

Abstract
The synthesis and characterization of new tris(carbene)borate ligand precursors containing substituted benzimidazol-2-ylidene and 1,3,4-triazol-2-ylidene donor groups, as well as a new tris(imidazol-2-ylidene)borate ligand precursor are reported. The relative donor strength of the tris(carbene)borate ligands have been evaluated by the position of ν(NO) in four-coordinate {NiNO}10 complexes, and follows the order: imidazol-2-ylidene > benzimidazol-2-ylidene > 1,3,4-triazol-2-ylidene. There is a large variation in ν(NO), suggesting these ligands to have a wide range of donor strengths while maintaining a consistent ligand topology. All ligands are stronger donors than Tp* and Cp*.
Introduction
The development of multidentate ligands incorporating multiple N-heterocyclic carbene (NHC) donors has been driven in part by the desire to develop new catalysts with increased stability. While the vast majority of multidentate NHC ligands are bidentate, higher denticity ligands, including tridentate1 and even tetradentate2 donors have been reported.3 Tridentate ligands composed solely of NHC donors are mainly limited to a small number of C3-symmetric tripodal ligands in which three imidazol-2-ylidene donors are connected through a central linker atom or group.4,5,6,7,8,9
Tripodal tris(carbene)borate ligands (Fig. 1) connect three imidazol-2-ylidene donors through a central borate linker.4 First generation tris(carbene)borate ligands (R = Me, Et; R′ = H) have been used to prepare octahedral manganese, iron and cobalt complexes,10,11 and more recently a copper(I) cluster.12 Our group has developed synthetic routes for second generation tris(carbene)borate ligands in which the steric bulk has been increased (R = tBu, Mes; R′ = H, Ph). As with the topologically related tris(pyrazolyl)borate ligands, the introduction of steric bulk allows for the synthesis of low coordinate transition metal complexes.13 However, tris(carbene)borate ligands are substantially stronger donors than tris(pyrazolyl)borates,14 and have been shown to stabilize high valent complexes of first row transition metals,15 including an isolable iron(V) nitride.16
Fig. 1.
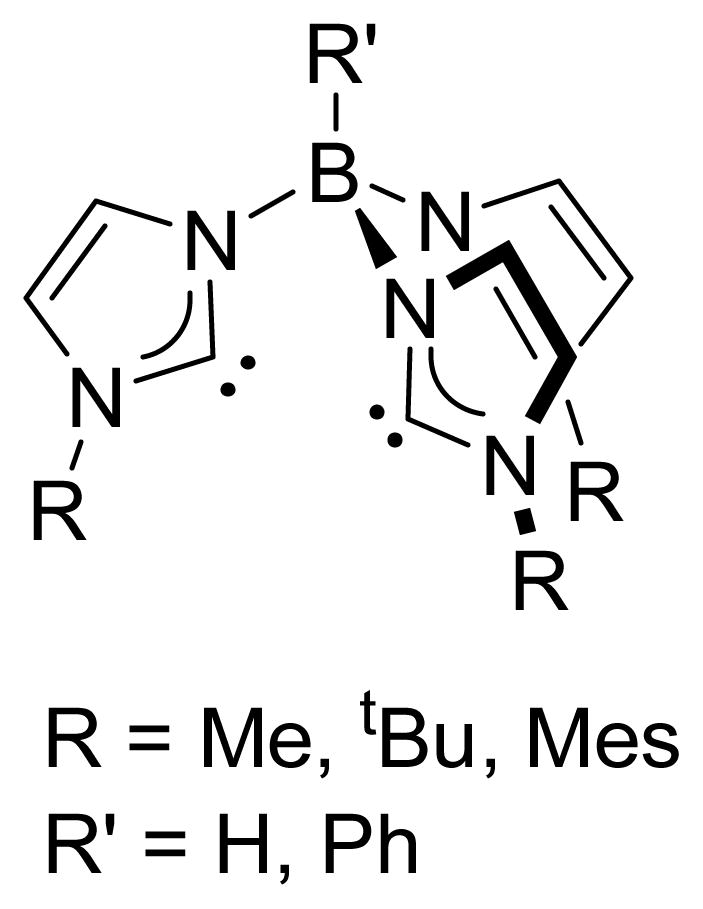
Tris(carbene)borate ligands based on imidazol-2-ylidene donors.
The donor strength of tris(pyrazolyl)borate ligands can be modified by suitable changes to the pyrazolyl ring substitutents.17 Similar flexibility in the donor strength of tris(carbene)borate ligands does not yet exist. In the case of monodentate NHC ligands, changing the N-substituents in imidazol-2-ylidenes does not typically have a major impact on the donor strength of the NHC ligand,18 and thus similar changes to the donors of tris(carbene)borate ligand are not expected to have a major influence on the ligand donor strength. However, recent work has shown that NHCs based on donors other than imidazol-2-ylidene can lead to large changes in donor strength. Thus, both experimental and computational studies of monodentate NHC ligands shows that carbenes derived from heterocycles other than imidazole can have substantially different donor strengths.19
In this paper we report the synthesis of new tris(carbene)borate ligands, including ligands based on benzimidazol-2-ylidene and 1,3,4-triazol-2-ylidene donors. Studies of monodentate NHC ligands have shown that the donor strength of these ligands decreases according to the order: imidazol-2-ylidene > benzimidazol-2-ylidene > 1,3,4-triazol-2-ylidene (Fig. 2) The donor properties of the new tris(carbene)borate ligands have been evaluated in four-coordinate {NiNO}10 complexes, allowing their donor properties to be compared with other monoanionic facially coordinating ligands. A number of proposals regarding the nature of bonding between Ni and NO in complexes of this type have been put forth,20 with the most comprehensive investigation describing the electronic structure as having multireference character; the dominant contributor being Ni(II) (SNi = 1) antiferromagnetically coupled to NO− (SNO = 1).21
Fig. 2.

Relative donor strength of selected N-heterocyclic carbene ligands.
Experimental
General Considerations
Manipulations involving air-sensitive materials were performed under a nitrogen atmosphere by standard Schlenk techniques or in an M. Braun Labmaster glove box. The quality of the glove box atmosphere was periodically checked with a toluene solution of “titanocene”.22 Glassware was dried at 150 °C overnight. Diethyl ether, pentane, tetrahydrofuran and toluene were purified by the Glass Contour solvent purification system. Deuterated benzene was dried first over CaH2, then over Na/benzophenone, before vacuum transfer into a storage container. Before use, aliquots of Et2O, THF and toluene were tested with a drop of Na/benzophenone in THF solution. N,N-Dimethylformamide azine dihydrochloride was prepared according to a literature procedure.23 Florisil for column chromatography was dried by heating at 100 °C under vacuum for 8 hours prior to use. The {NiNO}10 synthons Ni(PPh3)2(NO)Br24 and Ni(NO)I(THF)225 were prepared according to literature procedures. All other reagents were purchased from commercial vendors and used as received. 1H NMR data were recorded on a Varian Unity 400 spectrometer (400 MHz) at 22 °C. All resonances in the 1H NMR spectra are referenced to residual CHCl3 (δ 7.26 ppm), C6D5H (δ 7.16 ppm), C4D7H (δ 3.57 and 1.72 ppm) and CD2HCN (δ 1.94 ppm). Infrared spectra were recorded on Perkin–Elmer Spectrum One FTIR and Thermo Scientific Nicolet iS10 SMART iTR spectrophotometers, while UV-visible spectra were recorded on a Cary 100 spectrometer. Atmospheric pressure positive and negative ionization mass spectral data were collected using a Waters-Micromass ZQ2000 Mass Spectrometer. Cyclic voltammograms were recorded under a N2 atmosphere using a CH Instruments CHI600D potentiostat in 0.1 M Bu4PF6 in THF with a glassy carbon working electrode, a Pt wire counter electrode and a Ag+/Ag reference electrode. All electrochemical data are referenced to the Cp2Fe+/Cp2Fe couple. Elemental analyses were performed by Robertson Microlit Laboratories, Madison NJ.
Synthesis of compounds
Preparation of 1-(methylcyclohexyl)imidazole (1)
A 250 mL flask was charged with imidazole (4.81 g, 0.700 mol, 1.0 equiv) and stirred with freshly ground KOH (5.94 g, 0.106 mol, 1.5 equiv) in dimethylsulfoxide (80 mL) for three hours. The golden solution was cooled in ice, and then bromomethylcyclohexane (10.3 mL, 7.38 mmol, 1.05 equiv) added and maintained at 0 °C for four hours and left to stir overnight at room temperature. Hexanes (80 mL) and diethyl ether (40 mL) were added and the mixture washed in a separatory funnel with saline water (3 × 150 mL). The residual dimethylsulfoxide was removed by rewashing the combined organic fractions with three more portions of water (80 mL). The solvents were removed in vacuo and the product distilled with heating at reduced pressure to yield a hygroscopic white solid. The solid was dissolved in THF and dried over CaH2 (11.3 g, 97 %). The compound is spectroscopically identical to that reported in the literature.26
Preparation of 1-(methylcyclohexyl)benzimidazole (2)
A 1 L flask was charged with benzimidazole (19.1 g, 0.162 mol, 1 equiv) and stirred with freshly ground KOH (12.5 g, 0.223 mol, 1.37 equiv) in 220 mL dimethylsulfoxide for 1 d. The yellow solution was cooled in ice, bromomethylcyclohexane (23.3 mL, 0.166 mol, 1.02 equiv) added and the reaction allowed to warm to room temperature after four hours. After stirring for three days, dichloromethane (250 mL) was added to the resultant slurry. The mixture was washed with distilled water (3 × 250 mL). The solvents were removed in vacuo and the product sublimed with heating under reduced pressure to yield a white solid (32.1 g, 94 %). 1H NMR (CDCl3): δ 7.85 (s, 1H, Bz), 7.82 (d, 1H, JHH = 8.8, Bz), 7.40 (d, 1H, JHH = 7.2, Bz), 7.29 (m, 2H, Bz), 4.00 (d, 2H, JHH = 7.2, CH2), 1.87 (m, 1H, Cy), 1.75-1.64 (m, 5H, Cy), 1.27-1.14 (m, 3H, Cy), 1.06-0.97 (m, 2H, Cy). ESI+-MS 215 (C14H19N2+). Anal. Cald. for C14H18N2: 78.46, H 8.47, N 13.07. Found C 77.74, H 8.46, N 12.71. Multiple microanalysis trials were consistently low in carbon.
Preparation of 4-phenyl-1,2,4-triazole (3)
4-Phenyl-1,2,4-triazole was prepared by modification of a literature procedure.23 A 500 mL flask was charged with N,N-dimethylformamide azine dihydrochloride (20.9 g, 9.73 mmol, 1 equiv), freshly distilled aniline (8.87 mL, 9.73 mmol, 1 equiv) and benzene (100 mL). The reaction was heated at reflux overnight. After cooling to room temperature, the resultant mixture was dried in vacuo. Dichloromethane (100 mL) was added to the solid and the mixture washed with alkaline water (3 × 200 mL). The solvent was removed from the organic layer under reduced pressure and the white solid was purified by vacuum sublimation with heating to yield a white solid (8.61 g, 61 %). The spectral data are identical to that reported in the literature.27
Preparation of 4-(4-tert-butylphenyl)-1,2,4-triazole (4)
4-(4-tert-Butylphenyl)-1,2,4-triazole was prepared by modification of a literature procedure.23 A 500 mL flask was charged with N,N-dimethylformamide azine dihydrochloride (16.4 g, 7.61 mmol, 1 equiv), 4-tert-butylaniline (12.3 mL, 7.61 mmol, 1 equiv) and benzene (80 mL). The reaction was heated at reflux overnight. After cooling the resultant mixture was dried in vacuo. Dichloromethane (100 mL) was added to the solid and the mixture washed with alkaline water (3 × 200 mL). The solvent was removed from the organic layer under reduced pressure and the resulting white solid was purified by vacuum distillation while heating. A white solid was obtained upon cooling (14.9 g, 97 %). 1H NMR (CDCl3) δ 8.44 (s, 2H, Tz), 7.55 (d, JHH = 8.4, 2H, o/m-C6H4tBu), 7.32 (d, JHH = 8.8, 2H, o/m-C6H4tBu), 1.36 (s, 9H, tBu). ESI+-MS 202 (C12H16N3+). Anal. Cald. for C12H15N3: C 71.61, H 7.51, N 20.88. Found C 71.57, H 7.34, N 20.78.
Preparation of 4-mesityl-1,2,4-triazole (5)
4-Mesitylene-1,2,4-triazole was prepared by modification of a literature procedure.23 A 500 mL flask was charged with N,N-dimethylformamide azine dihydrochloride (20.6 g, 9.58 mmol, 1 equiv), 2,4,6-trimethylaniline (13.9 mL, 9.58 mmol, 1 equiv) and heated to 180 °C in chlorobenzene (100 mL) for 3 d. After cooling, hexanes (300 mL) was added to the vessel and the mixture cooled to −25 °C. The supernatant was decanted from the solid that had precipitated. Dichloromethane (100 mL) was added to the solid and the mixture washed with alkaline water (3 × 200 mL). The solvent was removed from the organic layer under reduced pressure to yield a white to off-white solid. Colored impurities were removed by recrystallization from CH2Cl2/Et2O at −25 °C. The white solid obtained was purified by vacuum sublimation with heating (10.4 g, 58%). The spectral data are identical to that reported in the literature.28
Preparation of [PhB(CyCH2ImH)3](OTf)2 (6)
N-Methylcyclohexylimidazole 1 (3.50 g, 21.3 mmol, 3.04 equiv) was added to a solution of PhBCl2 (1.11 g, 6.99 mmol, 1 equiv) in toluene (50 mL). After 30 min, TMSOTf (3.31 g, 14.9 mmol, 2.13 equiv) was added and the mixture was heated at 110 °C for 1 d. After cooling to room temperature hexanes (100 mL) was added, leading to the formation of a white precipitate. The mixture was cooled to −25 °C overnight and the resultant white solid collected by filtration and washed with hexanes. The solid was recrystallized from CH2Cl2/Et2O at −25 °C. The resulting white solid collected by vacuum filtration, washed with hexanes and dried in vacuo (5.14 g, 87 %). 1H NMR (CDCl3): δ 8.49 (s, 3H, Im-H), 7.46 (s, 3H, Im-H), 7.18 (s, 3H, Im-H), 7.16 (m, 5H, B(C6H5)), 4.10 (d, 6H, JHH = 7.6, CH2), 1.80-1.66 (m, 17H, Cy), 1.27-1.19 (m, 10H, Cy), 1.04-0.97 (m, 6H, Cy). ESI+-MS 879 (C38H54N6BF6O6S2+). Anal. Cald. for C38H53N6BF6O6S2: C 51.94, H 6.08, N 9.56. Found C 51.88, H 6.12, N 9.39.
Preparation of [PhB(PhTzH)3](OTf)2 (7)
4-Phenyl-1,2,4-triazole 3 (5.83 g, 40.0 mmol, 3.05 equiv) was added to a solution of PhBCl2 (2.15 g, 13.1 mmol, 1 equiv) in toluene (70 mL). After stirring for 30 min, TMSOTf (6.19 g, 27.6 mmol, 2.10 equiv) was added and the mixture was heated at 110 °C for 1 d. After cooling to room temperature, a white solid was collected by vacuum filtration, washed with diethyl ether and dried in vacuo (9.64 g, 94 %). 1H NMR (CDCl3): δ 9.83 (s, 3H, Tz-H), 8.72 (s, 3H, Tz-H), 7.80 (m, 6H, o-C6H5), 7.58 (m, 9H, m/p-C6H5), 7.50 (m, 5H, B(C6H5)). ESI+-MS 822 (C32H27N9BF6O6S2+). Anal. Cald. for C32H26N9BF6O6S2·0.25 C7H8: C 48.00, H 3.34, N 14.93. Found C 47.73, H 3.42, N 14.60.
Preparation of [PhB(p-tBuPhTzH)3](OTf)2 (8)
4-(4-tert-Butylphenyl)-1,2,4-triazole 4 (1.98 g, 9.8 mmol, 3.03 eqiuv) was added to a solution of PhBCl2 (0.513 g, 3.23 mmol, 1 equiv) in toluene (40 mL). After 30 min, TMSOTf (1.49 g, 6.70 mmol, 2.07 equiv) was added to the solidified reaction mixture and the reaction heated at 110 °C for 1 d. After cooling to room temperature, the solid was collected by vacuum filtration and washed with hexanes. The white solid was then collected and dried in vacuo (2.49 g, 78 %). 1H NMR (CDCl3): δ 9.76 (s, 3H, Tz-H), 8.67 (s, 3H, Tz-H), 7.71 (d, 6H, JHH = 8.8, o/m-C6H4tBu), 7.59 (d, 6H, JHH = 8.8, o/m-C6H4tBu), 7.49 (m, 5H, B(C6H5)), 1.33 (s, 27H, tBu). ESI+-MS 990 (C32H27N9BF6O6S2+). Anal. Cald. for C32H26N9BF6O6S2: C 46.78, H 3.19, N 15.34. Found C 46.58, H 3.27, N 15.07.
Preparation of [PhB(MesTzH)3](OTf)2 (9)
4-Mesitylene-1,2,4-triazole 5 (2.40 g, 12.8 mmol, 3.06 equiv) was partially dissolved by stirring in toluene (80 mL) overnight. Phenylboranedichloride, PhBCl2 (0.665 g, 4.18 mmol, 1 equiv) was added to the mixture and the reaction stirred for 8 h. Trimethylsilyl triflate, TMSOTf (1.91 g, 8.61 mmol, 2.06 eqiuv) was added to the cloudy solution and the reaction heated at 110 °C for 1 d. The resultant mixture was dried in vacuo to yield a white solid. The solid was dissolved in dichloromethane (15 mL) and diethyl ether (130 mL) added to induce precipitation. The solution was stored at −25 °C overnight and the resulting colorless crystals were dried in vacuo (3.70 g, 96 %). 1H NMR (CDCl3): δ 9.99 (s, 3H, Tz), 8.55 (s, 3H, Tz), 7.46 (m, 3H, m/p-B(C6H5)), 7.32 (m, 2H, o-B(C6H5)), 7.03 (s, 6H, m-Mes), 2.34 (s, 9H, p-Me), 2.16 (s, 18H, o-Me). ESI+-MS 948 (C41H45N9BF6O6S2+). Anal. Cald. for C41H44N9BF6O6S2·H2O: C 50.99, H 4.80, N 13.05. Found C 50.93, H 4.63, N 13.30.
Preparation of [HB(MeBzH)3](Br)2 (10)
A solution of N-methylbenzimidazole (3.54 g, 26.8 mmol, 3.08 eqiuv) and Me3N:BHBr2 (2.01 g, 8.69 mmol, 1 eqiuv) was heated in chlorobenzene (50 mL) at 150 °C for 1 d. After cooling to room temperature, the mixture was filtered to yield a white solid that was washed with diethyl ether and hexanes and then dried under reduced pressure. The white solid was dissolved in warm methanol (5 mL). Diethyl ether (50 mL) and hexanes (20 mL) were added to initiate precipitation and the mixture stored at −25 °C overnight. The solid that formed was separated from supernatant and the process repeated twice more. The white solid obtained was dried in vacuo (4.6 g, 92 %). 1H NMR (CDCl3): δ 10.82 (s, 3H, Bz), 7.75 (d, 3H, JHH = 8.4, Bz), 7.70 (d, 3H, JHH = 8.0, Bz), 7.59 (t, 3H, JHH = 7.4, Bz), 7.52 (t, 3H, JHH = 7.4, Bz), 5.70 (br s, 1H, B-H), 4.27 (s, 9H, Me). IR (ATR, cm−1): 2482 (w, B–H). ESI+-MS 568 (C24H25N6BBr2+). Anal. Cald. for C24H25N6BBr2·H2O: C 49.18, H 4.64, N 14.34. Found C 49.18, H 4.64, N 14.35.
Preparation of [HB(CyCH2BzH)3](Br)2 (11)
A solution of N-methylcyclohexylbenzimidazole 2 (2.54 g, 11.8 mmol, 3.02 eqiuv) and Me3N:BHBr2 (0.896 g, 3.90 mmol, 1 equiv) was heated in refluxing chlorobenzene (30 mL) for 1 d. After cooling to room temperature, hexanes (60 mL) were added and the solution heated at 50 °C for 15 min. On cooling, the white solid was separated by vacuum filtration, washed with hexanes and the residual solvent removed under reduced pressure The solid was purified by multiple recrystallizations from acetonitrile/Et2O at −25 °C. The white solid was then dried in vacuo (2.8 g, 88 %). 1H NMR (CDCl3): δ 10.47 (s, 3H, Bz), 8.05 (d, JHH = 6.8, 3H, Bz), 7.66 (d, JHH = 8.0, 3H, Bz), 7.56 (m, 6H, Bz), 5.83 (br s, 1H, B-H), 4.48 (d, JHH = 7.2, 6H, CH2), 1.97 (m, 3H, Cy), 1.74-1.62 (m, 14H, Cy), 1.15-1.06 (m, 16H, Cy). IR (ATR, cm−1): 2431 (w, B–H). ESI+-MS 815 (C42H56N6BBr2+). Anal. Cald. for C42H55N6BBr2·H2O: C 60.59, H 6.90, N 10.09. Found C 60.21, H 6.76, N 9.98.
Preparation of [HB(p-tBuPhTzH)3](Br)2 (12)
A solution of 4-(4-tert-butylphenyl)-1,2,4-triazole 3 (2.16 g, 10.7 mmol, 3.05 equiv) and Me3N:BHBr2 (0.809 g, 3.51 mmol, 1 equiv) in chlorobenzene (30 mL) was heated at 150 °C for 1 d. The solution was allowed to cool to 40 °C followed by dropwise addition of diethyl ether (30 mL) then pentane (30 mL) with stirring. The mixture was then stored at −25 °C overnight. The resulting solid was separated from supernatant and dried under reduced pressure. Dichloromethane (50 mL) was added to the solid and the solution washed with alkaline/saline water (1 × 10 mL). The organic layer was dried under reduced pressure to yield a white to off-white solid. Impurities were removed by recrystallization from acetonitrile/Et2O at −25 °C. The white solid obtained was dried in vacuo (1.61 g, 59%). 1H NMR (CDCl3): δ 10.99 (s, 3H, Tz-H), 8.61 (s, 3H, Tz-H), 7.94 (d, JHH = 8.8, 6H o/m-C6H4tBu), 7.61 (d, JHH = 8.8, 6H o/m-C6H4tBu), 5.28 (br s, 1H, B-H), 1.33 (s, 27H, tBu). IR (ATR, cm−1): 2518 (w, B–H). ESI+-MS 776 (C36H47N9BBr2+). Anal. Cald. for C36H46N9BBr2: C 55.76, H 5.98, N 16.26. Found C 56.03, H 6.27, N 16.46.
Preparation of [HB(MesTzH)3](Br)2 (13)
A toluene (100 mL) solution of 4-mesitylene-1,2,4-triazole 5 (3.44 g, 18.4 mmol, 3.10 equiv) and Me3N:BHBr2 (1.37 g, 5.93 mmol, 1 eqiuv) in a round bottom flask equipped with reflux condenser was heated at vigorous reflux for 3 d under nitrogen (heating in chlorobenzene at 150 °C results in the formation of impurities). The solution was allowed to cool and the solvent removed under reduced pressure. The resulting off-white solid was dissolved in dichloromethane (80 mL) and washed with alkaline/saline water (1 × 15 mL). The organic layer was collected and to the aqueous wash solution was added dichloromethane (30 mL) a second time. The organic layers were combined and dried under reduced pressure the resulting solid was purified by recrystallization from acetonitrile/Et2O at −25 °C. The white solid was dried in vacuo (3.25 g, 74 %). 1H NMR (CDCl3): δ 11.24 (s, 3H, Tz-H), 8.44 (s, 3H, Tz-H), 7.01 (s, 6H, m-Mes), 5.28 (br s, 1H, B-H), 2.33 (s, 9H, p-Me), 2.19 (s, 18H, o-Me). IR (ATR, cm−1): 2524 (w, B–H). ESI+-MS 734 (C33H41N9BBr2+). Anal. Cald. for C33H40N9BBr2: C 54.05, H 5.50, N 17.19. Found C 54.02, H 5.76, N 17.12.
Preparation of PhB(CyCH2Im)3NiNO (14)
A slurry of [PhB(CyCH2ImH)3](OTf)2 6 (304 mg, 0.345 mmol, 1 equiv) in toluene (5 mL) was cooled to −78 °C. To the mixture was added lithium diisopropylamide (112 mg, 1.04 mmol, 3.01 equiv). The solution was stirred for one hour at −78 °C, allowed to warm to room temperature and stirred for three hours. To the resulting yellow solution was added Ni(NO)I(THF)2 (124 mg, 0.345 mmol, 1 eqiuv). The reaction was left to stir for three hours at room temperature. The solution was filtered through Celite and the filtrate dried under reduced pressure. The residue was then extracted with hexanes (15 mL) and filtered. The filtrate was dried under vacuum to give an orange-red solid (72 mg, 31 %). 1H NMR (CDCl3): δ 7.86-7.85 (m, 2H, m-B(C6H5)),7.70-7.65 (m, 2H, o-B(C6H5)), 7.56-7.51 (m, 1H, p-B(C6H5)), 6.83 (d, JHH = 1.6, 3H, Im-H), 6.72 (d, JHH = 2.0, 3H, Im-H), 4.07 (d, 6H, JHH = 6.8, CH2), 1.73-1.71 (m, 17H, Cy), 1.20 (m, 10H, Cy), 1.09-1.03 (m, 6H, Cy). 13C NMR (CDCl3): δ 201.3 (carbene-C), 134.2, 127.5 127.4, 126.9, 122.8, 118.8, 57.0, 39.5, 30.7, 26.3, 25.8. IR (THF, cm−1): 1693 (s, N–O). ESI−-MS 668 (C36H53N7BNiO−). UV-Vis (THF): 441 nm (ε = 790 M−1 cm−1). Eox = −0.22 V (irr). Despite multiple attempts, we have been unable to obtain satisfactory microanalysis results.
Preparation of HB(MeBz)3NiNO (15)
To a solution of [HB(MeBzH)3](Br)2 10 (0.345 g, 0.607 mmol, 1equiv) in dichloromethane (7 mL) was added Ag2O (0.227 g, 0.979 mmol, 1.6 eqiuv). The reaction was heated at 45 °C overnight in air, dried under vacuum and an aliquot was analyzed by 1H NMR spectroscopy. Two sets of resonances associated with the deprotonated ligand are observed. This is possibly due to the formation of different silver complexes, but was not further investigated, (1H NMR (CD3Cl3)): Species A: δ 7.92 (s, 3H, Bz), 7.79 (d, JHH = 8.0, 3H, Bz), 7.44 (m, 3H, Bz), 7.35 (m, 3H, Bz), 3.86 (s, 9H, Me); Species B: δ 7.75 (d, 3H, Bz), 7.57 (d, JHH = 8.0, 3H, Bz), 7.43 (m, 3H, Bz), 7.34 (m, 3H, Bz), 3.69 (s, 9H, Me)). To the tris(carbene)borate ligand synthon was added THF (7 mL), the vessel charged with Ni(NO)Br(PPh3)2 (0.463 g, 0.668 mmol, 1.1 equiv) under nitrogen at room temperature, and stirred overnight. The solvent was removed under reduced pressure and the residue extracted with toluene and filtered through Celite. The toluene filtrate was dried and the residual solid was purified by column chromatography under nitrogen (Florisil, THF eluent). The orange-red colored fraction was collected and dried to give an orange-red solid (101 mg, 33 %). 1H NMR (C6D6): δ 7.86 (d, JHH = 8.0, 3H, Bz), 7.12-7.05 (m, 6H, Bz), 6.77 (d, JHH = 8.0, 3H, Bz), 5.79 (br s, 1H, B-H), 3.69 (s, 9H, Me). 13C NMR (CDCl3): δ 210.7 (carbene-C), 138.34, 136.81, 121.48, 120.78, 111.79, 109.06, 34.64. IR (THF, cm−1): 2462 (w, B–H), 1714 (s, N–O). ESI−-MS 492 (C24H22N7BNiO−). Anal. Calcd. for C24H22BN7NiO: C 58.35, H 4.49, N 19.85. Found C 58.04, H 4.48, N 19.74. UV-vis (THF): 433 nm (ε = 1180 M−1 cm−1). Ered = −1.43 V (irr).
Preparation of HB(CyCH2Bz)3NiNO (16)
To a solution of [HB(CyCH2BzH)3](Br)2 11 (341 mg, 0.418 mmol, 1 equiv) in acetonitrile (6 mL) was added Ag2O (194 mg, 0.837 mmol, 2 equiv). The reaction was heated at reflux for 1 d, dried under vacuum. The poor solubility of the residue prevented any further characterization. To the tris(carbene)borate ligand synthon was added THF (6 mL) and Ni(NO)I(THF)2 (0.105 g, 0.292 mmol, 0.7 equiv).29 The reaction was stirred overnight under nitrogen at room temperature. The solvent was removed under reduced pressure and the residue extracted with toluene and filtered through Celite. The toluene filtrate was dried, the resultant solid extracted with hexanes and filtered. The hexanes solution was dried under vacuum and the residue washed with acetone followed by MeOH to give a red-orange colored solid (87 mg, 40 % based on Ni). 1H NMR (C6D6): δ 7.91 (d, JHH = 7.6, 3H, Bz), 7.09-7.00 (m, 12H, Bz), 5.71 (br s, 1H, B-H), 4.33 (d, JHH = 7.2, 6H, CH2), 1.66 (br m, 7H, Cy), 1.57 (br m, 12H, Cy), 1.07 (br m, 14H, Cy). 13C NMR (CDCl3): δ 211.7 (carbene-C), 138.5, 136.6, 121.2, 120.4, 111.9, 109.5, 54.6, 38.8, 31.2, 26.3, 25.9. IR (THF, cm−1): 2463 (w, B–H), 1711 (s, N–O). ESI−-MS 739 (C42H52N7BNiO−). UV-vis 435 nm (ε = 1490 M−1 cm−1). Ered = −1.93 V (irr).
Preparation of HB(p-tBuPhTz)3NiNO (17)
To a solution of [HB(p-tBuPhTzH)3](Br)2 12 (59 mg, 0.07 mmol, 1 equiv) in acetonitrile (4 mL) was added Ag2O (35 mg, 0.15 mmol, 2.1 equiv). The reaction was heated at reflux for 1 d. The reaction mixture was dried under vacuum, following by addition of KBr (14 mg, 0.11 mmol) in THF (4 mL) and the reaction stirred under nitrogen overnight. The solvent was removed under reduced pressure and the residue extracted with toluene and filtered through Celite. The filtrate was dried under reduced pressure to yield “HB(p-tBuPhTz)3K” (49 mg). An aliquot of the residue analyzed by 1H NMR spectroscopy is consistent with threefold deprotonation (1H NMR (C6D6)): δ 7.88 (s, 3H, Tz-H), 7.00 (d, JHH = 8.4, 6H, o/m-C6H4tBu), 6.50 (d, JHH = 8.4, 6H, o/m-C6H4tBu), 1.11 (s, 27H, tBu). To the tris(carbene)borate ligand synthon was added a toluene (3 mL) solution of Ni(NO)I(THF)2 (27 mg, 0.07 mmol, 1 equiv) at room temperature. The reaction was stirred overnight, the solution filtered through Celite and dried in vacuo to give a maroon colored solid (29 mg, 52 % based on Ni). 1H NMR (C6D6): δ 7.55 (s, 3H, Tz-H), 7.39 (d, JHH = 8.8, 6H, o/m-C6H4tBu), 7.04 (d, JHH = 8.4, 6H, o/m-C6H4tBu), 1.03 (s, 27H, tBu). 13C NMR (CDCl3): δ 199.6 (carbene-C), 151.2, 141.3, 135.2, 126.4, 122.6, 34.7, 31.3. IR (THF, cm−1): 2544 (w, B–H), 1746 (s, N–O). ESI−-MS 700 (C36H43N10BNiO−). UV-vis (THF): 467 nm (ε = 500 M−1 cm−1). Eox = 0.38 V (irr); Ered = −1.78 V (irr). Despite multiple attempts, we have been unable to obtain satisfactory microanalysis data for this complex.
Preparation of HB(MesTz)3NiNO (18)
To a solution of [HB(MesTzH)3](Br)2 13 (184 mg, 0.250 mmol, 1 equiv) in acetonitrile (5 mL) was added Ag2O (93 mg, 0.40 mmol, 1.6 equiv). The reaction was heated at reflux for 1 d, the reaction mixture was then dried under vacuum and KBr (45 mg, 0.38 mmol, 1.5 equiv) in THF (5 mL) was added to the residue and stirred under nitrogen overnight. The solvent was removed under reduced pressure and the residue extracted with toluene and filtered through Celite. The filtrate was dried under reduced pressure to yield “HB(MesTz)3K” (89 mg). An aliquot of the residue analyzed by 1H NMR spectroscopy is consistent with threefold deprotonation (1H NMR (C6D6)): δ 7.40 (s, 3H, Tz-H), 6.49 (s, 6H, m-Mes), 1.98 (s, 9H, p-Me), 1.48 (s, 18H, o-Me). To the tris(carbene)borate ligand synthon was added a toluene solution (3 mL) of Ni(NO)I(THF)2 (52 mg, 0.14 mmol, 0.6 equiv.) The reaction was stirred overnight at room temperature. The solution was filtered through Celite and dried in vacuo to give a maroon colored solid (51 mg, 53 % based on Ni). 1H NMR (CDCl3): δ 7.84 (s, 3H, Tz-H), 6.91 (s, 6H, m-Mes), 2.27 (s, 9H, p-Me), 1.97 (s, 18H, o-Me). 13C NMR (CDCl3): δ 202.1 (carbene-C), 142.5, 138.6, 134.6, 133.2, 129.0, 20.9, 18.0. IR (THF, cm−1): 2537 (w, B–H), 1742 (s, N–O). ESI−-MS 658 (C33H37N10BNiO−). Anal. Calcd. for C33H37BN10NiO: C 60.12, H 5.66, N 21.25. Found C 59.81, H 5.40, N 21.00. UV-vis (THF): 451 nm (ε = 620 M−1 cm−1). Eox = 0.40 V (irr); Ered = −1.83 V (irr).
Solid Angle Calculations
Solid angle calculations were performed using the program Solid-G.30,31 The ligand coordinates used in the calculations were taken from X-ray crystal structure data of the corresponding {NiNO}10 complexes. The metal ligand bond lengths were set at a distance of 2.28 Å. In cases where there was more than one molecule in the asymmetric unit, solid angles were calculated for each molecule and the average value reported. For the truncated calculations, ORTEP-3 (version 2.02)32 was used to define a sphere of enclosure (4 Å, centered on Ni) and the resulting coordinates used for solid angle calculations.
Results and Discussion
Synthesis of heterocycles
N-Methylcyclohexylimidazole 1 was prepared by the reaction of deprotonated imidazole and bromomethylcyclohexane (Scheme 1). Conducting the reaction at low temperature and allowing for long reaction times provides the product in almost quantitative yield following distillation. This procedure is a substantial improvement over that previously reported,26 leading to substantially greater yields of product without the need for chromatography. The same procedure yields N-methylcyclohexylbenzimidazole as a colorless solid in almost quantitative yield. Both heterocycles have been spectroscopically characterized.
Scheme 1.

1-Aryl-1,3,4-triazoles were prepared by a similar procedure to that reported for the synthesis of 1-phenyl-1,3,4-triazole 3.23 This synthetic route was successfully extended to the synthesis of p-tBuPhTz 4 and MesTz 5 by reacting the dihydrochloride of N,N-dimethylformamide azine with para-tert-butylaniline and 2,4,6-trimethylaniline, respectively (Scheme 2).23 Increasing the bulk of the aryl group requires longer reaction times and higher temperatures to proceed to completion, e.g. for Ar = Ph, 1 d in refluxing benzene, while for Ar = Mes, 3 d heating in refluxing chlorobenzene. The colorless solids were purified by crystallization and characterized by 1H NMR spectroscopy. Both PhTz 323 and MesTz 528 have been previously reported, while p-tBuPhTz 4 is a new compound.
Scheme 2.

Synthesis of ligand precursors
Tris(carbene)borate ligand precursors were prepared by the same synthetic routes used for the synthesis of other tris(imidazol-2-ylidene)borate ligands.6,13 Thus, for example, tris(benzimidazolium)hydroborane dications HB(RBzH)3Br2 (R = Me 10, CyCH2 11) were prepared by reaction of the appropriate benzimidazole with Me3N:BHBr2 in hot chlorobenzene, as with the synthesis of tris(imidazolium)hydroborane dications (Scheme 3).13,33 As with the tris(triazolium)borane dications, the spectral data of these compounds is consistent with their proposed formulation. In the case of HB(MeBzH)3Br2 10, the resonance assigned to the acidic protons are observed at 10.82 ppm. Four resonances assigned to protons from the benzimidazolium group at 7.75, 7.70, 7.59 and 7.52 ppm, respectively, with the singlet at 4.27 ppm assigned to the methyl substituent.
Scheme 3.

Synthesis of HB(RBz)3NiNO complexes 15 and 16. Reagents and conditions: (i) 0.33 HBBr2:NMe3, C6H5Cl, reflux; (ii) 1. Ag2O, MeCN or CH2Cl2, Δ; 2. Ni(NO)I(THF)2 or Ni(NO)Br(PPh3)2, THF.
Tris(triazolium)hydroborane dications were prepared in high yield by heating a toluene solution containing three equivalents of the substituted triazole with Me3N:BHBr2 (Scheme 4).33 We note that a bis(1,3,4-triazole)hydroborane cation has been previously reported, although a different synthetic route was used.34 The resulting tris(triazolium)hydroborane dications 12 and 13 were characterized by 1H NMR spectroscopy, ESI-MS and microanalysis. The triazolium salts are colorless air-stable compounds that are less soluble in non-polar solvents than the related tris(imidazolium)hydroborane dications.13 Their solubility is influenced by the nature of the triazole substituent, with the relative solubility decreasing in the order p-tBuPh > Mes > Ph.
Scheme 4.

Synthesis of HB(RTz)3NiNO complexes 17 and 18. Reagents and conditions: (i) 0.33 HBBr2:NMe3, C6H5CH3 or C6H5Cl, Δ; (ii) 1. Ag2O, MeCN, Δ; 2. KBr, THF; 3. Ni(NO)I(THF)2, THF.
The spectral data for these compounds are consistent with their proposed formulation as tris(triazolium)hydroborane dications. Thus, for example, six resonances are observed in the 1H NMR spectrum of HB(MesTz)3Br2 13. The most notable feature of the spectrum is the downfield singlet at 11.24 ppm, which is assigned to the three acidic triazole protons. As expected for the 2+ charge, all the other resonances arising from the substitued triazole moeity are shifted downfield from the corresponding triazole. Thus, the remaining triazole ring protons resonate as a singlet at 8.55 ppm, while resonances assigned to the mesityl substituent are observed at 7.01 (m-H), 2.33 (p-Me) and 2.19 ppm (o-Me). A broad resonance attributed to the B-H group is observed at 5.28 ppm. Similar features are observed in the 1H NMR spectrum of HB(p-tBuPhTzH)3Br2 12.
We have also prepared a new tris(imidazolium)phenylborane dication starting from 1-methylcyclohexylimidazole, namely PhB(CyCH2ImH)3OTf2 6. This salt was prepared by the same method used for the synthesis of other tris(imidazolium)phenylborane triflate salts and has similar spectral properties.11,13 Colorless crystals of this salt were grown by diffusion of pentane into a CH2Cl2 solution at −25 °C. The solid state structure reveals the expected four-coordinate boron center that is bound to a phenyl and three imidazolium groups (Fig. 3). Two triflate anions are also observed in the solid state structure.
Fig. 3.

X-ray crystal structure of PhB(CyCH2ImH)3OTf2 6. Thermal ellipsoids shown at 50 % probability, hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): C(11)-N(11) 1.337(3); C(11)-N(21) 1.322(3); C(12)-N(12) 1.332(3); C(12)-N(22) 1.319(3); C(13)-N(13) 1.331(3); C(13)-N(23) 1.328(3; N(11)-C(11)-N(21) 109.8(2); N(12)-C(12)-N(22) 110.1(2); N(13)-C(13)-N(23) 110.2(2).
N-heterocyclic carbene formation
The new tris(carbene)borate ligands were generated by threefold deprotonation of the borane dications to yield the corresponding tris(carbene)borate ligands (Schemes 3 and 4). Threefold deprotonation of the borane dications to generate the corresponding tris(carbene)borate ligands was found to be very sensitive to the nature of the base. While the tris(methylcyclohexylimidazol-2-ylidene)phenylborate ligand was readily formed by deprotonation of the corresponding imidazole borane dication with LDA, as previously reported,13 similar attempts to generate the tris(benzimidazol-2-ylidene)hydroborate and tris(1,3,4-triazol-2-ylidene)hydroborate ligands were unsuccessful. A library of bases was canvassed, including LDA, NaH, MeMgBr, all of which provided intractable mixtures of products, as determined by 1H NMR spectroscopic analysis of the crude reaction mixtures.
The tris(benzimidazolium)hydroborane and tris(triazolium)hydroborane dications could be deprotonated by reaction with Ag2O in hot MeCN or CH2Cl2. While the insolubility of most of these materials prevented further characterization, subsequent reaction with {NiNO}10 synthons confirmed that tris(benzimidazol-2-ylidene)hydroborate synthons were formed (see below).
Unfortunately, the ligand synthons formed from the tris(triazolium)hydroborane dications and Ag2O did not cleanly transfer to nickel. These compounds were instead treated with KBr to species tentatively formulated as potassium tris(1,3,4-triazol-2-ylidene)hydroborates. Due to their greater solubility, deprotonation of the carbene precursors could be confirmed by 1H NMR spectroscopy. Thus, for example, four resonances are observed the 1H NMR spectrum of product prepared from [HB(p-tBuTz)3](Br)2: a singlet at δ 7.88 ppm, assigned to the remaining triazolylidene proton, doublets at δ 7.00 and 6.50 ppm, assigned to the protons of the p-tert-butylphenyl ring and a singlet at δ 1.11 ppm, assigned to the tert-butyl group. The resonance for the B-H group is not observed, presumably due to quadropolar broadening.
Synthesis and characterization of nickel nitrosyl complexes
The ligand synthons were not isolated, but instead were used in a one-pot reaction with the {NiNO}10 synthons Ni(NO)Br(PPh3)224 or Ni(NO)I(THF)2,25 leading to formation of new tris(carbene)borate {NiNO}10 complexes (Schemes 3 and 4). The nickel nitrosyl complexes were found to be diamagnetic, air stable complexes, characterized by a number of techniques including 1H and 13C{1H} NMR, UV-vis and IR spectroscopy, single crystal X-ray diffraction, CV, microanalysis and ESI-MS. When prepared using Ni(NO)Br(PPh3)2, the complexes were often contaminated with PPh3, PPh3O and PPh3AgBr byproducts. For some complexes, these byproducts could be removed by chromatography on a Florisil column in an air-free environment. However, other complexes, e.g. PhB(CyCH2Im)3NiNO 14, were found to decompose on the column, even when other solid supports were used. The use of Ni(NO)I(THF)225 as the {NiNO}10 synthon was not universally successful, and in some cases the use of a substochiometric amount of the reagent was necessary to avoid the formation of side products, e.g. HB(CyCH2Bz)3NiNO 16.
The complexes, PhB(CyCH2Im)3NiNO 14, HB(MeBz)3NiNO 15, HB(CyCH2Bz)3NiNO 16, HB(p-tBuPhTz)NiNO 17 and HB(MesTz)NiNO 18 were also crystallographically characterized.35 The molecular structures of these complexes (Fig. 4 and SI) reveal the expected four-coordinate metal center bound to the tripodal tris(carbene)borate ligand and terminal nitrosyl ligand. The complexes all have similar metrical parameters around the nickel center, regardless of the nature of the tripodal ligand. Thus, for example, the Ni–NO bond lengths in these complexes are in the narrow range 1.640(2)–1.646(2) Å, which compares well with the Ni–N bond length of 1.620(5) Å observed for HB(tBuIm)3NiNO.14 Similarly, the Ni–C bonds show little variation between the different tripodal ligands. There is greater variability in the Ni–N–O bond angle, which varies from 169.3(2) – 176.3(3), compared with 178.5(4) ° for HB(tBuIm)3NiNO.14 However, it is likely these differences are due to packing forces in the solid state, as evidenced by the variation in the Ni–N–O bond angle (169.3(2)–174.8(2) °) for the four independent molecules of HB(MeBz)3NiNO 15 in the unit cell. There do not appear to be any trends in metrical parameters that are ligand-dependent, and thus X-ray crystallography is unable to differentiate the donor abilities of the different tris(carbene)borate ligands, e.g. from N-O or Ni-C bond lengths.
Fig. 4.

X-ray crystal structures of representative nickel nitrosyl complexes. Thermal ellipsoids shown at 50% probability, most hydrogen atoms omitted for clarity. A: PhB(CyCH2Im)3NiNO 14. One of three independent molecules in the asymmetric unit. Selected bond lengths (Å) and angles (°): Ni(1) – N(7) 1.643(10); Ni(1) – C(1) 1.982(11); Ni(1) – C(11) 1.939(11); Ni(1) – C(21) 1.974(13); N(7) – Ni(1) – C(1) 122.8(5); N(7) – Ni(1) – C(11) 123.2(5); N(7) – Ni(1) – C(21) 130.3(5); Ni(1) – N(7) – O(1) 173.7(11). B: HB(MeBz)3NiNO 15. One of four independent molecules in the asymmetric unit. Selected bond lengths (Å) and angles (°): Ni(1)-N(7) 1.646(2); Ni(1)-C(15) 1.945(3); Ni(1)-C(1) 1.959(3); Ni(1)-C(8) 1.961(3); N(7)-Ni(1)-C(15) 118.85(11); N(7)-Ni(1)-C(1) 132.30(11); N(7)-Ni(1)-C(8) 123.90(11); O(1)-N(7)-Ni(1) 169.3(2). C: HB(p-tBuPhTz)3NiNO 17. Selected bond lengths (Å) and angles (°): Ni(1)-N(10) 1.640(2); Ni(1)-C(1) 1.979(3); Ni(1)-C(13) 1.989(3); Ni(1)-C(25) 1.985(3); N(10)-Ni(1)-C(1) 125.68(12); N(10)-Ni(1)-C(13) 122.51(12); N(10)-Ni(1)-C(25) 126.90(14); O(1)-N(10)-Ni(1) 176.3(3).
Infrared spectroscopy was used to evaluate the relative donor ability of the tris(carbene)borate ligands in solution, specifically from νNO (Table 1). Based on the bonding descriptions for structurally related Tp*NiNO, the impact of the tripodal ligand donor strength on νNO can be formulated in two ways. In the traditional formulation,20a the magnitude of νNO is related to the extent of π-backbonding from the Ni(0) center to the π* orbitals of coordinated NO+.36 Thus, increasing the electron-releasing ability of the tripodal ligand leads to a reduction in νNO. In the revised formulation, the electronic structure is described as having multireference character with NiI(S = 1/2)–NO0(S = 1/2), NiII(S = 0)–NO1− (S = 0) and NiII(S = 1)–NO1− (S = 1) configurations.21 In this description, the magnitude of νNO can be related to the relative stabilization of these different electronic configurations. Specifically, increasing the donor strength of the tripodal ligand will stabilize the Ni(II) configurations at the expense of the Ni(I) configuration. Since the Ni(II) configurations have populated N-O π* orbitals, this is expected to reduce the overall N-O bond order. Thus, both electronic structure descriptions predict that increasing the donor strength of the tripodal ligand will result in a lowering of νNO.
Table 1.
Infrared spectroscopic data for four-coordinate tris(carbene)borate {NiNO}10 complexes measured in THF.
The IR data reveal that, in general, the donor strength of these tris(carbene)borate ligands decreases in the order: imidazol-2-ylidene > benzimidazol-2-ylidene > 1,3,4-triazol-2-ylidene, which correlates well with the trend observed for monodentate NHC ligands.19a,c, 35 It has been previously shown in at least one instance that the nature of the fourth group on the boron atom (i.e. H− vs Ph−) has little impact on the donor strength of the tris(carbene)borate ligand.14 It is also notable that tris(carbene)borates provide an exceptional degree of electronic tunability, with νNO varying over a range of more than 50 cm−1. This should be compared with the smaller range of 25 cm−1 observed for Rh(CO)2(NHC)I complexes, where the monodentate N-heterocyclic carbene varied from relatively weakly donating 1,3-dimethylperimidin-2-ylidene to very strongly donating 1,2,3-trimethyl-4-isopropylpyrazolin-5-ylidene.19
Interestingly, and in contrast to monodentate NHC ligands, changing the ligand substituent influences the donor strength of tris(carbene)borate ligands.19 Thus, for example, alkyl substituted ligands have a greater donor ability than aryl, as reflected in the data for the imidazol-2-ylidene series. In fact, replacing alkyl substituents in the tris(imidazol-2-ylidene)borates by an aryl group attenuates donor strength sufficiently to make them similar to the alkyl-substituted tris(benzimidazol-2-ylidene)borate. It is likely that the three NHC donors in tris(carbene)borate ligands magnify these effects as compared to the monodentate analogues, allowing for fine-tuning of the tripodal ligand donor strength. Interestingly, for the imidazol-2-ylidene based ligands, the tris(carbene)borate donor strength does not follow the trend that would be predicted from the inductive properties of the alkyl donors, with the tert-butyl substituted ligand being the least donating. For this ligand it is likely that steric crowding resulting from this very bulky ligand reduces orbital overlap between nickel and the tris(carbene)borate. Evidence for this hypothesis comes from the average Ni-C bond length, which is longer for PhB(tBuIm)3NiNO (2.003(1) Å)14 than for PhB(CyCH2Im)3NiNO 14 (1.969(4) Å).
Comparing the donor properties of these tris(carbene)borate ligands with related facially-coordinating ligands is instructive (Table 2). Thus, although the tris(1,3,4-triazol-2-ylidene)borates are the weakest of the tris(carbene)borate donors, they are still more strongly donating than either Cp* and Tp*. In fact, the tris(1,3,4-triazol-2-ylidene)borates bridge a gap in donor strength between these classical ligands and the stronger tris(phosphino)borate39 and tris(thioimidazolyl)borate (TmR−) donors,36 while the tris(benzimidazol-2-ylidene)borates bridge a similar gap between these two ligands and the alkyl-substituted tris(imidazol-2-ylidene)borates. An important difference with these other ligand classes is that the ligand topology is consistent for all three types of tris(carbene)borate. Thus, the rigid tris(carbene)borates span a greater range of donor strengths than either the tris(phosphino)borate or tris(thioimidazolyl)borate ligands6 while maintaining a consistent topology.
Table 2.
Solution IR spectroscopic data for selected four-coordinate {NiNO}10 complexes.
Attempts to use other physical measurements to delineate the donor properties of the tris(carbene)borate ligands were not successful, e.g. 13C{1H} NMR, UV-vis, CV. Thus, for example, while the resonances attributed to the carbene carbon of the tris(carbene)borate ligand could be readily identified in the 13C{1H} NMR spectra, the chemical shifts are similar for all complexes and there is no systematic order related to the donor strength of the ligand.35 This is similar to observations reported for a large series of monodentate carbene ligands in Rh(CO)2(NHC)I complexes.19a
The range of substituents has also allowed us to explore the steric properties of the tris(carbene)borate ligands using solid angles. The solid angle can be visualized as follows: a light source is placed at the metal center that is circumscribed by a sphere of radius r. Since the ligand blocks the light source, it will cast a shadow of area A on the inside of the sphere, allowing the solid angle to be calculated in steradians as Ω = A/r2. A convenient way of expressing the solid angle is as the percentage of the sphere’s surface that is covered by the ligand shadow, G = 100Ω/4π (4π is the solid angle of a sphere in steradians).30
The percent coverage of a sphere (G) for the alkyl-substituted tris(carbene)borate ligands were calculated from the coordinates determined by X-ray crystallography (Table 3).35 As expected, these calculations reveal that the methyl-subtituted tris(carbene)borate ligand is the least encompassing ligand, but somewhat surprisingly, the tert-butyl and methylcyclohexyl substituted ligands appear to exert a similar steric effect. However, when G was determined with the ligand artificially truncated at a radius 4 Å from the metal center, the methylcyclohexyl and methyl substituents were found to be similar in size. Similar observations have been made for some monodentate NHC ligands.40 The truncated value of G, which approximates the steric profile of the ligand as experienced in the first coordination sphere of nickel, suggests that the methylcyclohexyl substituent provides an open environment in the immediate vicinity of the metal center, while the large value of G for the full ligand suggests that this substituent may also be sufficiently bulky to stabilize low coordination numbers.
Table 3.
Solid angle data (G)a for selected alkyl-substituted tris(carbene)borate ligands in {NiNO}10 complexes.
| Ligand | Full ligand | Truncated ligandb |
|---|---|---|
| HB(MeBz)3−c | 36.9 | 37.0 |
| HB(CyCH2Bz)3− | 56.3 | 36.9 |
| PhB(CyCH2Im)3−c | 52.8 | 39.0 |
| HB(tBuIm)3− | 53.2 | 53.2 |
| Tp* | 37.0 | 37.0 |
| PhB(CH2PPh2)3− | 55.4 | 54.1 |
Solid angles expressed as percentage coverage of a sphere. The G values were calculated using a metal ligand distance of 2.28 Å.
Ligand was truncated at a radius of 4 Å from the metal center.
Average values for the four independent molecules in the asymmetric unit.
For comparative purposes, G was also calculated for two other tripodal ligands using the X-ray crystal structure data of Tp*NiNO20b and PhB(CH2PPh2)3NiNO.39 The data suggest, not surprisingly, that the steric impact of tris(carbene)borate and tris(pyrazolyl)borate ligands are similar, as shown by the similar sizes of the methyl-substituted ligands HB(MeBz)3− and Tp*. The solid angle data also reveal that the very bulky HB(tBuIm)3− ligand is similar in size to the tris(phosphino)borate ligand PhB(CH2PPh2)3−.41 Interestingly, the solid angle of the tris(phosphino)borate ligand is dependent on the coordination number, and decreases to 45.96 % of a sphere (truncated at 4 Å) for the six-coordinate complex PhB(CH2PPh2)3Fe(κ2-OAc)(N2H4),42 highlighting the flexibility of this ligand.
Conclusions
The synthesis and donor properties of new tris(carbene)borate ligands incorporating a range of benzimidazol-2-ylidene and 1,3,4-triazol-2-ylidene donors has been reported. Changing the nature of the carbene donor results in predictable changes to the donor properties of these ligands, with the relative donor strength following the order imidazol-2-ylidene > benzimidazol-2-ylidene > 1,3,4-triazol-2-ylidene, as determined by IR spectroscopy of four-coordinate {NiNO}10 complexes. Moreover, in contrast to monodentate N-heterocyclic carbenes, the nature of the carbene N-substituent also has an impact on the donor strength of the ligand, with alkyl substituted ligands being stronger donors than aryl-substituted. A caveat to this observation is that very bulky substituents appear to hinder effective orbital overlap, thereby reducing the effective donor strength. Similarly to tris(pyrazolyl)borates, the consistent topology of the tris(carbene)borate ligand class allows for predictable changes to their steric properties. An interesting finding in this regard is that certain N-substituents, e.g. methylcyclohexyl, may provide an open environment close to the metal center, while still providing a sufficiently bulky profile to stabilize low coordination numbers. The combination of exceptional electronic tunability, and the consistent topology of the tris(carbene)borate ligand class is likely to allow for predictable variation in the properties of their transition metal complexes.
Supplementary Material
Acknowledgments
Funding from the DOE-BES (DE-FG02-08ER15996) and the Dreyfus Foundation is gratefully acknowledged. SBM thanks NSF-AMP (HRD 0929343) and NIH-RISE (R25 GM061222-11) for financial support. The Bruker X8 X-ray diffractometer was purchased via an NSF CRIF:MU award to The University of New Mexico, CHE-0443580. We thank Eileen Duesler (UNM) and Ranko P. Bontchev (Cabot Corporation) for assistance with X-ray crystallography.
Footnotes
Supporting Information Available. Further details on the solid angle calculations, X-ray crystallographic data for all structurally characterized compounds, including crystallographic information files (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selected reviews: Mata JA, Poyatos M, Peris E. Coord Chem Rev. 2007;251:84.Hu XL, Meyer K. J Organomet Chem. 2005;690:5474.
- 2.(a) Hahn FE, Langenhahn V, Luegger T, Pape T, Le Van D. Angew Chem, Int Ed. 2005;44:3759. doi: 10.1002/anie.200462690. [DOI] [PubMed] [Google Scholar]; (b) McKie R, Murphy JA, Park SR, Spicer MD, Zhou SZ. Angew Chem, Int Ed. 2007;46:6525. doi: 10.1002/anie.200702138. [DOI] [PubMed] [Google Scholar]; (c) Park SR, Findlay NJ, Garnier J, Zhou S, Spicer MD, Murphy JA. Tetrahedron. 2009;65:10756. [Google Scholar]; (d) Bass HM, Cramer SA, Price JL, Jenkins DM. Organometallics. 2010;29:3235. [Google Scholar]
- 3.Poyatos M, Mata JA, Peris E. Chem Rev. 2009;109:3677. doi: 10.1021/cr800501s. [DOI] [PubMed] [Google Scholar]
- 4.Biffis A, Lobbia GC, Papini G, Pellei M, Santini C, Scattolin E, Tubaro C. J Organomet Chem. 2008;693:3760. [Google Scholar]
- 5.Fränkel R, Birg C, Kernbach U, Habereder T, Noth H, Fehlhammer WP. Angew Chem Int Ed. 2001;40:1907. doi: 10.1002/1521-3773(20010518)40:10<1907::aid-anie1907>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 6.Smith JM. Comments Inorg Chem. 2008;29:289. [Google Scholar]
- 7.(a) Hu X, Tang Y, Gantzel P, Meyer K. Organometallics. 2003;22:612. [Google Scholar]; (b) Hu X, Castro-Rodriguez I, Meyer K. Organometallics. 2003;22:3016. [Google Scholar]; (c) Hu X, Castro-Rodriguez I, Olsen K, Meyer K. Organometallics. 2004;23:755. [Google Scholar]
- 8.(a) Rasika Dias HV, Jin W. Tetrahedron. 1994;35:1365. [Google Scholar]; (b) Nakai H, Tang Y, Gantzel P, Meyer K. Chem Commun. 2002:24. doi: 10.1039/b209071f. [DOI] [PubMed] [Google Scholar]; (c) Ellul CE, Reed G, Mahon MF, Pascu SI, Whittlesey MK. Organometallics. 2010;29:4097. [Google Scholar]
- 9.Chen F, Wang GF, Li YZ, Chen XY, Xue ZL. Inorg Chem Commun. 2012;21:88. [Google Scholar]
- 10.Fränkel R, Kernbach U, Bakola-Christianopoulou M, Plaia U, Suter M, Ponikwar W, Nöth H, Moinet C, Fehlhammer WP. J Organomet Chem. 2001;617–618:530. [Google Scholar]
- 11.Forshaw AP, Bontchev RP, Smith JM. Inorg Chem. 2007;46:3792. doi: 10.1021/ic070187w. [DOI] [PubMed] [Google Scholar]
- 12.(a) Tubaro C, Biffis A, Scattolin E, Basato M. Tetrahedron. 2008;64:4187. [Google Scholar]; (b) Biffis A, Tubaro C, Scattolin E, Basato M, Papini G, Santini C, Alvarez E, Conejero S. Dalton Trans. 2009:7223. doi: 10.1039/b906730b. [DOI] [PubMed] [Google Scholar]
- 13.(a) Nieto I, Cervantes-Lee F, Smith JM. Chem Commun. 2005:3811. doi: 10.1039/b505985b. [DOI] [PubMed] [Google Scholar]; (b) Cowley RE, Bontchev RP, Duesler EN, Smith JM. Inorg Chem. 2006;45:9771. doi: 10.1021/ic061299a. [DOI] [PubMed] [Google Scholar]
- 14.Nieto I, Bontchev RP, Ozarowski A, Smirnov D, Krzystek J, Telser J, Smith JM. Inorg Chim Acta. 2009;362:4449. [Google Scholar]
- 15.(a) Nieto I, Ding F, Bontchev RP, Wang H, Smith JM. J Am Chem Soc. 2008;130:2716. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]; (b) Scepaniak JJ, Fulton MD, Bontchev RP, Duesler EN, Kirk ML, Smith JM. J Am Chem Soc. 2008;130:10515. doi: 10.1021/ja8027372. [DOI] [PubMed] [Google Scholar]; (c) Scepaniak JJ, Young JA, Bontchev RP, Smith JM. Angew Chem Int Ed. 2009;48:3158. doi: 10.1002/anie.200900381. [DOI] [PubMed] [Google Scholar]
- 16.Scepaniak JJ, Vogel CS, Khusniyarov MM, Heinemann FW, Meyer K, Smith JM. Science. 2011;331:1049. doi: 10.1126/science.1198315. [DOI] [PubMed] [Google Scholar]
- 17.See, for example, Dias HVR, Lovely CJ. Chem Rev. 2008;108:3223. doi: 10.1021/cr078362d. and references cited therein.
- 18.(a) O’Brien CJ, Kantchev EAB, Chass GA, Hadei N, Hopkinson AC, Organ MG, Setiadi DH, Tang TH, Fang DC. Tetrahedron. 2005;61:9723. [Google Scholar]; (b) Urban S, Tursky M, Fröhlich R, Glorius F. Dalton Trans. 2009:6934. doi: 10.1039/b908981k. [DOI] [PubMed] [Google Scholar]; (c) Khramov DM, Lynch VM, Bielawski CW. Organometallics. 2007;26:6042. [Google Scholar]; (d) Bittermann A, Härter P, Herdtweck E, Hoffmann SD, Herrmann WA. J Organomet Chem. 2008;693:2079. [Google Scholar]; (e) Dröge T, Glorius F. Angew Chem Int Ed. 2010;47:6940. doi: 10.1002/anie.201001865. [DOI] [PubMed] [Google Scholar]
- 19.(a) Herrmann WA, Schütz J, Frey GD, Herdtweck E. Organometallics. 2006;25:2437. [Google Scholar]; (b) Huynh HV, Han Y, Jothibasu R, Yang JA. Organometallics. 2009;28:5395–5404. [Google Scholar]; (c) Gusev DG. Organometallics. 2009;28:6458. [Google Scholar]; (d) Schuster O, Yang L, Raubenheimer HG, Albrecht M. Chem Rev. 2009;109:3445. doi: 10.1021/cr8005087. [DOI] [PubMed] [Google Scholar]
- 20.(a) Harding DJ, Harding P, Adams H, Tuntulani T. Inorg Chim Acta. 2007;360:3335. [Google Scholar]; (b) Landry VK, Pang K, Quan SM, Parkin G. Dalton Trans. 2007:820. doi: 10.1039/b616674a. [DOI] [PubMed] [Google Scholar]
- 21.Tomson NC, Crimmin MR, Petrenko T, Roseburgh LE, Sproules S, Boyd WC, Bergman RG, DeBeer S, Toste FD, Wieghardt K. J Am Chem Soc. 2011;133:18785. doi: 10.1021/ja206042k. [DOI] [PubMed] [Google Scholar]
- 22.(a) Watt GW, Baye LJ, Drummond FO., Jr J Am Chem Soc. 1966;88:1138. [Google Scholar]; (b) Brintzinger HH, Bercaw JE. J Am Chem Soc. 1970;92:6182. [Google Scholar]
- 23.Naik AD, Marchand-Brynaert J, Garcia Y. Synthesis. 2008:149. [Google Scholar]
- 24.Feltham RD. Inorg Chem. 1964;3:116. [Google Scholar]
- 25.(a) Haymore B, Feltham RD. Inorg Synth. 1973;14:81. [Google Scholar]; (b) Varonka MS, Warren TH. Organometallics. 2010;29:717. [Google Scholar]
- 26.Verras A, Kuntz ID, Ortiz de Montellano PR. J Med Chem. 2004;47:3572. doi: 10.1021/jm030608t. [DOI] [PubMed] [Google Scholar]
- 27.Meyer D, Strassner T. J Org Chem. 2011;76:305. doi: 10.1021/jo101784v. [DOI] [PubMed] [Google Scholar]
- 28.Holm SC, Siegle AF, Loos C, Rominger F, Straub BF. Synthesis. 2010:2278. [Google Scholar]
- 29.The use of stoichiometric Ni(NO)I(THF)2 reagent led to the formation of inseparable green impurities.
- 30.Guzei IA, Wendt M. Dalton Trans. 2006:3991. doi: 10.1039/b605102b. [DOI] [PubMed] [Google Scholar]
- 31.Guzei IA, Wendt M. Solid-G. University of Wisconsin; Madison, WI: 2004. [Google Scholar]
- 32.Farrugia LJ. J Appl Cryst. 1997;30:565. [Google Scholar]
- 33.While both tris(triazolium)phenylborane and tris(benzimidazolium)phenylborane dications could also be prepared, attempts to generate tris(carbene)borates from these precursors were unsuccessful.
- 34.Papini G, Bandoli G, Dolmella A, Gioia Lobbia G, Pellei M, Santini C. Inorg Chem Commun. 2008;11:1103. doi: 10.1021/ic050193x. [DOI] [PubMed] [Google Scholar]
- 35.See the Supporting Information for more details.
- 36.Maffett LS, Gunter KL, Kreisel KA, Yap GPA, Rabinovich D. Polyhedron. 2007;26:4758. [Google Scholar]
- 37.Green JC, Underwood C. J Organomet Chem. 1997;528:91. [Google Scholar]
- 38.Landry VK, Pang K, Quan SM, Parkin G. Dalton Trans. 2007:820. doi: 10.1039/b616674a. [DOI] [PubMed] [Google Scholar]
- 39.MacBeth CE, Thomas JC, Betley TA, Peters JC. Inorg Chem. 2004;43:4645. doi: 10.1021/ic049936p. [DOI] [PubMed] [Google Scholar]
- 40.Dible BR, Cowley RE, Holland PL. Organometallics. 2011;30:5123. [Google Scholar]
- 41.Normalization of the solid angles to a metal-ligand distance of 2.28 Å makes no difference to the relative sizes of these two ligands. By the non-normalized data, the sizes of HB(tBuIm)3− (71.4 % coverage) and PhB(CH2PPh2)3− (71.5% coverage) are the same.
- 42.Saouma CT, Lu CC, Peters JC. Inorg Chem. 2012;51:10043. doi: 10.1021/ic301704f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.