

Abstract
Modern genomic sequencing technologies produce a large amount of data with reduced cost per base; however, this data consists of short reads. This reduction in the size of the reads, compared to those obtained with previous methodologies, presents new challenges, including a need for efficient algorithms for the assembly of genomes from short reads and for resolving repetitions. Additionally after abinitio assembly, curation of the hundreds or thousands of contigs generated by assemblers demands considerable time and computational resources. We developed Simplifier, a stand-alone software that selectively eliminates redundant sequences from the collection of contigs generated by ab initio assembly of genomes. Application of Simplifier to data generated by assembly of the genome of Corynebacterium pseudotuberculosis strain 258 reduced the number of contigs generated by ab initio methods from 8,004 to 5,272, a reduction of 34.14%; in addition, N50 increased from 1 kb to 1.5 kb. Processing the contigs of Escherichia coli DH10B with Simplifier reduced the mate-paired library 17.47% and the fragment library 23.91%. Simplifier removed redundant sequences from datasets produced by assemblers, thereby reducing the effort required for finalization of genome assembly in tests with data from Prokaryotic organisms.
Availability
Simplifier is available at http://www.genoma.ufpa.br/rramos/softwares/simplifier.xhtmlIt requires Sun jdk 6 or higher.
Keywords: NGS sequencing, ab initio assembly of genomes, redundant sequences
Background
NGS platforms utilize various different strategies for sequencing genomes and are capable of generating a large quantity of data with great accuracy and reduction in time necessary for sequencing and cost per base [1, 2]. However, they require considerable computational infrastructure for data processing [3]. The reduced size of the sequences that are initially generated presents challenges for the genome assembly process, including resolution of repetitive regions and a need to process extremely large numbers of reads, several magnitudes greater than produced by previous technologies [4].
Among strategies for obtaining complete genomes via NGS, there are hybrid approaches, involving the use of various assembly algorithms and combinations of data from various sequencing platforms to take advantage of complementarity among data sets. The redundancy of reads gives greater coverage of sequencing, which favors the generation of contigs that can represent regions in common or new regions of the genome, depending on the methodologies that are chosen [5]. The large number of contigs generated by these approaches demand considerable computational and human resources for their analysis, mainly for the identification of assembly errors and for elimination of the few wrong bases at the ends of contigs, which prevents overlapping extension due to mismatches [6–8].
To this end, we developed Simplifier; a stand-alone application that eliminates redundant sequences from groups of contigs generated by ab initio methodology, facilitating analysis of the data, reducing the time needed for the finalization and curation of the genome assemblies.
Software
Input and output
Data:
The data that we tested with this software were obtained by sequencing Escherichia coli DH10B http://www.ncbi.nlm.nih.gov/sra/SRX000353 with the SOLiD Version 3 sequencer; it provided a 50x50 mate-paired library with 28,627,096 reads and 37,365,488 (50 bp) fragments from http://solidsoftwaretools.com/gf/project/. We also processed genome data of Corynebacterium pseudotuberculosis strain 258 (Cp258), accession number CP003540.1, using the platform SOLiD Version 3 Plus, which provided a library of 50 bp fragments.
Sequence assembly pipeline:
The sequencing reads were submitted to an assembly pipeline, which began with the software SAET (Life Technologies) for the correction of sequencing errors, followed by Quality Assessment [8] for analysis of quality and application of the Phred 20 filter to the reads, to reduce the probability of assembly errors [9].
The processed data was then submitted to ab initio assembly, through the Bruijn approach, using the software Velvet [10] and overlap-layout-consensus with Edena [6]. The contigs abtained were aligned against the reference genome and visualized with the software Graphical Contig Analyser for All Sequencing Platforms – G4ALL (unpublished data), which identifies erroneous bases at the ends of the sequences (Figure 1).
Figure 1.
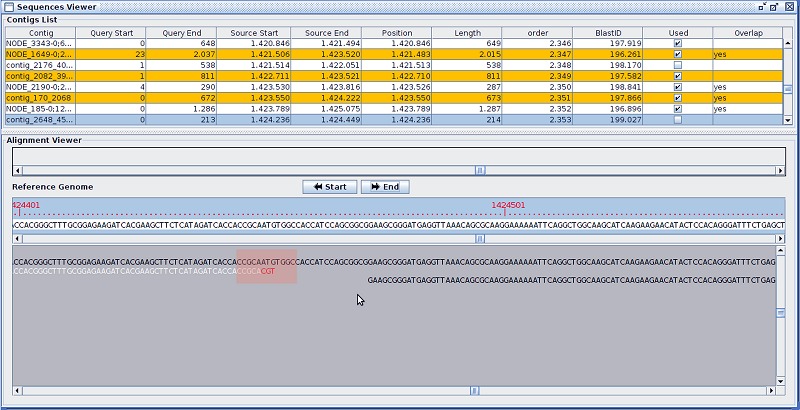
Interface of analysis of contigs of G4ALL. Superposition demonstrating that the last three bases of contig_2648 (white) are different from those of contig NODE_185 (black) and the reference. After elimination of these bases, there is redundancy between the sequences.
Simplifier:
Simplifier was implemented in JAVA (http://www.java.sun.com) utilizing the Swing library. The parallel processing was carried out with the use of threads, to improve the performance of the software. Simplifier receives multi fasta file containing contigs as input. Another file with relevant statistical informations such as N50 longest and shortest contig and number of bases. In the local version, these are entered with the command: java -jar Simplifier.jar -i <input file> -c <number of bases to be trimmed> -o <output file>. It then analyzes the information on how many bases at the ends of the contigs can be disregarded for the identification of redundant sequences. And finally it produces output file which contains contigs without redundancies from multifasta file produced by assemblers.
Steps for reducing the number of contigs:
When processing the data with Simplifier, the size of each sequence is calculated and stored in a list in decreasing order of contig size. Beginning with the smallest contig in the list, the program searches for larger ones can contain it, and thus, each contig is compared with all the others in the list. The redundant sequences recognized during the process are not considered in the subsequent comparisons, which reduces processing time.
The operator stipulates the maximum number of bases to be trimmed at the ends of the contigs; Simplifier trims from 0 up to the maximum number of bases defined. The search for redundancy is carried out in the following order: perfect matches, which are identified by the use of the sequences as a key of a hash table, trimming the 5' end, trimming the 3' end, and finally trimming both ends (Figure 2). After identifying redundancy, the sequence is saved to be used in the generation of the results file in multifasta format.
Figure 2.
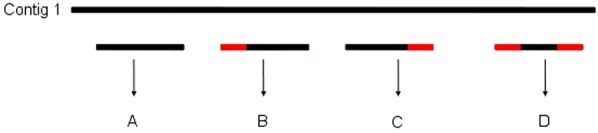
Criterion of elimination of redundancy of the contigs. Each contig of a group is compared with the others, considering the following possibilities: the – Contig completely redundant; B- Contig that when trimmed at the 5' end is redundant; CContig that when trimmed at the 3' end is redundant; D- Contig that when trimmed at both ends are redundant.
Utility
After applying a quality filter (phred 20) to the DH10B data, there were still 6,268,564 and 7,900,511 reads for the F3 and R3 mate-paired libraries, respectively, and 26,825,018 for the fragment library. In the assembly of the genomes, summing the contigs generated by Velvet and Edena, utilizing k-mer 29, resulted in 17,149 contigs for the mate-paired library, and 24,344 contigs for the fragments, utilizing k-mer 29 and 31, for Velvet and Edena, respectively. Using G4ALL, we observed mismatches in the bases at the ends of various contigs, mainly in the 3' region. Consequently, as a parameter of Simplifier, we defined three as the maximum number of bases to be trimmed at the ends of the sequences, in order to identify redundancies.
After processing contigs with Simplifier, there was a reduction of 17.47% (2,996 contigs) for mate-paired and 23.91% (5,821 contigs) for fragment libraries Table 1 (see supplementary material). In the two sets of data, few contigs less than or equal to 100 bp showed redundancy; most of the contigs eliminated by Simplifier were the size range of 101 to 499 bp; no contigs longer than 3 kb were eliminated Figure 3 and Table 2 (see supplementary material), resulting in an increase in N50 values (Table 1).
Figure 3.
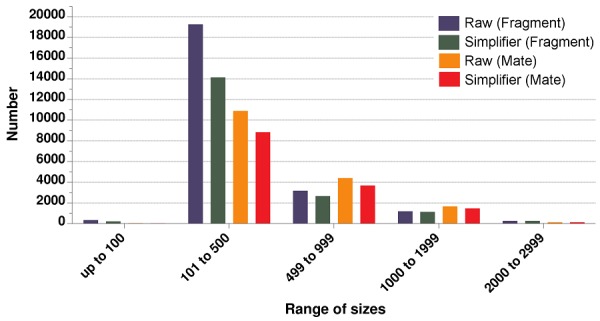
Relation between the frequency of contigs of Escherichia coli DH10B and its size range. The X-axis shows the size of the contigs; the Y-axis indicates the number of contigs in each range/library for the fragment and mate-paired libraries, with and without the use of Simplifier.
We ran BLAST against the NCBI non-redundant databank (utilizing a server with two Intel Xeon E5530 Quad Core 2.40 GHz processors and 24 Gb RAM) for identifying the products contained in the contigs; Simplifier reduced processing time up to 17 h for the fragments library, with about one million less hits compared to the original group that contained redundant contigs (Table 1). Simplifier used 62 minutes, 35 seconds processing time for the fragment library data (24,344 contigs) and 35 minutes, 41 seconds for the mate-paired data (17,149 contigs).
Application of Simplifier to the data for assembly of the Cp258, from SOLiD (library of 50 bp fragments) data, eliminated up to three bases at the ends from 8,004 contigs generated by Velvet and Edena. After 10 minutes, 49 seconds processing with Simplifier, the number of contigs was reduced to 5,272, which is a reduction of 34.14%; in addition, N50 increased from 1 kb to 1.5 kb.
Therefore, Simplifier enabled us to remove redundant Prokaryote sequences that were produced by genome assembly, before the manual curation step, which reduced the time and effort necessary for genome finalization. In addition, it allowed us apply different ab initio methods for genome assembly and then analyse only the unique sequences.
Supplementary material
Acknowledgments
This work was part of the Rede Paraense de Genômica e Proteômica supported by Fundação de Amparo a Pesquisa do Estado do Pará and Pronex Núcleo Amazônico de Excelência em Genômica de Microorganismos. M.P.S, A.S., V.A. and A.R.C. were supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). R.T.J.R. acknowledges support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Footnotes
Citation:Ramos et al, Bioinformation 8(20): 996-999 (2012)
References
- 1.SC Schuster. Nat Methods. 2008;5:16. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 2.JR Miller, et al. Genomics. 2010;95:315. [Google Scholar]
- 3.C Alkan, et al. Nat Methods. 2010;8:61. [Google Scholar]
- 4.ML Metzker. Nat Rev Genet. 2010;11:31. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 5.A Silva, et al. J Bacteriol. 2011;193:323. doi: 10.1128/JB.01211-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D Hernandez, et al. Genome Res. 2008;18:802. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.J JNijkamp, et al. Bioinformatics. 2010;26:i433. doi: 10.1093/bioinformatics/btq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.RT Ramos, et al. BMC Res Notes. 2011;4:130. doi: 10.1186/1756-0500-4-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.K Paszkiewicz, DJ Studholme. Brief Bioinform. 2010;11:457. doi: 10.1093/bib/bbq020. [DOI] [PubMed] [Google Scholar]
- 10.DR Zerbino, Birney Genome Res. 2008;18:821. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.