv) and 96:4 er with the latter species.
- (19).The minor E isomers are likely generated with appreciable enantioselectivity as well. As an example, we have been able to determine that E-15 is formed in 91:9 er.
- (20).See the Supporting Information for details.
- (21).Broadly applicable, efficient and highly enantioselective catalytic protocols for aldol additions to ketones remain lacking. For key reports, see: List B, Shabat D, Zhong G, Turner JM, Li A, Bui T, Anderson J, Lerner LA, Barbas CF. J. Am. Chem. Soc. 1999;121:7283. Denmark SE, Fan Y, Eastgate MD. J. Org. Chem. 2005;70:5235. doi: 10.1021/jo0506276. For a recent review on Cu-catalyzed ketone aldol processes, see: Shibasaki M, Kanai M. Chem. Rev. 2008;108:2853. doi: 10.1021/cr078340r. For Ag-catalyzed aldol adition to α-ketoesters, see: Akullian LC, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2006;128:6532. doi: 10.1021/ja061166o.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.