

Abstract
Admixture is an important evolutionary force that can and should be used in efforts to apply genomic data and technology to the study of complex disease genetics. Admixture linkage disequilibrium (ALD) is created by the process of admixture and, in recently admixed populations, extends for substantial distances (of the order of 10 to 20 cM). The amount of ALD generated depends on the level of admixture, ancestry information content of markers and the admixture dynamics of the population, and thus influences admixture mapping (AM). The authors discuss different models of admixture and how these can have an impact on the success of AM studies. Selection of markers is important, since markers informative for parental population ancestry are required and these are uncommon. Rarely does the process of admixture result in a population that is uniform for individual admixture levels, but instead there is substantial population stratification. This stratification can be understood as variation in individual admixtures and can be both a source of statistical power for ancestry-phenotype correlation studies as well as a confounder in causing false-positives in gene association studies. Methods to detect and control for stratification in case/control and AM studies are reviewed, along with recent studies showing individual ancestry-phenotype correlations. Using skin pigmentation as a model phenotype, implications of AM in complex disease gene mapping studies are discussed. Finally, the article discusses some limitations of this approach that should be considered when designing an effective AM study.
Keywords: complex diseases, admixture linkage disequilibrium (ALD), admixture mapping (AM), biogeographical ancestry (BGA), structure, phenotype-ancestry correlation
Introduction
Genetic analysis of phenotypes and diseases has traditionally followed two approaches: family-based linkage analysis and population-based association studies. While in linkage analysis it is the co-segregation of alleles in families that is measured, population-based studies use non-random associations between phenotypes and alleles in populations to identify causative genes. Linkage analysis has proven to be immensely successful as a means of identifying genes for a number of single gene diseases with simple Mendelian inheritance (eg see OMIM database). Complex diseases are multifactorial, polygenic and often characterised by late age of onset, incomplete penetrance, locus heterogeneity and environmental exposures and, despite significant efforts, have not been amenable to family-based mapping.
Linkage disequilibrium (LD) is an important aspect of genetic association studies and is generated in a population through mutation, selection, drift, non-random mating and admixture [1]. Allelic associations due to LD are significant and are correlated with physical distance within small genomic regions but decay over time due to recombination [2-4]. LD-based association studies have been successful in both fine scale mapping [5,6] and initial disease gene mapping in homogeneous populations that have undergone recent bottlenecks (eg Hirschsprung disease in Mennonites [7], Bardet- Beidle syndrome in Bedouins [8]). Allelic associations can result either from direct functional effects of the alleles tested or indirectly through non-random associations between the allele measured and nearby functional alleles. Since functional alleles in most genes are still unknown and are indeed an object of the research, LD is an important feature of how genes can be screened for alleles that alter disease risk. Thus, there has been substantial focus on the extent of LD across the genome and the definition of statistical methods for disease gene mapping using LD [9-11]. In large cosmopolitan populations, however, LD may be difficult to detect when the mutation is old, since the amount of remaining LD may be small. Additionally, false-positive associations due to population stratification are important confounders in LD-based association studies.
Admixture studies and their use in disease gene mapping
Intermixture between previously isolated populations leads to the creation of admixed populations. The process of admixture itself creates LD between all loci, linked and unlinked, that have different allele frequencies in the parental populations. The magnitude of admixture linkage disequilibrium (ALD) in an admixed population depends on the allele frequency differential between the parental populations, the level of admixture, the admixture dynamics, the time since admixture and the recombination rate between the loci [12]. While ALD between unlinked markers decays rapidly (within two to four generations), ALD between linked markers decays more slowly. The exponential decrease in ALD with genetic distance facilitates the differentiation of ALD that is high between markers that are close together and genetically linked, from ALD generated at unlinked loci. Thus, if the parental populations differ in a trait or disease due to different frequencies of risk alleles, it should be possible to identify the loci containing these alleles using admixture mapping (AM) [12-14].
Many US residents can trace their genetic ancestry to more than one continent. The European colonial period that started in the late 1400s brought together in the New World populations that had been geographically isolated, namely, Europeans, West Africans and Native Americans. Given the recent and common origin of all human populations, this admixture had only a small average effect on the gene pools of these new populations. In other words, for most genomic regions, the pre-colonial (or parental) populations had similar allele frequencies and, at these, admixture was of little consequence. At some other loci, however, there had been some change in allele frequency in the time since the separation of parental populations and it is at these loci where admixture has had an important effect. Since populations like African Americans, African Caribbeans and Mexican Americans were formed in the recent past, allelic associations in these populations that were created by admixture extend over large distances. Admixed populations represent a useful resource for mapping complex-disease genes by using this long-range ALD [12], which requires fewer markers to screen the genome than other populations or approaches. Understanding the genetic consequences of admixture is important because it can be both a confounding factor and a source of statistical power in gene identification studies.
Two models of admixture dynamics have been described to represent the extremes of the process by which an admixed population is formed: the continuous gene flow (CGF) model and the hybrid isolation (HI) model [15,16]. In the HI model, admixture occurs immediately in a single generation without further contribution from either parental population, hence, ALD is generated in a single generation and gradually decays in successive generations through independent assortment and recombination between loci. Few false-positive results are thus expected in an association study under the HI model. Alternatively, the CGF model represents a situation where admixture occurs at a steady rate in each generation, with contributions from one (or all) of the parental populations into the admixed population. ALD under the CGF model increases in each generation, since new admixture is constantly occurring. A point will be reached, however (when the admixture proportion = 0.5), where continued admixture will actually decrease the ALD, since added gene flow will result in the conversion of the admixed population into the introgressing parental population. Figure 1 shows the amount of ALD expected under these two models for linked and unlinked loci. For both models, association between markers is inversely correlated with the genetic distance between them. Simulation studies have shown that populations that have a demographic history more consistent with the CGF model of admixture retain ALD over larger chromosomal regions and show significant associations between unlinked marker loci [15]. While associations between unlinked markers could potentially lead to false-positives, conditioning upon parental admixture allows the distinction between associations arising due to true linkage and those due to CGF stratification to be made, thereby providing greater power for detecting ALD over larger chromosomal distances [15].
Figure 1.
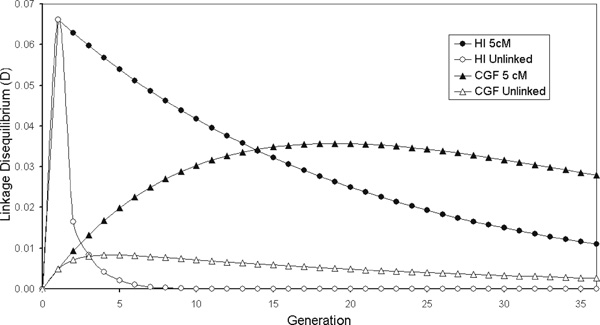
The amount of admixture linkage disequilibrium (ALD) expected under the continuous gene flow (CGF) and hybrid isolation (HI) models of admixture for unlinked loci and loci linked at 5 cM. The results shown are for two loci with δ = 0.54 and 0.49, and with 50 per cent admixture in the first generation for the HI model and 1.9 per cent admixture for 36 generations under the CGF model (equivalent to 50 per cent total). ALD under the HI model decreases for both linked and unlinked loci, whereas ALD under the CGF model for both linked and unlinked loci increases initially and then decreases (adapted from Pfaff et al., 2001 [15])
There are several ways in which admixture can be an important resource in the elucidation of genetic factors that contribute to the risk of common disease. Common diseases often have environmental components to their risk, and the clinical phenotype results from currently unknown interactions between environmental factors and underlying genotypes. Decomposing the sources of variation is thus important in order accurately to understand the aetiology of the trait. It is possible to distinguish between the genetic and environmental explanations for ethnic differences in disease risk (and investigating the mode of inheritance), by studying the relationship of disease risk to individual admixture [14,17-19]. For example, recent studies have demonstrated a strong relationship between proportional West African ancestry and the risk of systemic lupus erythematosus in admixed populations in Trinidad [18]. Several common diseases (eg hypertension, diabetes, obesity, prostate cancer and osteoporosis) have differences in risk among population groups (see Table 1). In situations where these differences have a genetic basis, genes underlying these differences can be identified by testing for locus ancestry by conditioning on parental admixture. As detailed by Shriver et al., this approach has a greater statistical power than family linkage studies for mapping polygenic traits [14]. Estimates of biogeographical ancestry (BGA), the proportional ancestry levels of an individual, can be used in conjunction with measured environmental effects for investigating the roles of environmental and inherited risks underlying complex traits [18-20]. It is important to recognise that associations between individual admixture and disease risk might reflect correlations between BGA and socio-cultural variables and exposures. For example, hypothetically, if BGA and years of education were to be correlated, hypertension might be correlated with BGA, even though the causal risk factor was years of education or vice versa.
Table 1.
Diseases with possible genetic components based on ethnic differences in disease rates and hence amenable to admixture mapping
| Disease | High-risk groups | Low-risk groups | Relative risk ratio | Reference(s) |
|---|---|---|---|---|
| Obesity | African women | Europeans | 2:4 | [64,65] |
| Native Americans | ||||
| South Asians (central adiposity,) | ||||
| Pacific Islanders, Aboriginal Australians | ||||
| Non-insulin dependent diabetes (NIDDM) | South Asians, West Africans, Peninsular Arabs, Pacific Islanders and Native Americans | Europeans | 4:7 | [66,67] |
| Hypertension | African Americans, West Africans | Europeans | 2:3 | [68,69] |
| Coronary heart disease | South Asians | West African men | 2:4 | [70,71] |
| End-stage renal disease | Native Americans and African populations | Europeans | N/A | [72] |
| Dementia | Europeans | African Americans, Hispanic Americans | N/A | [73] |
| Autoimmune diseases: | ||||
| Systemic lupus | West Africans | Europeans | N/A | [55] |
| erythematosus | Native Americans | Europeans | ||
| Skin cancer | Europeans | N/A | [74] | |
| Lung cancer | Africans | European Americans, Chinese, Japanese | [75,76] | |
| Prostate cancer | Africans and African Americans | [77] | ||
| Multiple sclerosis | Europeans | Chinese, Japanese, African Americans, Turkmens, Uzbeks, Native Siberians, New Zealand Maoris | N/A | [78] |
| Osteoporosis | European Americans | African Americans | N/A | [79] |
N/A = not available.
Marker choice for admixture mapping
Admixture-based methods rely on using suitable markers and estimates of allele frequencies from appropriately identified parental populations. Since ALD is fairly new and extends over larger distances, fewer markers are required for AM studies. Markers informative for ancestry have been used in several contexts and have been referred to as 'ideal,' [21] 'private' [22] and 'unique' [23]. Informativeness of such markers can be measured as the allele frequency differential (δ), which is the absolute value of the difference of a particular allele between populations [12,24]. Microsatellites and insertion/deletion polymorphisms with δ > 0.3 were recently called 'ethnic-difference markers' (EDMs) [25] suitable for mapping by admixture linkage disequilibrium (MALD). Additionally, markers with high δ and very high log likelihood allelic ratio (LLAR) between populations have been designated 'population specific alleles' (PSAs) [26]. This report followed from earlier work where markers with large allele frequency difference were identified to be appropriate for admixture studies [27,28], and most (> 95 per cent) of the arbitrarily identified biallelic markers had δ < 50 per cent [24]. Thus, the authors proposed that ideal PSAs should have δ > 50 per cent and also indicated that for multiallelic loci, a composite δ could be estimated as one half the summation of the absolute value of allelic frequency differences for all alleles at that locus [26]. It has also been shown that markers with lower δ values, of approximately 30 per cent, can provide up to 80 per cent power for detecting associations at distances of 5 cM with a large enough sample size (N = 1,000) [15].
Pfaff et al. [15], suggested referring to markers suitable for admixture studies as 'ancestry informative markers' (AIMs), given that the central feature of these markers is the ancestry information content (f) [29]. The present authors agree that the term AIM more accurately describes these markers and does so using language that is less likely to be misunderstood and misinterpreted [14,17,28]. Marker information content 'f' denotes the locus-specific Fst and is a value representative of the differentiation between two populations at a single locus. This is equivalent to Wahlund's standardised variance for allele frequency. Simulation studies for estimating the information content of markers with varying levels of f have shown that for 1,000 markers with average information content for ancestry at 40 per cent between two ancestral subpopulations, approximately 80 per cent of the information about ancestry can be extracted from an initial genome screen [13,29]. After initial identification of regions showing admixture, more markers can be typed in these regions to increase extraction of information to nearly 100 per cent.
It is well established, however, that only 5-15 per cent of the total genetic variation results from differences among human populations [30-32]. Moreover, most alleles are shared between populations, and alleles common in one population are also common in other populations. Thus, most genetic markers are unaffected by admixture and it is imperative to choose markers that show high levels of d (and f) between the parental populations. Recent studies by several groups have focused on identifying panels of markers suitable for admixture studies. One notable study screened 744 microsatellite markers for composite d values and LLAR in four different populations and identified a genome spanning set of 315 markers (average spacing 10 cM, δ ≥ 0.3) for mapping in African Americans and 214 markers (average spacing of 16 cM, δ ≥ 0.25) for mapping in Hispanics [33]. A DNA pooling method was used to identify 151 AIMs (microsatellites and short insertion/deletion polymorphisms), with δ > 0.3 for mapping in Mexican American populations to distinguish between European-American and Native-American contributions [25]. Ninety-seven AIMs were identified for mapping in African-American populations [25] that show limited variation within Africa [34]. The authors' group has reported AIMs over the past few years [14,17,26,35,36]. Additional resources are available for obtaining marker frequency, and genotype and haplotype information, from The SNP Consortium (TSC; http://snp.cshl.org), the National Center for Biotechnology Information's 'dbSNP' website (http://www.ncbi.nlm.nih.gov/SNP), the Marshfield Database (http://research.marshfieldclinic.org/genetics/Default.htm) and the ongoing HapMap project.
Admixed populations and admixture proportions
Since the amount of ALD created is proportional to the level of admixture in a population, it is important briefly to review studies on admixture levels across populations. Those populations that are likely to be useful for admixture studies include African Americans, Mexican Americans, Cubans and Puerto Ricans in the USA, African Caribbeans, various Latin American populations, various groups in Central and South America and the Caribbean islands, Anglo Indians in India and 'coloured' populations of South Africa. Various statistical approaches have been used to estimate admixture proportions in these populations and have been reviewed in detail elsewhere [37]. These include a least squares method, a weighted least squares method [16,38,39] and likelihood methods [38,40]. A recent review of admixture studies and admixture proportions of various Latin American populations is provided by Sans [41]. African Americans are a well-studied group with substantial European and West African contributions and a smaller Native American contribution [27,35,42,43]. A survey of current literature indicates that European admixture ranges from 3.5 per cent in the Gullah Sea Islanders of South Carolina [35], to 28 per cent in New Orleans [35]. Admixture estimates in African-American populations can be highly variable across the USA, which is likely to reflect local variation in the demographic histories and social norms.
US Hispanics form a complex socio-political conglomerate including Puerto Ricans, Cubans, Spanish Americans, Mexican Americans. Various groups from Central and South America can also be studied using ancestry AM. The proportional contributions from parental Europeans are estimated to be the largest, followed by a substantial Native American ancestry and varying amounts of West African ancestry [16,17,44]. In a sample of Mexican Americans from Arizona, the admixture estimates obtained using a weighted least squares method showed 29 ± 4 per cent Native American, 68 ± 5 per cent European and 3 ± 2 per cent West African contribution [16]. A recent study reports the following estimates for a Hispanic population from the San Luis Valley, Colorado: 62.7 ± 2.1 per cent European, 34.1 ± 1.9 per cent Native American, 3.2 ± 1.5 per cent West African [17]. In Puerto Ricans from New York City, the estimates obtained were 53.3 ± 2.8 per cent European, 29.1 ± 2.3 per cent West African, 17.6 ± 2.4 per cent Native American [17]. In a separate Mexican-American population sample from California, European ancestry was estimated to be 60 per cent and Native American contribution was estimated at 40 per cent [25]. As with African-American populations, there is substantial variation across populations. From these results, it is evident that, when studying any new admixed population sample, it is important to accurately determine the proportional contributions and not to rely on previously obtained estimates from a similar population. Additionally, it is instructive to have information on the levels of stratification related to admixture that are present in the population under consideration [15].
Ancestry-phenotype correlations; phenotype and complex disease gene mapping
Traits and diseases more prevalent in one population than in others are amenable to admixture analysis and some examples are listed in Table 1. Most of the diseases shown in this Table have a complex aetiology affected by multiple genes and environmental factors. Earlier studies [45,46] focused on admixed populations as units of analysis in exploring relationships between ancestry and phenotypes [12]. These authors showed that non-insulin-dependent (Type 2) diabetes mellitus prevalence is correlated with admixture proportions among a selection of populations with varying levels of Native American ancestry. Data like these provide compelling evidence for frequency differences in risk modifying alleles, but such data have not been collected for many diseases. Another related approach is to test for individual admixture-phenotype correlations within an admixed population. Correlations between ancestry and phenotypes have been detected and reported by various authors [14,17-19,44,45,47].
A prerequisite for testing ancestry/phenotype correlations is the presence of stratification related to admixture, which will be evident in variation in individual ancestry levels. Figure 2 shows the distribution of BGA estimates from three examples of Hispanic population samples, Puerto Ricans from New York, Mexicans from Tlapa, Mexico and Hispanics from the San Luis Valley, Colorado [17]. Substantial variation is observed in all three samples. With the San Luis Valley group, more variability is observed on the European-Native American axis, while the New York group is more variable on the European-West African axis. Following the argument of Chakraborty and Weiss [48], admixture proportions should be correlated with diseases/traits that differ in populations due to underlying genetic differences. In each of these population samples, strong positive correlation was observed between individual ancestry and skin pigmentation measured as melanin index 'M' or lightness index 'L' (Figures 3A, 3B and 3C). A significant negative correlation was also observed between the proportion of West African ancestry and bone mineral density (BMD) in the Puerto Rican sample [17]. Proportion West African ancestry and skin pigmentation (measured as melanin index) in individuals is also correlated in African Americans from Washington DC and African Caribbeans from the UK, but not in European Americans from State College, Pennsylvania (Figure 4) [14]. Recently, correlations have been observed between proportion West African ancestry and lower insulin sensitivity, higher fasting insulin and acute insulin response to glucose in a combined sample of African-American and European-American children [20]. In a separate sample of African-American females, West African admixture is associated with body mass index, fat mass, fat-free mass and BMD [19]. It is important to keep in mind that ancestry-pheno-type correlations are dependent on both the existence of functional alleles at different frequencies in parental populations, and significant stratification related to admixture. Although most admixed populations tested to date are structured, there is variation in the amount of stratification present, and this structure should be tested for explicitly when investigating a new population [15,42,49].
Figure 2.
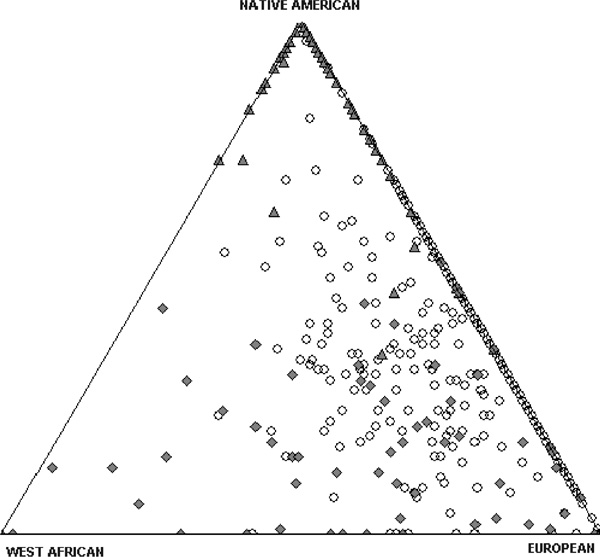
Triangle plot showing biogeographical ancestry of three Hispanic populations. Each vertex represents a parental population, which for this plot are Europeans, West Africans and Native Americans. The three populations shown are Hispanics from the San Luis Valley (blank circles), Puerto Ricans from New York City (grey diamonds) and Mexicans from Tlapa, Mexico (grey triangles) (adapted from Bonilla, 2003 [17])
Figure 3.
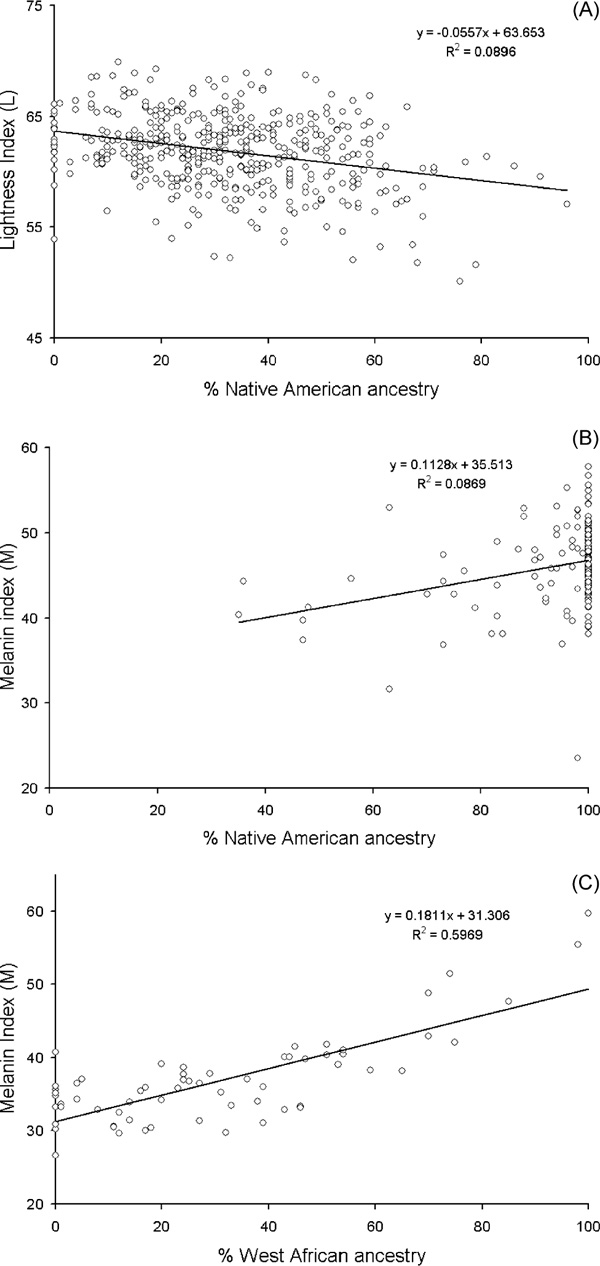
The relationship between proportional ancestry and skin pigmentation in three Hispanic populations. For all populations, proportional ancestry was estimated using the maximum likelihood (ML) method (adapted from Bonilla, 2003) [17]. (A) Percent Native American ancestry versus lightness index (L) in Hispanics from the San Luis Valley, Colorado (ancestry estimated using 22 AIMs). (B) Percent Native American ancestry versus melanin index in Mexicans from Tlapa, Mexico (ancestry estimates using 29 AIMs). (C) Percent African ancestry versus melanin index (M) in Puerto Ricans from New York City (ancestry estimated using 35 AIMs)
Figure 4.
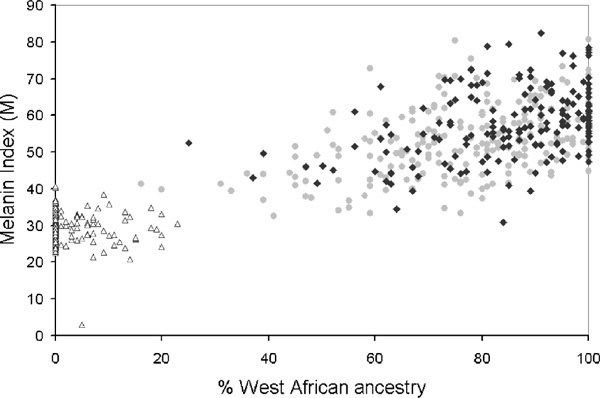
The relationship between percent African ancestry and skin pigmentation in three populations. Percent African Ancestry (obtained using 34 AIMs and calculated by the maximum likelihood (ML) method) and the melanin index (M) are shown for three populations, European Americans from State College, Pennsylvania (diamonds), African Americans from Washington, DC and State College, Pennsylvania (squares) and African Caribbeans from Britain (triangles). (With permission from Shriver et al., 2003 [14])
Methods developed for admixture analyses/study design
Theoretical and experimental studies have explored the parameters that characterise and affect admixture studies [15,24,28,35,42,50,51]. The acronym MALD was proposed [28,50] to designate the mapping method proposed originally by Chakraborty and Weiss, which exploited the long range allelic associations created through ALD [12]. Parameters critical for MALD include the genetic distance between markers and disease locus (θ); number of generations since admixture (t); proportion of admixture (m) from one parental population; the allele frequency differential (δ) between parental populations; and sample size (N) [12,28,52]. Simulation studies suggest that sample sizes of 200-300 patients, typed for 200-300 evenly spaced markers, each having allele frequency differentials >0.3, have a >95 per cent chance of locating the causative gene, when there has been no new admixture from the parental population in the last four generations and no other sources of population structure or sample heterogeneity [28,50].
Other approaches proposed for using admixture include a method based on the transmission disequilibrium test (TDT) [53] that assesses excess transmission of alleles derived from high-risk ancestors to affected offspring of parents who are heterozygous at the marker locus, containing one allele from each of two ancestral populations [52]. A second TDT-based likelihood approach was developed that compared the transmission of haplotypes with non-transmission in affected offspring in an admixed population following a multipoint method. It obtained a likelihood statistic to determine the significance of various models under different scenarios [54].
One fundamental limitation of MALD as initially described and in its early extensions, is the effects of stratification on causing false-positive association [12,24,28]. The TDT is one means of correcting for this stratification. Another is by conditioning on parental admixture [29]. Marker data at all loci are combined to estimate ancestry of alleles at each locus. When allelic ancestry at marker loci is known, this approach is analogous to a linkage analysis, hence the term AM is more appropriate than MALD for describing this method and to distinguish it from LD approaches [13,14,29]. The underlying variation in ancestry of chromosomes of mixed descent is modelled to extract all of the information about linkage that is generated by admixture. For example, where a locus is assumed to account for variation in skin pigmentation between two parental groups, eg West Africans and Europeans, individuals can be classified according to whether they have 0, 1 or 2 alleles of West African descent at this locus. By comparing these three groups for mean pigmentation level, holding all other factors constant, variation in pigmentation can be observed depending upon the number of alleles of West African ancestry in an individual. Controlling for parental admixture eliminates association of the trait with ancestry at unlinked loci. By removing the background effects of ancestry, it is possible to observe the locus-specific effects on a trait/disease [14,17]. Allelic ancestry at a locus is inferred from the marker by using the conditional probability of each allelic state given the ancestry-specific allele frequencies. A complex hierarchical model with many nuisance parameters is used to model the distribution of admixture in the population. This is implemented using the ADMIXMAP program (at http://www.lshtm.ac.uk/eph.eu/GeneticEpidemiologyGroup/htm), which follows a Bayesian approach with Markov chain simulation, and incorporates the admixture of each individual's parents and the random variation of ancestry on chromosomes inherited from each of the parents in the model [13,14,29].
Variation in individual admixture introduces population stratification, which in turn can inflate the number of significant associations that are observed [53,55,56] and is a potential confounder in association studies [29,57-59]. Various statistical approaches have been developed to detect and control for stratification within a population sample [14,15,17,42,60-62]. For example, the Dt/D0 test examines the relationship between the observed LD and the predicted ALD between unlinked marker pairs for detecting structure within the sample. Using individual ancestry as a conditioning variable in analysis of variance tests, it is possible to eliminate association of the trait with unlinked alleles [14,17]. The Bayesian approaches implemented by McKeigue et al. and Pritchard et al [13,61]. offer an advantage over classical maximum likelihood based methods [44,63] by allowing for missing genotype and ancestry data and modelling admixture hierarchically. Methods have been developed to control for parental admixture [29] and to account for uncertain BGA estimation [59].
Recent studies and future directions
Several theoretical and practical studies indicate that AM approaches promise to be suitable for identifying genes causing complex diseases. Methodological advancements have been made to offset the potential problems arising from association between unlinked loci by conditioning on parental admixture [13,29], and to detect and correct for population stratification [59,60]. Use of Bayesian AM [13,29,59] can take into consideration various uncertainties, including missing data values for estimating admixture proportions, and can overcome problems arising out of mis-specification of parental allele frequencies and promises to be an effective tool for admixture studies. This method, which is different from the classical disequilibrium-based approach that is more commonly used, is perhaps more suitable for disease gene mapping in admixed populations and has already been successfully used for mapping [14]. Table 2 summarises recent studies showing associations between ancestry and phenotypes/diseases and instances where AM was used to identify genes. Currently, the primary impediment to exhaustive AM genome scans is the lack of verified AIM panels. Sufficient numbers of markers are available as candidate AIMs, but effort and resources are required to confirm these markers and to generate accurate parental allele frequencies. Efforts are currently underway in several laboratories to identify more AIMs for this purpose. It seems inevitable that more such studies will be carried out in the near future to utilise the immense potential of this approach.
Table 2.
Diseases showing ancestry-phenotype correlation
| Phenotype | Population studied | Association observed | Test statistic reported | Reference |
|---|---|---|---|---|
| Non-insulin-dependent diabetes mellitus (NIDDM) | Mexican Americans and Pima Indians | Amerindian ancestry with NIDDM | Kendall's τ = 0.848 ± 0.221, [p ⊬ 8.1 × 105] | [48] |
| NIDDM | Mexican Americans | Amerindian ancestry with NIDDM | 0.943c (p < 0.001) | [63] |
| 1) Body mass index (BMI) | Pima Indians | European admixture with BMI, plasma glucose, 2-hour glucose | 0.455b (95% CI: 0.301-0.688) | [47] |
| 2) Plasma glucose | ||||
| 3) NIDDM | ||||
| NIDDM | Mexican Americans | Native American ancestry with NIDDM prevalence | N/A | [45] |
| Skin pigmentation (reflectrometry) | 1) African Americans | Melanin index versus % African ancestry | 1) 0.21a, (p < 0.0001) | [14] |
| 2) Afro-Caribbeans | 2) 0.16a (p < 0.0001) | * Mapped phenotype to two loci: TYR and OCA as candidates which influence normal pigmentation variation | ||
| 3) European Americans | 3) 0.001a (p = NS) | |||
| Systemic lupus erythematosus (SLE) | Caribbeans (without Indian or Chinese ancestry) | SLE and African Ancestry | 28.4 | [18] |
| (95% CI: 1.7-485 after SES adjustmentb) | ||||
| Insulin-related phenotypes | African American | African admixture (ADM): | [20] | |
| Europeans | ||||
| Americans | ||||
| 1) Insulin sensitivity (SI), | 1) with SI | 1) (p < 0.001)a | ||
| 2) Fasting insulin (FA), | 2) with FA | 2) (p < 0.01)a | ||
| 3) Acute insulin response (AIR) | 3) with AIR | 3) (p < 0.001)a | ||
| Oxygen capacity | Quechua natives | Positive: | 0.8a | [80] |
| Spanish admixture with large VO2 at high altitudes | ||||
| Bone mineral density (BMD) | Puerto Ricans from New York | Positive: | 0.065a (p = 0.042) | [17] |
| European admixture with lower BMD | ||||
| Skin pigmentation (Lightness index) | Hispanics from the San Luis Valley, Colorado | Positive: | 0.0821a (p < 0.001) | [17] |
| Proportional European ancestry with increased Lightness |
a = R2; b = risk ratio; c = rank-order correlation.
Acknowledgements
We thank Dr Paul McKeigue and Dr Esteban Parra for helpful discussions on the subject. We also acknowledge helpful comments from an unknown reviewer. This work was supported in part by grants from NIH/NIDDK (DK53958) and NIH/NHGRI (HG02154) to M.D.S.
References
- Hartl DL, Clark AG. Principles of Population Genetics. Sinauer Associates, Sunderland, MA; 1997. [Google Scholar]
- Jorde LB. 'Linkage disequilibrium as a gene-mapping tool'. Am J Hum Genet. 1995;56:11–14. [PMC free article] [PubMed] [Google Scholar]
- Huttley GA, Smith MW, Carrington M, O'Brien SJ. 'A scan for linkage disequilibrium across the human genome'. Genetics. 1999;152:1711–1722. doi: 10.1093/genetics/152.4.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M. 'Patterns of linkage disequilibrium in the human genome'. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. Erratum in: Nat. Rev. Genet., Vol. 3, p. 566. [DOI] [PubMed] [Google Scholar]
- Kerem E, Reisman J, Corey M. et al. 'Prediction of mortality in patients with cystic fibrosis'. N Engl J Med. 1989;326:1187–1191. doi: 10.1056/NEJM199204303261804. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Vonsattel JP, Shrinidhi J. et al. 'Evidence for the GluR6 gene associated with younger onset age of Huntington's disease'. Neurology. 1992;53:1330–1332. doi: 10.1212/wnl.53.6.1330. [DOI] [PubMed] [Google Scholar]
- Puffenberger EG, Kaufmann ER, Bolk S. et al. 'Identity-by-descent and association mapping of a recessive gene for Hirschsprung disease on human chromosome 13q22'. Hum Mol Genet. 1994;3:1217–1225. doi: 10.1093/hmg/3.8.1217. [DOI] [PubMed] [Google Scholar]
- Sheffield VC, Carmi R, Kwitek-Black A. et al. 'Identification of a Bardet-Beidle syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping'. Hum Mol Genet. 1994;3:1331–1335. doi: 10.1093/hmg/3.8.1331. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. 'The future of genetic studies of complex human diseases'. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M. 'Linkage disequilibrium in humans: Models and data'. Am J Hum Genet. 2001;69:1–14. doi: 10.1086/321275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KM, Clark AG. 'Linkage disequilibrium and the mapping of complex human traits'. Trends Genet. 2002;18:19–24. doi: 10.1016/S0168-9525(01)02550-1. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Weiss KM. 'Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci'. Genetics. 1988;85:9119–9123. doi: 10.1073/pnas.85.23.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM, Carpenter J, Parra EJ, Shriver MD. 'Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: Application to African-American populations'. Ann Hum Genet. 2000;64:171–186. doi: 10.1046/j.1469-1809.2000.6420171.x. [DOI] [PubMed] [Google Scholar]
- Shriver MD, Parra EJ, Dios S. et al. 'Skin pigmentation, biogeographical ancestry and admixture mapping'. Hum Genet. 2003;112:387–399. doi: 10.1007/s00439-002-0896-y. [DOI] [PubMed] [Google Scholar]
- Pfaff CL, Parra EJ, Bonilla C. et al. 'Population structure in admixed populations: Effects of admixture dynamics on the pattern of linkage disequilibrium'. Am J Hum Genet. 2001;68:198–207. doi: 10.1086/316935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC. 'The genetic structure of admixed populations'. Genetics. 1991;127:417–428. doi: 10.1093/genetics/127.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla C. 'Admixture in three Hispanic populations: Ancestry proportions, population structure, and gene mapping'. The Pennsylvania State University, University Park, PA, USA; PhD Thesis, Department of Anthropology, [Google Scholar]
- Molokhia M, Hoggart CJ, Patrick AL. et al. 'Relation of risk of systemic lupus erythematosus to West African admixture in a Caribbean population'. Hum Genet. 2003;112:310–318. doi: 10.1007/s00439-002-0883-3. [DOI] [PubMed] [Google Scholar]
- Fernández JR, Shriver MD, Beasley TM. et al. 'Association of African genetic admixture with resting metabolic rate and obesity among African American women'. Obesity Res. 2003;11(7):904–911. doi: 10.1038/oby.2003.124. [DOI] [PubMed] [Google Scholar]
- Gower BA, Fernandez JR, Beasley TM. et al. 'Using genetic admixture to explain racial differences in insulin-related phenotypes'. Diabetes. 2003;52:1047–1051. doi: 10.2337/diabetes.52.4.1047. [DOI] [PubMed] [Google Scholar]
- Reed TE. 'Number of gene loci required for accurate estimation of ancestral population proportions in individual human hybrids'. Science. 1973;244:575–576. doi: 10.1038/244575a0. [DOI] [PubMed] [Google Scholar]
- Neel JV. 'Developments in monitoring human populations for mutation rates'. Mutat Res. 1974;26:319–328. doi: 10.1016/S0027-5107(74)80029-1. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Kamboh MI, Ferrell RE. 'Unique alleles in admixed populations: A strategy for determining hereditary population differences of disease frequencies'. Ethn Dis. 1991;1:245–256. [PubMed] [Google Scholar]
- Dean M, Stephens JC, Winkler C. et al. 'Polymorphic admixture typing in human ethnic populations'. Am J Hum Genet. 1994;55:788–808. [PMC free article] [PubMed] [Google Scholar]
- Collins-Schramm HE, Phillips CM, Operario DJ. et al. 'Ethnic-difference markers for use in mapping by admixture linage disequilibrium'. Am J Hum Genet. 2002;70:737–750. doi: 10.1086/339368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver MD, Smith MW, Jin L. et al. 'Ethnic-affiliation estimation by use of population-specific DNA markers'. Am J Hum Genet. 1997;60:957–964. [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Kamboh MI, Nwankwo M, Ferrell RE. 'Caucasian genes in American blacks: New data'. Am J Hum Genet. 1992;50:145–155. [PMC free article] [PubMed] [Google Scholar]
- Stephens JC, Briscoe D, O'Brien SJ. 'Mapping by admixture linkage disequilibrium in human populations: Limits and guidelines'. Am J Hum Genet. 1994;55:809–824. [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM. 'Mapping genes that underlie ethnic differences in disease risk: Methods for detecting linkage in admixed populations, by conditioning on parental admixture'. Am J Hum Genet. 1998;63:241–251. doi: 10.1086/301908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular Population Genetics. Columbia University Press, New York, NY; 1987. [Google Scholar]
- Cavalli-Sforza L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton University Press, Princeton, NJ; 1994. [Google Scholar]
- Deka R, Shriver MD, Yu LM. et al. 'Intra- and inter-population diversity at short tandem repeat loci in diverse populations of the world'. Electrophoresis. 1995;16:1659–1664. doi: 10.1002/elps.11501601275. [DOI] [PubMed] [Google Scholar]
- Smith MW, Lautenberger JA, Shin HD. et al. 'Markers for mapping by admixture linkage disequilibrium in African-American and Hispanic Populations'. Am J Hum Genet. 2001;69:1080–1094. doi: 10.1086/323922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-Schramm HE, Kittles RA, Operario DJ. et al. 'Markers that discriminate between European and African ancestry show limited variation within Africa'. Hum Genet. 2002;111:566–569. doi: 10.1007/s00439-002-0818-z. [DOI] [PubMed] [Google Scholar]
- Parra EJ, Marcini A, Akey J. et al. 'Estimating African American admixture proportions by use of population specific alleles'. Am J Hum Genet. 1998;63:1839–1851. doi: 10.1086/302148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey J, Zhang G, Jin L, Shriver MD. 'Interrogating a high-density SNP map for signatures of natural selection'. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R. 'Gene admixture in human populations: Models and predictions'. Yearb Phys Anthropol. 1986;29:1–43. doi: 10.1002/ajpa.1330290502. [DOI] [Google Scholar]
- Elston RC. 'The estimation of admixture in racial hybrids'. Ann Hum Genet. 1971;35:9–17. doi: 10.1111/j.1469-1809.1956.tb01373.x. [DOI] [PubMed] [Google Scholar]
- Long JC, Smouse PE. 'Intertribal gene flow between the Ye'cuana and Yanomama: Genetic analysis of an admixed village'. Am J Phys Anthropol. 1983;61:411–422. doi: 10.1002/ajpa.1330610403. [DOI] [PubMed] [Google Scholar]
- Chikhi L, Bruford MW, Beaumont MA. 'Estimation of admixture proportions: A likelihood-based approach using Markov chain Monte Carlo'. Genetics. 2001;158:1347–1362. doi: 10.1093/genetics/158.3.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans M. 'Admixture studies in Latin America: From the 20th to the 21st century'. Hum Biol. 2000;72:155–177. [PubMed] [Google Scholar]
- Parra EJ, Kittles RA, Argyropoulos G. et al. 'Ancestral proportions and admixture dynamics in geographically defined African-Americans living in South Carolina'. Am J Phys Anthropol. 2001;114:18–29. doi: 10.1002/1096-8644(200101)114:1<18::AID-AJPA1002>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Destro-Bisol G, Maviglia R, Caglia A. et al. 'Estimating European admixture in African Americans by using microsatellites and a microsatellite haplotype (CD4/Alu)'. Hum Genet. 1999;104:149–157. doi: 10.1007/s004390050928. [DOI] [PubMed] [Google Scholar]
- Hanis CL, Chakraborty R, Ferrell RE, Schull WJ. 'Individual admixture estimates: Disease associations and individual risk of diabetes and gallbladder disease among Mexican-Americans in Starr County, Texas'. Am J Phys Anthropol. 1986;70:433–441. doi: 10.1002/ajpa.1330700404. [DOI] [PubMed] [Google Scholar]
- Gardner LI Jr, Stern MP, Haffner SM. et al. 'Prevalence of diabetes in Mexican Americans. Relationship to percent of gene pool derived from Native American sources'. Diabetes. 1984;33:86–92. doi: 10.2337/diabetes.33.1.86. [DOI] [PubMed] [Google Scholar]
- Long JC, Williams RC, McAuley JE. et al. 'Genetic variation in Arizona Mexican Americans: Estimation and interpretation of admixture proportions'. Am J Phys Anthropol. 1991;84:141–157. doi: 10.1002/ajpa.1330840204. [DOI] [PubMed] [Google Scholar]
- Williams RC, Long JC, Hanson RL. et al. 'Individual estimates of European genetic admixture associated with lower body-mass index, plasma glucose, and prevalence of type 2 diabetes in Pima Indians'. Am J Hum Genet. 2000;66:527–538. doi: 10.1086/302773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Weiss KM. 'Frequencies of complex diseases in hybrid populations'. Am J Phys Anthropol. 1986;70:489–503. doi: 10.1002/ajpa.1330700408. [DOI] [PubMed] [Google Scholar]
- Pfaff CL. 'Estimating admixture dynamics: Implications for mapping genes'. The Pennsylvania State University, University Park, PA, USA; 2001. PhD Thesis, Department of Anthropology, [Google Scholar]
- Briscoe D, Stephens JC, O'Brien SJ. 'Linkage disequilibrium in admixed populations: Applications in gene mapping'. J Hered. 1994;85:59–63. [PubMed] [Google Scholar]
- Lautenberger JA, Stephens JC, O'Brien SJ, Smith MW. 'Significant admixture linkage disequilibrium across 30 cM around the FY locus in African Americans'. Am J Hum Genet. 2000;66:969–978. doi: 10.1086/302820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM. 'Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations'. Am J Hum Genet. 1997;60:188–196. [PMC free article] [PubMed] [Google Scholar]
- Ewens WJ, Spielman RS. 'The transmission/disequilibrium test: History, subdivision, and admixture'. Am J Hum Genet. 1995;57:455–464. [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Elston RC. 'Multipoint linkage disequilibrium mapping with particular reference to the African-American population'. Genet Epidemiol. 1999;17:79–101. doi: 10.1002/(SICI)1098-2272(1999)17:2<79::AID-GEPI1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Molokhia M, McKeigue PM. 'Risk for rheumatic disease in relation to ethnicity and admixture'. Arthritis Res. 2000;2:115–125. doi: 10.1186/ar76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybicki BA, Iyengar SK, Harris T. et al. 'The distribution of long range admixture linkage disequilibrium in an African-American population'. Hum Hered. 2002;53:187–196. doi: 10.1159/000066193. [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ. 'Genetic dissection of complex traits'. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- Thomas DC, Witte JS. 'Point: population stratification: A problem for case-control studies of candidate-gene associations?'. Cancer Epidemiol Biomarkers Prev. 2002;11:505–512. [PubMed] [Google Scholar]
- Hoggart CJ, Parra EJ, Shriver MD. et al. 'Control of confounding of genetic associations in stratified populations'. Am J Hum Genet. 2003;72:1492–1504. doi: 10.1086/375613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. 'Genomic control for association studies'. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. 'Association mapping in structured populations'. Am J Hum Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich RE, Goldstein DB. 'Detecting association in a case-control study while correcting for population stratification'. Genet Epidemiol. 2001;20:4–16. doi: 10.1002/1098-2272(200101)20:1<4::AID-GEPI2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Ferrell RE, Stern MP. et al. 'Relationship of prevalence of non-insulin-dependent diabetes mellitus to Amerindian admixture in the Mexican Americans of San Antonio, Texas'. Genet Epidemiol. 1986;3:435–454. doi: 10.1002/gepi.1370030608. [DOI] [PubMed] [Google Scholar]
- McKeigue PM, Shah B, Marmot MG. 'Relation of central obesity and insulin resistance with high diabetes prevalence and cardiovascular risk in South Asians'. Lancet. 1991;337:382–386. doi: 10.1016/0140-6736(91)91164-P. [DOI] [PubMed] [Google Scholar]
- Hodge AM, Zimmet PZ. 'The epidemiology of obesity'. Baillieres Clin Endocrinol Metab. 1994;8:577–599. doi: 10.1016/S0950-351X(05)80287-3. [DOI] [PubMed] [Google Scholar]
- Songer TJ, Zimmet PZ. 'Epidemiology of type II diabetes: An international perspective'. Pharmacoeconomics. 1995;8(Suppl 1):1–11. doi: 10.2165/00019053-199500081-00003. [DOI] [PubMed] [Google Scholar]
- Martinez NC. 'Diabetes and minority populations. Focus on Mexican Americans'. Nurs Clin North Am. 1993;28:87–95. [PubMed] [Google Scholar]
- Douglas JG, Thibonnier M, Wright JT Jr. 'Essential hypertension: Racial/ethnic differences in pathophysiology'. J Assoc Acad Minor Phys. 1996;7:16–21. [PubMed] [Google Scholar]
- Gaines K, Burke G. 'Ethnic differences in stroke: Black-white differences in the United States population. SECORDS Investigators. Southeastern Consortium on Racial Differences in Stroke'. Neuroepidemiology. 1995;14:209–239. doi: 10.1159/000109798. [DOI] [PubMed] [Google Scholar]
- Zoratti R. 'A review on ethnic differences in plasma triglycerides and high-density-lipoprotein cholesterol: Is the lipid pattern the key factor for the low coronary heart disease rate in people of African origin?'. Eur J Epidemiol. 1998;14:9–21. doi: 10.1023/A:1007492202045. [DOI] [PubMed] [Google Scholar]
- McKeigue PM, Miller GJ, Marmot MG. 'Coronary heart disease in south Asians overseas: A review'. J Clin Epidemiol. 1989;42:597–609. doi: 10.1016/0895-4356(89)90002-4. [DOI] [PubMed] [Google Scholar]
- Ferguson R, Morrissey E. 'Risk factors for end-stage renal disease among minorities'. Transplant Proc. 1993;25:2415–2420. [PubMed] [Google Scholar]
- Hargrave R, Stoeklin M, Haan M, Reed B. 'Clinical aspects of dementia in African-American, Hispanic, and white patients'. J Nat Med Assoc. 2000;92:15–21. [PMC free article] [PubMed] [Google Scholar]
- Boni R, Schuster C, Nehrhoff B, Burg G. 'Epidemiology of skin cancer'. Neuroendocrinol Lett. 2002;23(Suppl 2):48–51. [PubMed] [Google Scholar]
- Schwartz AG, Swanson GM. 'Lung carcinoma in African Americans and whites. A population-based study in metropolitan Detroit, Michigan'. Cancer. 1997;79:45–52. doi: 10.1002/(SICI)1097-0142(19970101)79:1<45::AID-CNCR7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Wu AH, Koo LC. et al. 'Lung cancer in women living in the Pacific Basin area'. Nat Cancer Inst Monogr. 1985;69:197–201. [PubMed] [Google Scholar]
- Hoffman RM, Gilliland FD, Eley JW. et al. 'Racial and ethnic differences in advanced-stage prostate cancer: The Prostate Cancer Outcomes Study'. J Nat Cancer Inst. 2001;93:388–395. doi: 10.1093/jnci/93.5.388. [DOI] [PubMed] [Google Scholar]
- Rosati G. 'The prevalence of multiple sclerosis in the world: An update'. Neurol Sci. 2001;22:117–139. doi: 10.1007/s100720170011. [DOI] [PubMed] [Google Scholar]
- Bohannon AD. 'Osteoporosis and African American women'. J Womens Health Gend Based Med. 1999;8:609–615. doi: 10.1089/jwh.1.1999.8.609. [DOI] [PubMed] [Google Scholar]
- Brutsaert TD, Parra EJ, Shriver MD. et al. 'Spanish genetic admixture is associated with larger VO2max decrement from sea level to 4,338 meters in Peruvian Quechua'. J Appl Physiol. 2003;95(2):519–528. doi: 10.1152/japplphysiol.01088.2002. [DOI] [PubMed] [Google Scholar]