

Abstract
Most candidate genes and genetic abnormalities linked to autism spectrum disorders (ASD) are thought to play a role in developmental and experience-dependent plasticity. As a possible index of plasticity, we assessed the modulation of motor corticospinal excitability in individuals with Asperger’s Syndrome (AS) using transcranial magnetic stimulation (TMS). We measured the modulatory effects of Theta Burst Stimulation (TBS) on motor evoked potentials (MEPs) induced by single-pulse TMS in individuals with AS as compared with age-, gender-, and IQ-matched neurotypical controls. The effect of TBS lasted significantly longer in the AS group. The duration of the TBS-induced modulation alone enabled the reliable classification of a second study cohort of subjects as AS or neurotypical. The alteration in the modulation of corticospinal excitability in AS is thought to reflect aberrant mechanisms of plasticity, and might provide a valuable future diagnostic biomarker for the disease and ultimately offer a target for novel therapeutic interventions.
Keywords: Autism Spectrum Disorders, Theta Burst Stimulation, Transcranial Magnetic Stimulation, Plasticity
Introduction
Autism spectrum disorders (ASD) have become the most prevalent of the developmental disorders, affecting an estimated 1 in every 110 births (Baird, et al., 2006; Baron-Cohen, et al., 2009) yet their etiology remains unknown. Several investigators have proposed that aberrant cortical plasticity may play a role in the pathogenesis of ASD (Markram, Rinaldi, and Markram, 2007; Tsai, 2005; Dolen and Bear, 2009). Consistent with this hypothesis, many of the genes associated with ASD are involved in various aspects of synaptic development and plasticity (Morrow, et al., 2008.) Additionally, several animal models of ASD exhibit altered cortical plasticity as characterized by various different measures (for a review see Tordjman, et al., 2007). In humans, some neuroanatomical, brain imaging and neurophysiologic studies in ASD subjects have demonstrated anomalies in cortical excitability and connectivity (Geschwind & Levitt, 2007; Rubenstein & Merzenich, 2003; Belmonte et al., 2004), that might be consistent with alterations of mechanisms of plasticity (Oberman and Pascual-Leone, 2008).
In the present study, we used transcranial magnetic stimulation (TMS) to explore this issue further. Repetitive TMS (rTMS) enables the safe and noninvasive characterization of cortical reactivity mechanisms in humans (Kobayashi and Pascual-Leone, 2003). The aftereffects of rTMS have been proposed to relate to activity-dependent changes in the effectiveness of synaptic connections between cortical neurons, reflecting plasticity mechanisms of the brain (see Fitzgerald et al. 2006, Hoogendam et al. 2010, and Ziemann et al. 2008 for review). The theta burst stimulation (TBS) protocool has been proposed as a putative measure of synaptic plasticity (Huang et al., 2005). When applied over the motor cortex and depending on the stimulation parameters, TBS can induce a transient suppression of corticospinal excitibility (following continuous TBS, cTBS), or an enhancement (following intermittent TBS, iTBS). Suppression of corticospinal excitability by cTBS and its enhancement by iTBS appear to be mediated by cortical processes (Di Lazzaro et al., 2011), are considered indices of LTD and LTP-like mechanisms (Huerta and Volpe, 2009; Huang et al., 2005), and have been shown to involve GABAergic and Glutamatergic NMDA receptor pathways respectively (Stagg et al., 2009; Huang et al., 2007). Here we used single-pulse TMS to assess corticospinal excitability in 20 individuals with Asperger’s Syndrome (AS) and 20 age-, gender- and IQ-matched neurotypical controls before and after cTBS and iTBS. We hypothesized that in individuals with AS the effects of cTBS and iTBS upon TMS-induced MEPs would be significantly enhanced, possibly reflecting a human correlate of the alterations in LTD and LTP mechanisms found in animal models of ASD (Rinaldi et al., 2007; Bassell and Warren, 2008). Following the results of our first experiment, in order to evaluate the reliability of this TMS measure and its diagnostic potential, we evaluated a separate cohort of 15 individuals with AS and 15 matched controls participants with the same cTBS protocol.
Materials and Methods
Participants
We studied two cohorts of participants with AS and matching neurotypical controls. Data from cohort one was collected in Boston, Massachusetts and was composed of 20 individuals with AS [16 M, 4 F; age 18–64 (M= 34.3 years, SD=16.4); mean IQ 118.2 (SD=17.3)] and 20 age, gender, and full-scale IQ matched, typically developing individuals [16 M, 4 F; mean age: 34.9 years (SD=16.2); mean IQ 112.0 (SD=13.0)]. All participants in cohort one completed the cTBS protocol. A subset of these individuals (nine with AS and nine of their matched typically developing participants) also underwent the iTBS protocol [AS: 7 M, 2 F; mean age 40.7 years (SD=18.02); mean IQ 117.2 (SD=21.8) TD: 8 M, 2 F; mean age 41.3 years (SD=17.4); mean IQ 111.5 (SD=12.92)]. For those who participated in both the cTBS and iTBS protocols, the two sessions were separated by at least 1 week. Not all participants consented to come back for the iTBS session. Data from the second cohort was collected in Barcelona, Spain and was composed of 15 individuals with AS [(14 M, 1 F; mean age 42.4 years (SD=7.36); mean IQ 110.4 (SD=18.75)] and 15 age, gender, and IQ matched typically developing individuals [(14 M, 1 F; mean age 42.4.1 (SD=7.36); mean IQ 115.3 (SD=16.41)]. All participants from this cohort also completed the cTBS paradigm. All participants gave informed consent to the study, which was reviewed and approved by the institutional review boards at each participating institution. Participants were recruited through local community advertisement and local Asperger’s Associations and clinics.
All AS participants in both cohorts had an IQ over 80 based on the Weschler Abbreviated Scale of Intelligence (WASI) and a formal clinical diagnosis from an independent clinician prior to participation in the study. All met DSM-IV-TR criteria for Asperger’s Syndrome and met criteria for ASD on the Autism Diagnostic Observation Schedule – Module 4 (ADOS) (Mean Social and Communication score: 10.2 (SD= 4.6). Additionally, the Autism Diagnostic Interview Revised (ADI-R) was completed on 11 participants whose parents were available for interview. For these individuals the mean Social score was 18.2 (SD = 5.1), Communication score was 20.0 (SD = 2.6), and Repetitive Behaviors was 6.0 (SD = 2.3). Cognitive and clinical evaluation was identical for both cohorts, with Spanish translated versions of the ADOS and WASI used for the participants in Cohort Two.
Participants in the neurotypical group were healthy controls with no neurological or psychiatric disorders. This group was matched with respect to chronological age, gender, and full scale IQ with the AS group. All participants were given a comprehensive neurological exam by a board certified neurologist to confirm normal gross motor and fine motor functioning. Lastly, all participants were screened following published recommendations (Rossi et al., 2009) to ensure that they did not have any condition that would put them at greater risk for an adverse event related to TMS (e.g. a personal or family history of epilepsy).
Stimulation and Recording
Study procedures were identical across the two study locations. The experimenters who collected the data at each location were trained by Dr. Pascual-Leone and used the same equipment and procedures described herein. Continuous TBS (cTBS) and intermittent TBS (iTBS) were applied as described in Huang et al., 2005. The cTBS paradigm consisted of three pulses of 50 Hz stimulation repeated at 200 ms intervals for 40 seconds (for a total of 600 pulses) at an intensity of 80% of active motor threshold (AMT). In the iTBS paradigm participants received a two-second train of TBS repeated every 10 seconds for a total of 190 seconds (600 pulses) also at an intensity of 80% of active motor threshold (AMT). (Figure 1). Corticospinal excitability was assessed prior to and following cTBS or iTBS by measuring peak-to-peak amplitude of MEPs induced in the contralateral first dorsal interosseus (FDI) muscle in response to single pulse TMS at a rate of approximately 0.1Hz (a random jitter of ± 1 s was introduced to avoid any train effects). Three batches of 10 MEPs were recorded prior to cTBS or iTBS and used as a baseline. Following cTBS or iTBS, batches of 10 MEPs were measured at periodic intervals for a total of 120 minutes to track changes in MEP amplitude over time.
Figure 1.
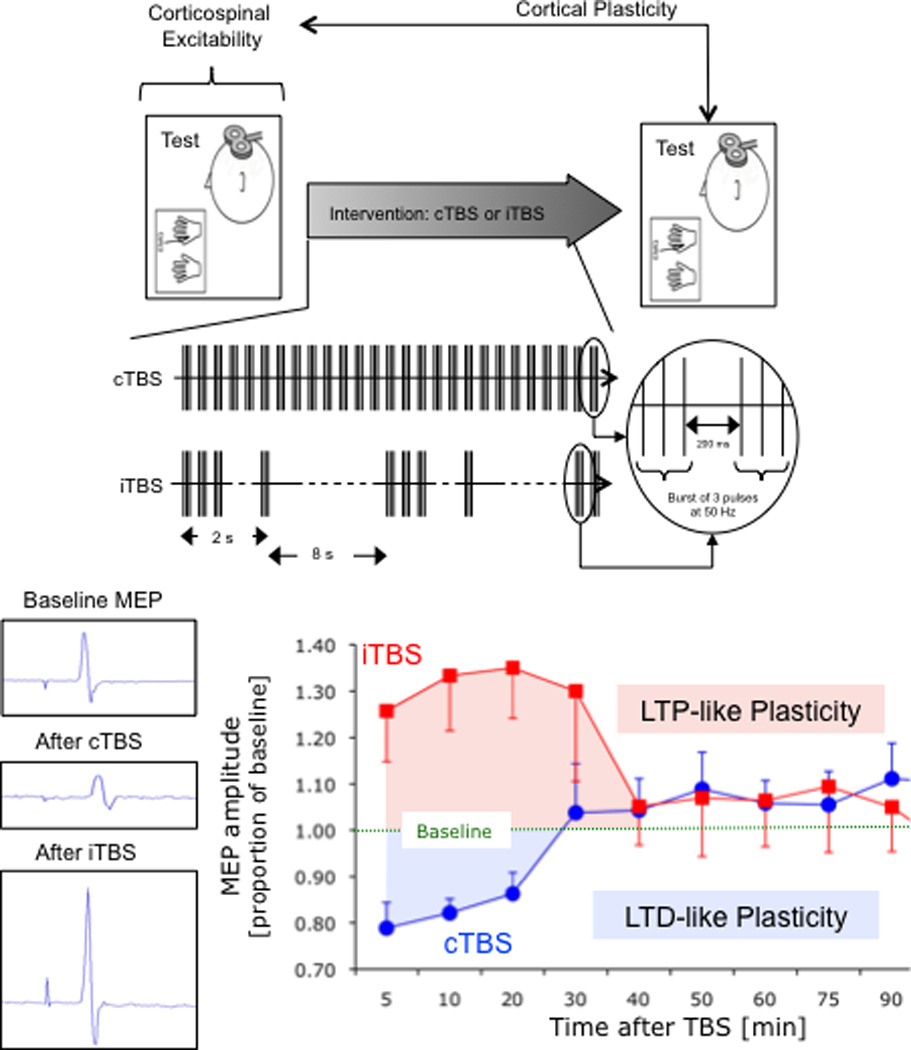
Schematic summary of applied methodology. TBS involves applying bursts of high frequency transcranial magnetic stimulation (3 pulses at 50 Hz) repeated at intervals of 200ms. After TBS is applied to the motor cortex in an intermittent fashion (iTBS), single-pulse TMS-induced MEPs show increased amplitude for a period of 20–30 minutes, whereas continuous TBS (cTBS) leads to a suppression of the TMS-induced MEPs for approximately the same amount of time (Huang et al., 2005).
In order to measure TMS induced MEPs, Ag-AgCl EMG surface electrodes were placed in a belly-tendon montage over the FDI muscle of participants’ right hand. Raw signals were amplified and band-pass-filtered between 20 and 2000 Hz. EMG signals were sampled at a rate of 5000 Hz. All stimulation (single pulse TMS and TBS) was delivered using a hand-held figure-eight coil attached to a Magstim Super Rapid stimulator. The coil was placed tangentially to the scalp with the handle pointing posteriorly. All stimulation was applied over the hand area of the left motor cortex and individually localized for each participant based on the optimal position for eliciting MEPs in the right FDI. The stimulation intensity for baseline and post TBS single pulses was set at 120% of each individual’s resting motor threshold (RMT) while the TBS itself was delivered at 80% of AMT. RMT and AMT were defined following recommendation from the International Federation of Clinical Neurophysiology. RMT was defined as the minimum single pulse TMS intensity required to induce an MEP in the contralateral FDI of greater than 50 µV peak-to-peak amplitude on more than five out of ten consecutive trials while the target muscle was at rest. AMT was definied as the minimum single pulse TMS intensity required to induce an MEP in the contralateral FDI of greater than 200 µV peak-to-peak amplitude on more than five out of ten consecutive trials while the target muscle was held at approximately 20% of the maximal contraction. In order to precisely target the stimulation site (primary motor cortex) and keep the brain target constant throughout the stimulation session, we used a frameless stereotactic neuronavigation system (Brainsight, Rogue Inc).
Data Analysis
For all experiments across both cohorts data were analyzed using SPSS version 17 by a blinded experimenter.
MEP amplitude at a given time point was defined as the mean amplitude of the 10 MEPs to single TMS pulses recorded in a given 2 minute time window. As an index of the duration of the TBS-induced modulation of cortico-spinal excitability, we defined, for each participant, the time point at which the average MEP amplitude at a given time following TBS returned to within the 95% confidence interval of the baseline amplitude and did not return to outside that interval on subsequent time point measures. MEP amplitudes were standardized forming a ratio of MEP amplitudes following TBS relative to average baseline MEP amplitude for each individual.
For the first cohort, our primary outcome measure was time to return to baseline, thus a t-test was used to compare the duration of the suppression (to cTBS) or facilitation (to iTBS) of MEP amplitude following cTBS and iTBS respectively. We also evaluated the degree of suppression at all 11 timepoints as a secondary measure of group difference. In order to further assess the predictive value of cTBS to discriminate between individuals with AS and neurotypical controls, the data from the first cohort was used as a learning set, and data collected from a separate sample of individuals was used as a validation set. These two cohorts were taken from two different samples, one collected in Boston, MA and the other collected in Barcelona, Spain. We chose to analyse the data separately rather than combining the data because we felt that we had sufficient power to analyse the two samples separately, and this provided us with an opportunity to test the validity and generalizability of the finding. From the data from the first cohort, a receiver operating characteristic (ROC) curve was created and the area under the curve at the various time points was determined by calculating the c-statistic. Based on this statistic a timepoint was chosen at which returning to baseline would optimally differentiate between the first cohort groups. This value was then applied to the new cohort’s data and diagnostic sensitivity and specificity values were obtained.
Results
All participants tolerated the TMS study without any side-effects or complications.
Consistent with prior findings (Theoret et al., 2005), AS and control groups did not differ significantly in resting motor threshold (RMT) (ASD Mean = 42.6, SD = 6.0; Control M = 46.9, SD = 6.6; p = 0.14) or in baseline MEP values prior to either cTBS (p = 0.48) or iTBS (p=0.51). Consistent with our hypothesis, the AS group showed greater and longer lasting modulation of their MEPs following both forms of TBS. The average time to return to baseline MEP values following cTBS was 35.5 minutes (SD = 13.2) for the controls, while the AS group did not return to baseline levels until an average of 87 minutes (SD = 26.3) (Figure 2). Similarly, for iTBS, the average time taken to return to baseline was 37.2 minutes (SD = 35.3) in the control group and 77.8 minutes (SD = 31.3) in the AS group. These differences were significant for both forms of TBS (cTBS: t(19) = 8.20, p<0.001; iTBS: t(8) = 3.04, p<0.05) and were not correlated with age, IQ, ADOS score, or ADI score (all ps >0.05). In addition, following cTBS, the AS group was significantly different in baseline-corrected MEPs as compared to the control group beginning at 20 minutes post TBS and lasting until 50 minutes post TBS (all p-values < 0.004 Bonferroni corrected). For the iTBS paradigm, the groups were not significant at any timepoint after Bonferroni correction was applied.
Figure 2.
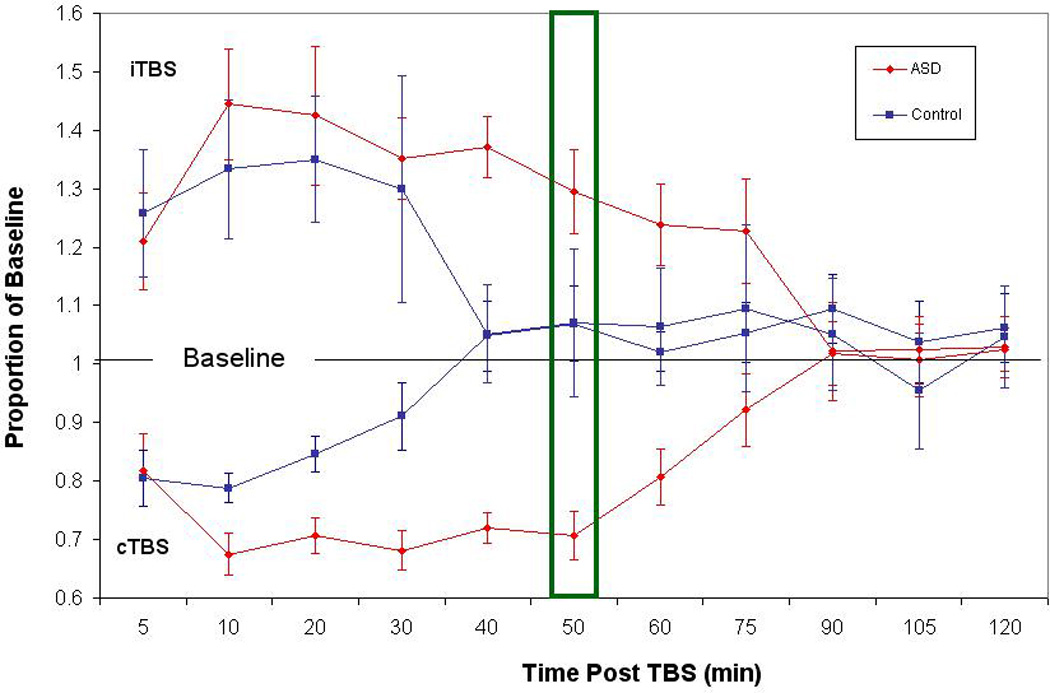
Baseline corrected MEP amplitude following cTBS and iTBS. Average baseline-corrected MEP amplitude for the control group (in blue) and AS group (in red) at 11 time points from 5 minutes to 120 minutes post cTBS. In error bars indicate standard error of the mean for each time point. Values are represented as proportion of baseline amplitude with a line at 1.0 (representing baseline amplitude). The box at the 50 minute time point represents the criterion determined by the ROC curve to be the point of maximal sensitivity and selectivity between the groups.
We chose to use the cTBS paradigm to test the diagnostic potential of this TMS protocol in a different cohort. The cTBS paradigm was chosen for this second cohort based on several factors. Firstly, the cTBS paradigm was found to be more reliable in the first cohort as compared to the iTBS paradigm. Secondly, to simplify the study, we only wanted to include a single TBS session and we felt that the cTBS protocol, being a suppressive protocol, would be theoretically safer (i.e. less likely to induce a seizure).
Using data from the first cohort, we calculated a ROC curve, which resulted in a c-statistic (Area Under the Curve) of 0.966 and a standard error of .023. The ROC curve also indicated that the timepoints of maximal sensitivity and selectivity were at 50 (sensitivity = 1, selectivity = 0.75) and 60 minutes (sensitivity = 0.85, selectivity = 0.1) respectively. Erring on the side of sensitivity for this analysis (assuming a type I error of flagging a healthy individual as being part of the AS group would be less costly than a type II error of missing an individual that should have been flagged as being part of the AS group), we assigned 50 minutes as our criterion for minimal duration of effect to be classified as belonging to the AS group. Figure 3 shows the new cohort of individuals classified according to this cut-off point and their clinical diagnostic status. The suggested diagnostic test reveals a sensitivity of 0.93 (95% CI: 0.66, 1.0) and a specificity of 0.8 (95% CI: 0.51, 0.95).
Figure 3.
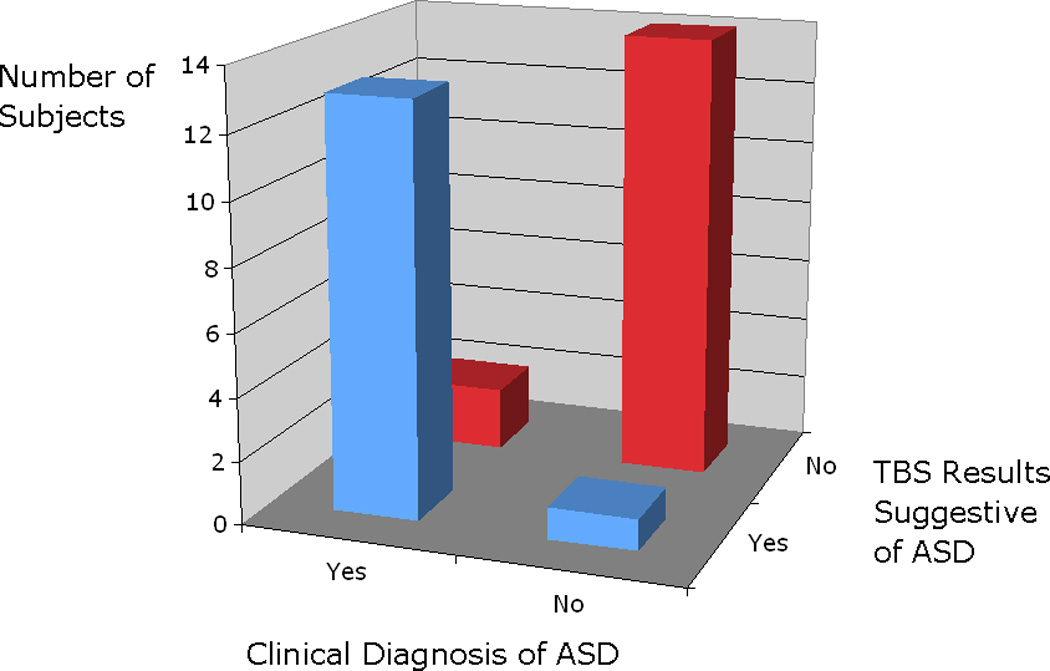
Graph of accuracy of classification of subjects into ASD versus not, according to results of the TBS modulation of TMS responses. Classification is based on whether the individual was back to baseline at the timepoint of 50 minutes or longer.
It is important to note that despite the heterogeneity of our sample (e.g., the broad age-range, the possible differences in genetic predisposition and the fact that environmental exposures were likely different in the two cohorts), we found consistent disturbances in cortical plasticity responses to TBS in practically all AS subjects. Figure 4 displays data from all individual subjects obtained from both cohorts and demonstrates a strong dissociation between cTBS induced effects in neurotypical and AS participants.
Figure 4.
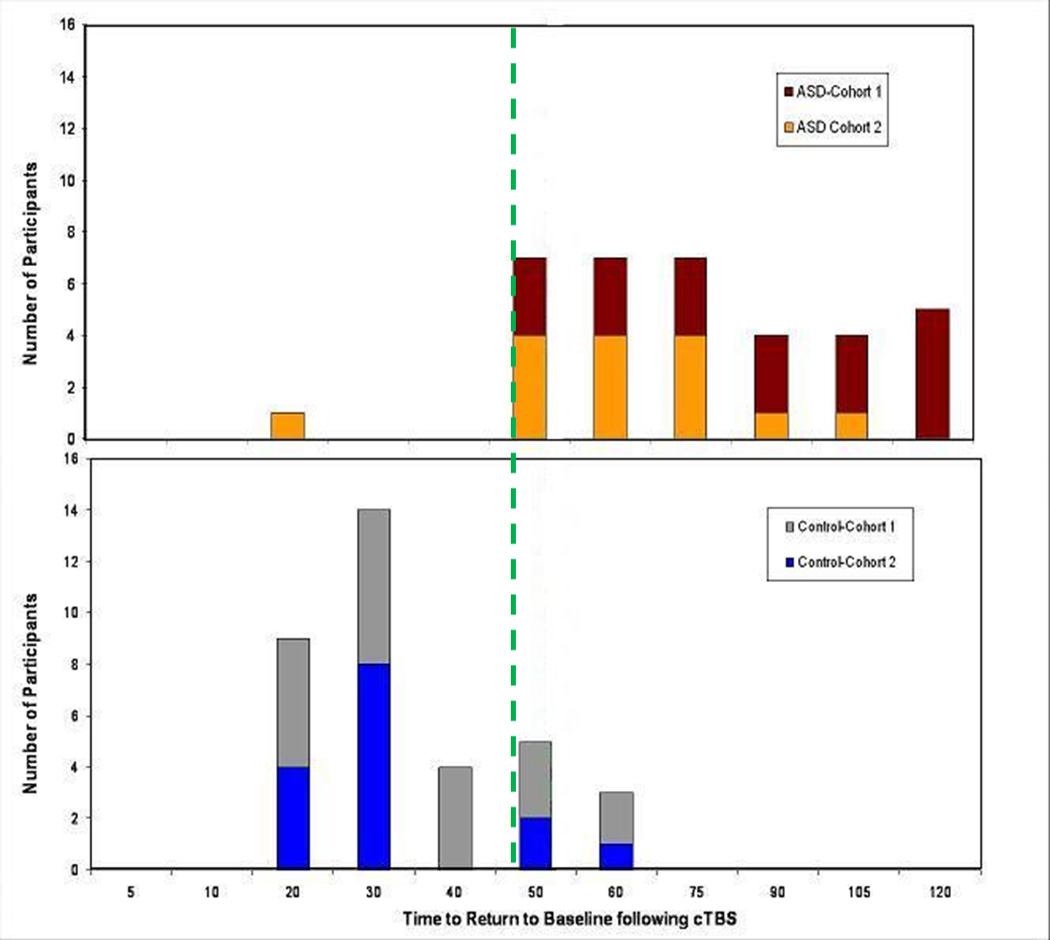
Summary of individual results: Distribution of the number of subjects from each study group (AS top graph; neurotypical controls in bottom graph) and their time to return to baseline the TMS-induced potentials following theta burst stimulation. Note that there is an almost complete separation of the results for all subjects in both the first cohort (brown, gray) or second cohort (orange, blue) with cTBS. 34 out of 35 AS subjects show a duration of the modulatory effects of cTBS of 50 minutes or longer while 27 out of 35 neurotypical control participants show a duration of less than 50 minutes. The dashed green line at 50 min following the TBS is the calculated time for greatest diagnostic value of the test.
Discussion
Our findings reveal altered modulation of corticospinal excitability in ASD. Specifically we found that the modulation induced by TBS was significantly longer lasting in ASD than in neurotypical, control subjects. The cellular and molecular substrates for TBS-induced modulation of TMS evoked motor potentials are unclear, though studies suggest that LTD- and LTP-like mechanisms of synaptic plasticity are involved (Stagg et al., 2009; Huang et al., 2007).
Plasticity is an intrinsic property of the brain, which allows for adaptive changes in neural architecture to take place over the course of the lifetime (Pascual-Leone et. al 2011). This can occur for example by altering the functional weighting of synaptic connections (e.g. by strengthening or weakening these), by modifying the structure of these connections (e.g. by synaptic pruning or the addition of new synapses), or by promoting neurogenesis (Pascual-Leone et al., 2011). Aberrations in these mechanisms could conceivably lead to a pathological phenotype in one of two (not mutually exclusive) ways: Normal mechanisms could serve to compound the pathological consequences of a specific genetic mutation or sustained environmental insult. Alternatively, aberrant plasticity mechanisms could act on a previously normal brain to induce a disease phenotype. The timing of plastic brain changes may also be important. Mistimed alterations in plasticity may set the stage for a processes that otherwise would have been behaviorally innocuous, to become pathogenic (Gogolla et al., 2009).
It is plausible that the neurological and behavioral phenotype of ASD is associated with altered brain plasticity. Differences in brain volume and cortical connectivity (Courchesne, et al., 2001; Herbert et al., 2004) for example may stem from underlying abnormalities in plasticity. Indeed, many of the genes that have been linked to ASD, such as BDNF, are known to play critical roles in cortical reactivity, plasticity and connectivity (Lu, 2003; Kleim, et al., 2006). In addition, disorders that clinically resemble ASD are associated with single-gene mutations affecting genes related to protein synthesis-dependent LTP and LTD (e.g. Fragile X syndrome, Tuberous sclerosis complex, and PTEN hamartoma syndrome) (Dolen and Bear, 2009). Lastly, several animal models of ASD have revealed abnormal plasticity mechanisms (for a review see Markram, Rinaldi, and Markram, 2007). These findings have lead researchers (Markram, Rinaldi and Markram, 2007; Oberman and Pascual-Leone, 2008) to suggest that plasticity abnormalities underlie the clinical symptoms of ASD, however, empirical studies directly linking measures of plasticity at both the system-level and molecular level to the clinical symptoms of ASD are lacking, so such claims are purely speculative at this point.
Our results demonstrate that the duration of effect of TBS is significantly longer in humans with AS. Future studies to clarify the neural substrate of such findings are needed. It is conceivable that the enhanced duration of excitability of the targeted cells is a consequence of hyperplasticity of the local network. Alternatively, it is plausible that the observed response is a consequence of hypoplasticity in the compensatory response of distal cells. Follow-up studies using real-time integration of TMS with electroencephalography (EEG) to record local as well as global response to TBS may shed light on this question.
The molecular mechanisms underlying this effect are also unclear based on the current findings. Recent reports find both enhanced expression of metabotropic glutamate receptor 5 (MGluR5) (Fatemi and Folsom, 2011) and decreased expression of GABAA and GABAB receptors in ASD (Fatemi et al., 2009a,b; 2010). Both MGluR5 and GABA receptors play critical roles in modulating reactivity at the synaptic level and thus may contribute to the physiological mechanism underlying TBS-induced modulation of corticospinal excitability. Alterations in MGluR5 and GABA receptors may play an important pathophysiologic role in our findings. Follow-up studies directly testing the relationship between GABA and MGluR5 receptor expression (perhaps through Magnetic Resonance Spectrosopy) and measures of cortical reactivity in humans with ASD are needed.
Independent of the underlying mechanisms though, the potential clinical utility of our findings is supported by the measure’s ability to accurately classify a separate cohort of individuals as either AS or neurotypical. Nonetheless, this also must be taken as preliminary, as other neuropsychiatric conditions were not included in this analysis. Therefore, it is unclear whether one could accurately discriminate between AS and another similar neuropsychiatric condition with our methodology.
It could be argued that the differential results of TBS modulation in AS and neurotypical controls are simply the consequence of a differential impact of TMS on the targeted brain region in the different subject groups. However, we believe this to be unlikely. First, there was no difference between groups in terms of baseline motor excitability. Second, stimulation intensity both pre and post TBS, as well as the stimulation intensity of the TBS itself, was determined individually for each subject based on their own motor threshold, and there were no group differences between AS and neurotypical participants. Third, the difference across groups was primarily in the duration of the TBS induced modulation, rather than in the pattern or amplitude of the initial effect. Fourth, there was no difference in head or brain sizes between our adult AS participants and the neurotypical controls, and anatomical MRIs in all our study subjects confirmed no difference in the distance from the coil to the targeted cortical stimulation site (p=0.09) across groups. There was also no correlation of the TBS results with the individual measures of distance from coil to stimulation target. Finally, in a previous TMS study (Theoret et al., 2005) there were no abnormalities in input/output curves, intracortical inhibition and facilitation, motor thresholds, or silent periods in a group of individuals with ASD. Therefore, we believe that the differential effects of TBS in AS as compared with neurotypical controls reveal fundamental differences in the mechanism governing the modulation of corticospinal excitability.
In the current study, we focused on primary motor cortex in the left hemisphere. Thus, it is unclear whether other cortical regions would show similar abnormalities in the modulatory effects of TBS or whether there would be a laterality effect in these individuals. The left primary motor cortex was chosen in this study for two reasons. First, MEPs are the standard index used to quantify the effect of TBS protocols. Other indices of cortical excitability outside the motor cortex (e.g. based on electroencephalographic measures) have not yet been well validated for this application. We chose the left hemisphere as it is typically the dominant hemisphere for both right and left handed individuals. Secondly, although motor abnormalities are not considered core symptoms of AS, many studies have reported motor deficits in individuals with ASD including alterations in motor milestone development (Teitelbaum et al., 1998), clumsiness, motor incoordination, disturbances in reach-to-grasp movement (Ghaziuddin & Butler, 1998; Miyahara et al., 1997; Mari et al., 2003), deficits in gross and fine motor movement (Noterdaeme et al., 2002), and impaired postural control (Kohen-Raz et al., 1992; Minshew et al., 2004).
If our findings prove to be specific to different neurodevelopmental disorders, future TBS studies might be used to establish valuable neurophysiologic biomarkers in clinical populations. As more evidence is garnered about aberrant responses to the modulatory effects of TBS in different neurodevelopmental disorders, it should be possible to assess the full diagnostic utility of such tests. In addition, real-time integration of TMS with EEG will allow investigators to apply these measures to cortical brain regions other than motor cortex (Thut et al., 2005; Ives et al., 2006; Thut & Pascual-Leone, 2010a,b). Finally, if our results are replicated and it is determined that there is a relationship to behavioral symptoms, therapeutic interventions aimed at regulating such alterations may be worth pursuing.
Acknowledgements
Work on this study was supported by grants from the National Center for Research Resources: Harvard-Thorndike Clinical Research Center at BIDMC (NCRR MO1 RR01032) and Harvard Clinical and Translational Science Center (UL1 RR025758); NIH grant K24 RR018875 and a grants from Autism Speaks and the Nancy Lurie Marks Family Foundation to A.P.-L. . L.Oberman was supported by NIH fellowship F32MH080493. We thank Paul Wang, Joseph Gonzalez-Heydrich, Alexander Rotenberg, Jonathan Picker, Albert Galaburda, Mike Greenberg, Christopher Walsh, Shiva Gautam, Murray Mittleman, and Carla Shatz for valuable comments on the data and the manuscript. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the Nancy Lurie Marks Family Foundation, National Center for Research Resources or the National Institutes of Health.
References
- Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, et al. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: The Special Needs and Autism Project (SNAP) The Lancet. 2006;368(9531):210–215. doi: 10.1016/S0140-6736(06)69041-7. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Scott FJ, Allison C, et al. Prevalence of autism-spectrum conditions: UK school-based population study. Br J Psychiatry. 2009;194(6):500–509. doi: 10.1192/bjp.bp.108.059345. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MK, Allen G, Beckel-Mitchener A, Boulanger LM, Carper RA, Webb SJ. Autism and abnormal development of brain connectivity. J Neurosci. 2004;24(42):9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, et al. Modulation of motor cortex neuronal networks by rTMS: comparison of local and remote effects of six different protocols of stimulation. J Neurophysiol. 2011;105(5):2150–2156. doi: 10.1152/jn.00781.2010. [DOI] [PubMed] [Google Scholar]
- Dolen G, Bear MF. Fragile x syndrome and autism: from disease model to therapeutic targets. J Neurodev Disord. 2009;1(2):133–140. doi: 10.1007/s11689-009-9015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, et al. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both Fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec (Hoboken) 2011;294(10):1635–1645. doi: 10.1002/ar.21299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, et al. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum, 2009. 2009b;8(1):64–69. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, et al. mRNA and protein levels for GABAAalpha4, alpha5, beta1 and GABABR1 receptors are altered in brains from subjects with autism. J Autism Dev Disord. 2010;40(6):743–750. doi: 10.1007/s10803-009-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, et al. GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord, 2009. 2009a;39(2):223–230. doi: 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PB, Fountain S, Daskalakis ZJ. A comprehensive review of the effects of rTMS on motor cortical excitability and inhibition. Clin Neurophysiol. 2006;117:2584–2596. doi: 10.1016/j.clinph.2006.06.712. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17(1):103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Ghaziuddin M, Butler E. Clumsiness in autism and Asperger syndrome: a further report. J Intellect Disabil Res. 1998;42:43–48. doi: 10.1046/j.1365-2788.1998.00065.x. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Leblanc JJ, Quast KB, Sudhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord. 2009;1(2):172–181. doi: 10.1007/s11689-009-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert MR, Ziegler DA, Makris N, Filipek PA, Kemper TL, Normandin JJ, et al. Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol. 2004;55(4):530–540. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- Hoogendam JM, Ramakers GM, Di Lazzaro V. Physiology of repetitive transcranial magnetic stimulation of the human brain. Brain Stimul. 2010;3(2):95–118. doi: 10.1016/j.brs.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Chen RS, Rothwell JC, Wen HY. The after-effect of human theta burst stimulation is NMDA receptor dependent. Clin Neurophysiol. 2007;118(5):1028–1032. doi: 10.1016/j.clinph.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Edwards MJ, Rounis E, Bhatia KP, Rothwell JC. Theta burst stimulation of the human motor cortex. Neuron. 2005;45(2):201–206. doi: 10.1016/j.neuron.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Volpe BT. Transcranial magnetic stimulation, synaptic plasticity and network oscillations. J. Neuroeng Rehabil. 2009;2:7. doi: 10.1186/1743-0003-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives JR, et al. Electroencephalographic recording during transcranial magnetic stimulation in humans and animals. Clin Neurophysiol. 2006;117(8):1870–1875. doi: 10.1016/j.clinph.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Kleim JA, Chan S, Pringle E, Schallert K, Procaccio V, Jimenez R, et al. BDNF val66met polymorphism is associated with modified experience-dependent plasticity in human motor cortex. Nat Neurosci. 2006;9(6):735–737. doi: 10.1038/nn1699. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Pascual-Leone A. Transcranial magnetic stimulation in neurology. Lancet Neurol. 2003;2(3):145–156. doi: 10.1016/s1474-4422(03)00321-1. [DOI] [PubMed] [Google Scholar]
- Kohen-Raz R, Volkmar FR, Cohen DJ. Postural control in children with autism. J Autism Dev Disord. 1992;22:419–432. doi: 10.1007/BF01048244. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10(2):86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Castiello U, Marks D, Marraffa C, Prior M. The reach-to-grasp movement in children with autism spectrum disorder. Philos Trans R Soc Lond B Biol Sci. 2003;385:393–403. doi: 10.1098/rstb.2002.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Rinaldi T, Markram K. The intense world syndrome--an alternative hypothesis for autism. Front Neurosci. 2007;1(1):77–96. doi: 10.3389/neuro.01.1.1.006.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshew NJ, Sung K, Jones BL, Furman JM. Underdevelopment of the postural control system in autism. Neurology. 2004;63:2056–2061. doi: 10.1212/01.wnl.0000145771.98657.62. [DOI] [PubMed] [Google Scholar]
- Miyahara M, Tsuji M, Hori M, Nakanishi K, Kageyama H, Sugiyama T. Brief report: motor incoordination in children with Asperger syndrome and learning disabilities. J Autism Dev Disord. 1997;27:595–603. doi: 10.1023/a:1025834211548. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321(5886):218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noterdaeme M, Mildenberger K, Minow F, Amorosa H. Evaluation of neuromotor deficits in children with autism and children with a specific speech and language disorder. Eur Child & Adoles Psych. 2002;11:219–225. doi: 10.1007/s00787-002-0285-z. [DOI] [PubMed] [Google Scholar]
- Oberman L, Pascual-Leone A. Cortical plasticity: A proposed mechanism by which genomic factors lead to the behavioral and neurological phenotype of autism spectrum and psychotic spectrum disorders. Behavioral and Brain Sciences. 2008;31:241–320. [Google Scholar]
- Pascual-Leone A, Freitas C, Oberman L, Horvath JC, Halko M, Eldaief M, et al. Characterizing Brain Cortical Plasticity and Network Dynamics Across the Age-Span in Health and Disease with TMS-EEG and TMS-fMRI. Brain Topogr. 2011;24(3–4):302–315. doi: 10.1007/s10548-011-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi T, et al. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A. 2007;104(33):13501–13506. doi: 10.1073/pnas.0704391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, et al. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2009;120(12):2008–2039. doi: 10.1016/j.clinph.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg CJ, et al. Neurochemical effects of theta burst stimulation as assessed by magnetic resonance spectroscopy. J Neurophysiol, 2009. 2009;101(6):2872–2877. doi: 10.1152/jn.91060.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum P, Teitelbaum O, Nye J, Fryman J, Maurer RG. Movement analysis in infancy may be useful for early diagnosis of autism. Proc Natl Acad of Sci USA. 1998;95:13982–13987. doi: 10.1073/pnas.95.23.13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoret H, Halligan E, Kobayashi M, Fregni F, Tager-Flusberg H, Pascual-Leone A. Impaired motor facilitation during action observation in individuals with autism spectrum disorder. Curr Biol. 2005;15(3):R84–R85. doi: 10.1016/j.cub.2005.01.022. [DOI] [PubMed] [Google Scholar]
- Thut G, Pascual-Leone A. A review of combined TMS-EEG studies to characterize lasting effects of repetitive TMS and assess their usefulness in cognitive and clinical neuroscience. Brain Topogr. 2010a;22(4):219–232. doi: 10.1007/s10548-009-0115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thut G, Pascual-Leone A. Integrating TMS with EEG: How and what for? Brain Topogr. 2010b;22(4):215–218. doi: 10.1007/s10548-009-0128-z. [DOI] [PubMed] [Google Scholar]
- Thut G, et al. A new device and protocol for combining TMS and online recordings of EEG and evoked potentials. J Neurosci Methods. 2005;141(2):207–217. doi: 10.1016/j.jneumeth.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Tordjman S, Drapier D, Bonnot O, Graignic R, Fortes S, Cohen D, et al. Animal models relevant to schizophrenia and autism: validity and limitations. Behav Genet. 2007;37(1):61–78. doi: 10.1007/s10519-006-9120-5. [DOI] [PubMed] [Google Scholar]
- Tsai SJ. Is autism caused by early hyperactivity of brain-derived neurotrophic factor? Med Hypotheses. 2005;65(1):79–82. doi: 10.1016/j.mehy.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Paulus W, Nitsche MA, Pascual-Leone A, Byblow WD, Berardelli A, Siebner HR, Classen J, Cohen LG, Rothwell JC. Consensus: motor cortex plasticity protocols. Brain Stimul. 2008;1:164–182. doi: 10.1016/j.brs.2008.06.006. [DOI] [PubMed] [Google Scholar]