

Abstract
Methylobacteria are ubiquitous in the biosphere which are capable of growing on C1 compounds such as formate, formaldehyde, methanol and methylamine as well as on a wide range of multi-carbon growth substrates such as C2, C3 and C4 compounds due to the methylotrophic enzymes methanol dehydrogenase (MDH). MDH is performing these functions with the help of a key protein mxaF. Unfortunately, detailed structural analysis and homology modeling of mxaF is remains undefined. Hence, the objective of this research is the characterization and three dimensional modeling of mxaF protein from three different methylotrophs by using I-TASSER server. The predicted model were further optimize and validate by Profile 3D, Errat, Verifiy3-D and PROCHECK server. Predicted and best evaluated models have been successfully deposited to PMDB database with PMDB ID PM0077505, PM0077506 and PM0077507. Active site identification revealed 11, 13 and 14 putative functional site residues in respected models. It may play a major role during protein-protein, and protein-cofactor interactions. This study can provide us an ab-initio and detail information to understand the structure, mechanism of action and regulation of mxaF protein.
Keywords: Methylobacteria, mxaF protein, homology modeling, functional site
Background
Methylotrophic bacteria are a diverse group of organisms that possess a great number of specialized enzymes that enable them to grow on reduced carbon substrates without carbon– carbon bonds and use these as energy as well as a carbon source. They play an important role in biogeochemical cycling and possess a potential for use in bioremediation [1]. The members of Methylobacterium have a property to oxidize methanol due to the presence of methanol dehydrogenase (MDH), a pyrroloquinoline quinone (PQQ)-linked protein with an α2β2 tetramer structure [2], known as Methylotrophy, a capacity to aerobically utilize single carbon (C1) compounds as a sole source of carbon and energy by a bacterial metabolic pathway used to assimilate carbon or retrieve energy [3]. Until recently, the ability to oxidize methanol by gram-negative bacteria has been attributed almost exclusively to the MDH enzyme encoded by mxaF1. The study of amino acid sequences of mxaF protein show that several key amino acids that are required for the MDH enzyme activity are located in the deduced MxaF peptide [4]. The mxaF genes are well conserved among different classes of proteobacteria (alpha, beta, and gamma) in terms of both gene clustering and protein sequence identity [3, 5], suggesting a monophyletic origin for the mxa (mox) encoded methanol oxidation machinery. Based on its conservation, mxaF has served as a genetic marker for environmental detection of Methylotrophy [5]. Protein structure prediction is one of most intensely studied subjects in modern computational biology. It could be achieved at both the secondary structure level and the three-dimensional level. These are intimately related to each other. The accuracy of secondary structure prediction has increased gradually over the years as we gain a better understanding of the principles of sequence-structure relationships and the effect of evolution on the proteomes of organisms [7]. But the prediction of the threedimensional structure of a protein from its amino acid sequence is a challenge that has fascinated researchers in different disciplines for many years.Because Proteins play a key role in almost all biological process like maintaining the structural integrity of the cell, transport and storage of small molecules, catalysis, regulation, signaling and the immune system. Their 3- D structure and functional properties depend intricately upon their structure. As a result there has been much effort, both experimental and computational, in determining protein structures.
Due to the high impact of mxaF protein, it is necessary to extract structural information from sequences, has become increasingly important for bioinformatics research. So a detailed structural analysis of mxaF, a large subunit of methanol dehydrogenase protein has yet to be reported. In this study it was achieved by homology modeling. A comparative and detailed structural analysis of methanol dehydrogenase protein was assessed among facultative, restricted facultative and obligate methylotroph strains. The 3-D structure of methanol dehydrogenase was developed for three methylotrophic strains follow Serine pathway for methylotrophic metabolism and was compared.
Methodology
The amino acid sequence of MxaF protein of three different types of methylobacteria was retrieved from the NCBI database (CAI30806, CAA69318 and AAR88789) [8]. The interrogatory sequences from mxaF of methylotrophs were skim to find out the related protein structure to be used as a template by the BLAST program [9] against Protein Data Bank database.
Primary structural analysis:
Expasy's ProtParam server [10], has been applied for study of physiochemical characterization like theoretical isoelectric point (pI), molecular weight, molecular formula, total number of positive and negative residues, instability index [11], extinction coefficient [12], aliphatic index [13] and grand average hydropathy (GRAVY) [14]. The sulphide (S-S) bond pattern is predicted by using the tool CYS_REC [15]. The predicted results were shown in Table 1 (see supplementary material).
Secondary structural analysis:
For the enumeration of the secondary structural features of MxaF protein sequences, PSIPRED view [16], a new highly accurate secondary structure prediction method was employed. PSIPRED incorporates two feed-forward neural networks which perform an analysis on output obtained from PSI-BLAST (position specific Iterated BLAST) [17]. Results are shown in Table 2 (see supplementary material).
Homology modeling, Structure refinement and identification of functional site:
3-D model of MxaF protein sequence of three different methylobacterium was generated by I-TASSER, a web based server. Further the models were evaluated by VARIFY 3D [18], Profile 3D [19] and Errat [20], to check the correctness of the overall fold/structure, errors over localized regions and stereochemical parameters such as bond lengths and angles. Visualization and protein contact map of target proteins were carried out by Accelry's Discovery Studio software. Structural validation of target proteins model were done by PROCHECK which determine stereochemical aspects along with main chain and side chain parameters with comprehensive analysis. The shows that various The MxaF residues falling under allowed, favoured and in disallowed regions was predicted by Ramachandran plot perform by PROCHECK [21]. Structure based protein functional site of mxaF of three methylotrophs were predicted by Q-site Finder [22].
Discussion
The primary structure of MxaF protein of Methylosinus trichosporium, Hyphomicrobium zavarzinii and Methylobacterium podarium was speculated and compared Table 3 (see supplementary material). The computed isoelectric point (pI) for the computed pI value of all three strains are 5.13, 5.78 and 5.46 and is less than 7 (pI<7), reveals the acidic nature of protein [23]. Total predicted negative residues of mxaF protein are more in comparison to positive residues. These results also supported the acidic nature of target proteins of above Methylotrophic strains. Among those Methylotrophic strains, the extinction coefficient of H. zavarzinii is high, that indicates the presence of high concentration of Cys, Trp and Tyr which helps in the quantitative study of protein-protein and proteinlegend interactions in solution. The sulphide (S-S) bonding pattern has been shown that the M. tricosporium and H. zavarzinni have two cystein at position 145, 174 but M. podarium have three cystein at position 3, 146, 175.
These results unveil that the mxaF protein residue of these methylotrophs has high enthalpy at folded state because disulphide bond increase the enthalpy of the folded state by stabilizing local interaction [24]. The thermal stability of mxaF protein was determined by Instability Index. The predicted Instability index of mxaF of target strains were 16.74, 22.65 and 27.18 reveals that mxaF protein is thermostable because Instability index of a protein smaller than 40 make it stable while more than 40 make it unstable [11]. The mxaF protein of above strain are stable at wide range of temperature due to the Higher aliphatic index (51.73, 51.73 57.62) since higher AI indicate increased stability while lower AI indicate increased flexibility in the protein structure [23]. GRAVY (Grand average hydropathy) value of mxaF protein is -0.812, -0.970 and -0.728 expresses the hydrophilicity of protein of target strains due to their lower value because lower value indicates possible better interaction with water. The GRAVY value of H. zavarzinii was lower (-0.970) in comparison to other strains, indicates the better solubility of mxaF protein.
Most algorithms for protein secondary structure prediction currently in use are based on machine learning techniques in which PSIPRED view has been shown to be capable of achieving an average Q3 score of 76.5%, a highest level of accuracy published for any method to date [25]. The secondary structure of mxaF proteins of target methylotrophs were predicted and analyzed by PSIPRED view and were shown in Table 2 (see supplementary material). The results were expressed that all the residues lies under the strands and coil and have only little differences among them. There is no alpha helix found in the predicted structure. It reveals the unfavored structural property of protein in non-polar solvent. These results also can help in experimental verification of a predicted folding motif because it may be gained by measurements of protein secondary structural elements of which the motif is composed [26].
Three dimensional structure of mxaF protein of three methylotrophic strains were predicted and compared. The comparative protein structure analysis for methylotrophs is still untouched and unavailable. The tertiary structure prediction was performed by I-TASSER server by using the best align template (4aahA). The template was selected to analyze 3-D structure because a high level of sequence identity should guarantee a more accurate alignment between the target sequence and template structure [27]. Out of five generated similar models of the target sequence, the best one have been chosen to employing the criteria of good alignment with template, C-Score, TM score and RMSD values Table 3 (see supplementary material). The developed 3-D model of mxaF protein of methylotrophs was deposited to the PMDB database and their PMDB accession number is given in Table 3. The Predicted models were visualized through Accelry's Discovery Studio visualize 2.5 (Figure 1). The generated contact map of mxaF protein of methylotrophs explains the reduced representation of the target structure that helps in to the superimposition and similarity with other protein. The quality of predicted structures of mxaF were further assessed and confirmed by VARIFY 3D [28] Profile 3D [19] and Errat [20]. The scores (from -1 to +1) were added and plotted for individual residues. The residues falling in the area where the orange line crosses 0.0 have low prediction accuracy and less stable conformation whereas, most of the residues fall above 0.15-0.4 so we can say that the model is of good quality. The stereochemical quality and accuracy of the predicted model of mxaF were evaluated after the refinement process using Ramachandran Map calculation with the PROCHECK program [21]. The Ramachandran plot has been shown a tight clustering of phi~ -50 and psi~ -50. In the plot analysis, the residues were classified according to its regions in the quadrangle. The red regions in the graph point out the most allowed regions whereas the yellow regions represent allowed regions. Glycine is restrained by triangles and other residues are represented by squares.
Figure 1.
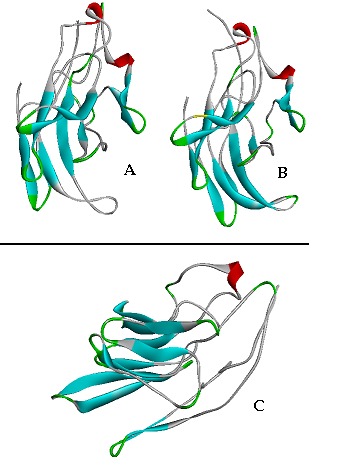
(A Hyphomicrobium zavarzinii; (B Methylobacterium podarium; (C Methylosinus trichosporium
The analysis report of Ramachandran plot concluding phi and psi angles to contribute in conformation of amino acids excluding glycine and proline. PROCHECK analysis of mxaF protein reveals in Ramachandran plot concluding phi and psi angles to contribute in conformation of amino acids excluding glycine and proline with 85.3%, 83.2%, 85.7% residue in most favoured region, 11.3%, 15.4%, 12.3% (16 amino acid) in additional allowed region, 2.7%, 1.3%, 1.9% generously allowed region and 0.7%, 0.0%, 0.0% residue in disallowed region in M. trichosporium, H. zavarzinii and M. podarium respectively.Q-Site Finder server was employed for the prediction of functional sites in the modeled mxaF proteins. Server were detected the 11, 13 and 14 putative functional site residues with significant matches in the modeled protein of H. zarzvinni, M. podarium and M. trichosporium respectively. The putative residues are given in Table 4 (see supplementary material), which could be important for protein interactions and/or activity of mxaF. The predicted 3-D structures of the target methylotrophs have been shown good stereochemistry among the strains, indicating reasonable good quality. The Overall 3-D structure of mxaF is well conserved among the methylotrophs who differ at the level of their nutrition.
Conclusion
Precise evaluation and modeling of proteins is a major goals and key aspect of computational Biology. The methylotrophs play a vital role in biogeochemical cycling and have potentiality for use in bioremediation due to the mxaF a major sub-unite of MDH protein. So the structural exploration and 3-D model was generated for the first time of three different methylotrophs which varying at nutrition level. It offers an alternative way to obtain structural information well before the structure of the new protein is determined by X-ray crystallography or NMR. Physicochemical and functional studies performed for characterization of mxaF in reaching conclusions about the biochemistry and biological function of the modeled protein. Structure prediction and functional analysis of mxaF will give an insight to the location of these proteins along with site of utilization of methanol. The present study would aid in detailed molecular mechanism of biostimulation of contaminant biodegradation and biotransformation through mxaF protein of methylotrophs.
Supplementary material
Footnotes
Citation:Singh et al, Bioinformation 8(21): 1042-1046 (2012)
References
- 1.SF Altschul, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.C Anthony, et al. Biochem J. 1994;304:665. [Google Scholar]
- 3.L Chistoserdova, et al. J Bacteriol. 2007;189:4020. doi: 10.1128/JB.00045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.L Chistoserdova, et al. J Bacteriol. 2003;185:2980. doi: 10.1128/JB.185.10.2980-2987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.P De Marco, et al. FEMS Microbiol Lett. 2004;234:75. doi: 10.1016/j.femsle.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 6.D Eisenberg, et al. Methods in Enzymol. 1997;277:396. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 7.SC Gill, PH Von Hippel. Anal Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 8.RK Gundampati, et al. J Mol Model. 2012;18:653. doi: 10.1007/s00894-011-1078-4. [DOI] [PubMed] [Google Scholar]
- 9.K Guruprasad, et al. Prot Eng. 1990;4:155. [Google Scholar]
- 10. http://blast.ncbi.nlm.nih.gov/Blast.cgi.
- 11. http://www.ncbi.nlm.nih.gov/entrez/
- 12.AJ Ikai, et al. J Biochem. 1980;88:1895. [PubMed] [Google Scholar]
- 13.Jae Hoon Jeong, et al. Mol Cells. 2002;13:369. [PubMed] [Google Scholar]
- 14.T John Pelton, R Larry Mclean. Analytical Biochemistry. 2000;277:167. doi: 10.1006/abio.1999.4320. [DOI] [PubMed] [Google Scholar]
- 15.J Kyte, RF Doolittle. J Mol Biol. 1982;157:105. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 16.RA Laskowski, et al. J App Cryst. 1993;26:283. [Google Scholar]
- 17.J Liam, et al. Bioinformatics. 1999;16:404. [Google Scholar]
- 18.Mc Donald, et al. Appl Environ Microbiol. 2008;74:1305. doi: 10.1128/AEM.02233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.RP Singh, et al. J Applied Sciences in Environmental Sanitation. 2011;6:357. [Google Scholar]
- 20.BJ Rost, et al. Struct Biol. 2001;134:204. [Google Scholar]
- 21.PN Sekhar, et al. In silico biology. 2006;6:0041. [Google Scholar]
- 22.M Suyama, et al. J Mol Evol. 1997;1:S163. doi: 10.1007/pl00000065. [DOI] [PubMed] [Google Scholar]
- 23.WJ Wedemeyer, et al. Biochemistry. 2000;39:7032. doi: 10.1021/bi005111p. [DOI] [PubMed] [Google Scholar]
- 24. http://us.expasy.org/tools/protparam.html.
- 25. http://linux1.softberry.com/berry.phtml?topic.
- 26. http://globin.bio.warwick.ac.uk/psipred.
- 27. http://toolkit.tuebingen.mpg.de/modeller/verify3d/
- 28.A Laurie, R Jackson. Bioinformatics. 2005;21:1908. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.