

Abstract
The authors present ELB, an easy to programme and computationally fast algorithm for inferring gametic phase in population samples of multilocus genotypes. Phase updates are made on the basis of a window of neighbouring loci, and the window size varies according to the local level of linkage disequilibrium. Thus, ELB is particularly well suited to problems involving many loci and/or relatively large genomic regions, including those with variable recombination rate. The authors have simulated population samples of single nucleotide polymorphism genotypes with varying levels of recombination and marker density, and find that ELB provides better local estimation of gametic phase than the PHASE or HTYPER programs, while its global accuracy is broadly similar. The relative improvement in local accuracy increases both with increasing recombination and with increasing marker density. Short tandem repeat (STR, or microsatellite) simulation studies demonstrate ELB's superiority over PHASE both globally and locally. Missing data are handled by ELB; simulations show that phase recovery is virtually unaffected by up to 2 per cent of missing data, but that phase estimation is noticeably impaired beyond this amount. The authors also applied ELB to datasets obtained from random pairings of 42 human X chromosomes typed at 97 diallelic markers in a 200 kb low-recombination region. Once again, they found ELB to have consistently better local accuracy than PHASE or HTYPER, while its global accuracy was close to the best.
Keywords: linkage disequilibrium, gametic phase, haplotype, Bayesian estimation
Introduction
The human genome is highly polymorphic, with more than one heterozygous nucleotide per 500 sites [1]. Over the past few years, it has become increasingly easy to document much of this polymorphism in population samples, using dense maps of single nucleotide polymorphism (SNP) or short tandem repeat (STR, or microsatellite) markers [2]. Applications of such data include assessing population structure and migration levels [3,4], detecting selection and founder effects on disease alleles [5,6] and mapping genes associated with disease [7].
Due to the diploid nature of the human genome, the most accessible information consists of multi-locus genotypes, for which phase information is absent. Haplotype data, equivalent to genotype data plus gametic phase, is advantageous for many applications, such as linkage disequilibrium (LD) mapping [8-10], even though the additional information content of haplotype over genotype data is not very large. Indeed, the authors demonstrate below that it is often possible to infer the former from the latter with few errors. Instead, the advantages of haplotype data arise because they are much more amenable to analysis, in large part because haplotype segments are inherited uniparentally [11].
Laboratory techniques are available for resolving gametic phase [12,13]. These techniques are generally reliable, but costly in terms of time and money. Statistical tools for inferring the gametic phases of individuals drawn from a population tend to be cheap and fast compared with laboratory techniques. Statistical methods rely on the fact that the shared ancestry of individuals within the population means that they tend to share haplotype segments, so that relatively few of the large number of possible haplotypes will be observed in a sample. Thus, resolving a query genotype into the haplotype pair with the highest population frequency can be highly accurate. Population frequencies of haplotype pairs are usually unknown, but under the assumption of the Hardy-Weinberg equilibrium (HWE) they can often be estimated well enough that the resulting accuracy of phase inferences remains good, although typically less good than for laboratory techniques. Family data can also be exploited to resolve phase, although if the number of polymorphic loci is large, it is necessary to obtain and type deep pedigrees to infer the full phase information.
In the following, 'to phase' will be used as shorthand for 'to assign gametic phase', and an individual with more than one heterozygous site will be referred to as 'ambiguous'. There are currently four principal statistical algorithms for phasing samples of multi-locus genotypes. The first, which the authors call CEM (Clark Empirical Method) [14], attempts to minimise the total number of distinct haplotypes inferred from the observed genotypes. A list of observed haplotypes is initiated from the individuals that are heterozygous at no more than one locus and hence are non-ambiguous. The remaining genotypes are then checked in turn to see if they are compatible with any haplotype in the list. If so, the corresponding phase is assigned and the complementary haplotype is added to the list (if not already there). This process is iterated until no more ambiguous individuals can be resolved. The CEM algorithm has several weaknesses: it cannot start if all sampled individuals are ambiguous; some individuals may not be phased; and the resulting phase allocation can depend on the order in which individuals are examined.
A second statistical approach is based on implementing an expectation-maximisation (EM) algorithm to obtain maximum likelihood estimates of population haplotype frequen-cies [15-17]. The algorithm starts by attributing arbitrary initial values to the haplotype frequencies, and then iterates between calculating the expected genotype frequencies, given the current haplotype frequencies and the assumption of HWE, and generating new haplotype frequencies via a gene counting method. After convergence, each individual can be phased, for example by maximising the likelihood based on the final haplotype frequencies and the HWE assumption. This method leads to reasonably good haplotype frequency estimates and phase reconstructions [18-21] but it is limited to a small number of polymorphic loci since all haplotypes consistent with the genotypes must be enumerated, the size of this task increases exponentially with the number of loci.
PHASE [22] implements a pseudo-Gibbs sampler which explores the space of possible phases more efficiently than the EM algorithm. PHASE starts by assigning an arbitrary initial gametic phase to each individual and then repeats the following steps: 1) select an individual at random; 2) select five heterozygous loci and erase the existing phase allocations at these loci; 3) re-assign the phases according to the joint probabilities of the resulting pair of 5-locus haplotypes, given the current haplotype assignments of the rest of the sample. The probability used in step 3 relies on the HWE assumption, together with an approximation to the conditional haplotype probabilities arising under the coalescent model [23]. In addition to drawing on the frequencies of the haplotypes already present in the sample (as do the CEM and the EM algorithms) PHASE also takes account, via this coalescent approximation, of haplotypes that are similar, but not identical, to observed haplotypes. After a burn-in period, the proportion of steps at which a particular phase assignment is made is interpreted as the posterior probability of that phase allocation.
HTYPER [24] is also based on a pseudo-Gibbs sampler, but differs from PHASE in two principal respects: it does not take into account haplotypes similar to the observed haplotypes, and it builds up the full gametic phase in a hierarchical manner, by first resolving smaller segments which are then progressively combined.
When samples are simulated under the coalescent model, with or without recombination, PHASE outperforms the other algorithms [22,24], while HTYPER gives slightly better results than the EM algorithm [24] and CEM is consistently worst. PHASE has been reported to be inferior to other algorithms in some settings involving real data, or data simulated under another scheme than the standard coalescent [20]. PHASE is usually the slowest of the algorithms, and is often orders of magnitude slower than HTYPER.
In HTYPER, the gametic phase is built locally over short segments, and larger segments are phased by a series of ligations of these short segments, without making use of overlapping information. PHASE chooses five loci to update at a time, but the loci are not contiguous and the new assignment is chosen on the basis of the entire haplotypes. Thus, neither of these algorithms explicitly accommodates the effects of recombination, although both are somewhat robust to small amounts of recombination. The authors' new ELB algorithm, introduced below, is designed to be fast and more robust to recombination than other algorithms.
Methods
An adaptive window approach: The Excoffier-Laval-Balding (ELB) algorithm
Suppose that we have a sample of n individuals drawn from some population and genotyped at S loci whose chromosomal order is assumed known. Adjacent pairs of loci are assumed to be tightly linked, but S may be large so that the two extremal loci are effectively unlinked. In this case, as recognised previously [22,24,25], reconstructing the gametic phase in one step can be inefficient, because recombination may have created too many distinct haplotypes for their frequencies to be well estimated. Locally, however, recombination may be rare, and to exploit this situation, in ELB the updates of the phase at a heterozygous locus are based on 'windows' of neighbouring loci. The algorithm adjusts the window sizes and locations in order to maximise the information for the phase updates.
ELB starts with an arbitrary phase assignment for all individuals in the sample. Associated with each heterozygous locus are two windows, containing the locus itself and all neighbouring loci to the left (respectively, right), up to and including the nearest heterozygous locus to the left (right). Note that the use of two windows per heterozygous locus is not necessary, but the authors found that it led to better mixing than an algorithm with one window per locus, and the extra space and computation overhead are not important.
At each iteration of the algorithm, an individual is chosen at random and its heterozygous loci are successively visited in random order. At each locus visit, one of the two current windows is chosen according to its information content (see below). Two attempts are then made to update that window, by proposing, and then accepting or rejecting, (i) the addition of a locus at one end of the window and (ii) the removal of a locus at the other end. The locus being visited is never removed from the window, and each window always includes at least one other heterozygous locus. The two update proposals are made sequentially, so that the window can either grow by one locus, shrink by one locus, or, if both changes are accepted, the window 'slides' by one locus either to the right or the left. If both proposals are rejected, the window remains unchanged. Next, the phase at the locus being visited is updated based on the current haplotype pairs, within the chosen window, of the other individuals in the sample.
In summary, the algorithm proceeds by repeating the following steps:
1) Choose an individual: at random among all individuals.
2) Choose a heterozygous locus: at random among those not yet visited since step 1) was last performed.
3) Choose and update a window: choose one of the two windows currently associated with the individual and locus chosen in 1) and 2), and attempt to update it by successively proposing, and then accepting or rejecting, the addition and the removal of a locus from the window (details below).
4) Update the phase: based on haplotype counts in the rest of the sample and given their current phase allocations within the window chosen in step 3) (details below).
5) Repeat 2) to 4) until each of the individual's heterozygous loci has been visited.
Phase updates
Let h11 and h22 denote the two haplotypes within the window given the current phase assignment, and let h12 and h21 denote the haplotypes which would result from the alternative phase assignment at the locus being visited. Ideally, we would wish to choose between the two haplotype assignments, h11/h22 and h12/h21, with probabilities proportional to their (joint) population frequencies. These are unknown, and in practice they are too small for direct estimation to be feasible. To overcome the latter problem, we use the HWE assumption, so that we now seek to choose between h11/h22 and h12/h21 with probabilities proportional to p11p22 and p12p21, where pij, i,j = 1, 2, denotes the population frequency of hij. Although the pij are also unknown, we can estimate them using the nij, the haplotype counts among the other n - 1 individuals in the sample, given their current phase assignments within the window.
The maximum-likelihood estimate of pij pi'j' is proportional to nij ni'j', but this is unsatisfactory for our purposes since its use would imply that the haplotype pair hij/hi'j' will never be assigned if either hij or hi'j' is not observed among the other individuals under their current phase assignments. Instead, we adopt a Bayesian posterior mean estimate of pij pi'j', based on a symmetrical Dirichlet prior distribution for the pij with parameter α > 0, and hence we obtain:
| (1) |
Larger values of α imply a greater chance of choosing a haplotype pair that includes an unobserved haplotype.
Increasing α thus allows more flexibility to choose new haplotypes, but this is a 'noisy' solution: all unobserved haplotypes are treated the same. A recent mutation event can create haplotypes that are rare, but similar to a more common haplotype, however, whereas haplotypes that are very dissimilar to all observed haplotypes are highly implausible. This phenomenon is particularly prevalent for STR loci, with their relatively high mutation rates.
To encapsulate the effect of mutation, when making a phase assignment we wish to give additional weight to an unobserved haplotype for each observed haplotype that is 'close' to it. PHASE achieves this via a coalescent approximation, which is costly to compute. We adopt here a simpler, ad hoc scheme in which we define 'close' to mean 'differs by one locus', and in the phase update step 4) we choose h11/h22 rather than h12/h21 with probability:
| (2) |
where nij_1 is the sample count of haplotypes that are close to hij within the current window. Since ε is a parameter reflecting the effect of mutation, it should, for example, be larger for STR than for SNP data.
Window updates
We choose one of the two windows around the locus being visited with probability proportional to its information content defined as:
This favours windows in which the possible haplotypes are frequent in the rest of the sample.
The value of R = max{r; 1/r}, where r = p11p22/p12p21, gives a measure of linkage disequil. LD within the window. Broadly speaking, at each choice between two windows, we would generally prefer the window that gives the largest value to R. Based on (2), a natural estimate of r is:
but this estimate leads to difficulties, since larger windows tend to have smaller counts, and hence more extreme estimates, amounting to a 'bias' towards larger windows. This bias could be counteracted by increasing α but we prefer to adjust α to optimise the phase updates probability (2). Instead, we add a constant to both numerator and denominator, leading to:
| (3) |
Thus, at each attempt to update the length of a window in step 3) above, we choose between windows according to their values: window 2 replaces window 1 with probability
| (4) |
Even a large value for γ can fail to prevent a window from growing too large when two consecutive heterozygous loci in an individual are separated by many homozygous loci. The window must then be large in order to contain the necessary minimum of two heterozygous loci. To circumvent the problem of small haplotype counts which may then result, when updating an individual's phase allocation we ignore homozygous loci that are separated from the nearest heterozygous locus by more than five intervening homozygous loci.
Parameter assignment
The phase and window updates described above rely on three parameters: α, ε and γ. Small simulation studies were conducted separately for SNP and STR data to investigate good values for these parameters. For both data types, a small number of values were considered between 0 and 0.1 for α, between 0 and 0.5 for ε and between 0 and 1 for γ. For SNP data, it was found that the performance of ELB was not highly sensitive to α and ε within these ranges, but that too low a value for γ could seriously impair performance in the presence of little or no recombination because of the problem of very large windows. The values α = 0.01; ε = 0.01 and γ = 0.01 gave good results for moderate to high levels of recombination, whereas a larger value of γ was sometimes needed in the presence of little or no recombination. For STR data, the values α = 0.01; ε = 0.1 and γ = 0 were found to perform well; ELB had little sensitivity to any of these parameters. A larger ε value than that used for SNPs is appropriate because of the larger mutation rate of STRs, and this higher ε value makes it unlikely that the denominator in equation (3) becomes very small, so that the protection of a positive γ value becomes unnecessary.
The results reported below and shown later in Figures 1 to 4 adopted the two sets of parameter values stated above, according to whether the data were STR or SNP. Beyond this, the authors' results have not been artificially enhanced by optimising the parameter values individually for each analysis. Conversely, there may be scope for further improvement of these results by realistic fine-tuning of the parameter values, for example based on prior information about the recombination rate.
Figure 1.
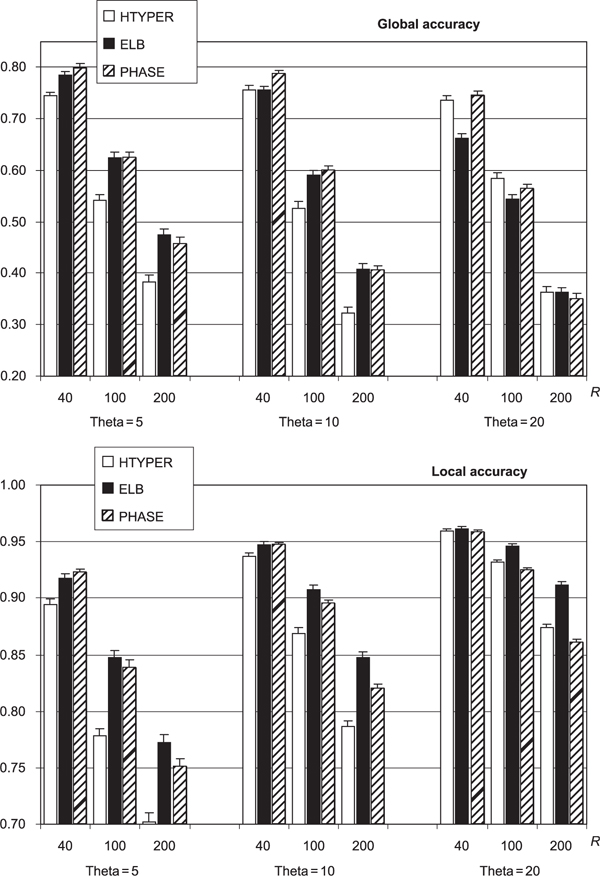
Mean global and local accuracy of ELB, PHASE (Ver. 1) and HTYPER algorithms when inferring gametic phase in 100 simulated single nucleotide polymorphism datasets. The lines at the top of each histogram bar show the standard error of the mean. PHASE was run with: burn-in 5,000 steps; thinning interval 100; number of samples 5,000. ELB was run with: burn-in 400,000 steps; thinning interval 1,000; number of samples 2,000; α = 0.01; ε = 0.01; γ = 0.01: HTYPER results are those reported after 20 independent runs, as recommended by its authors.
Figure 4.
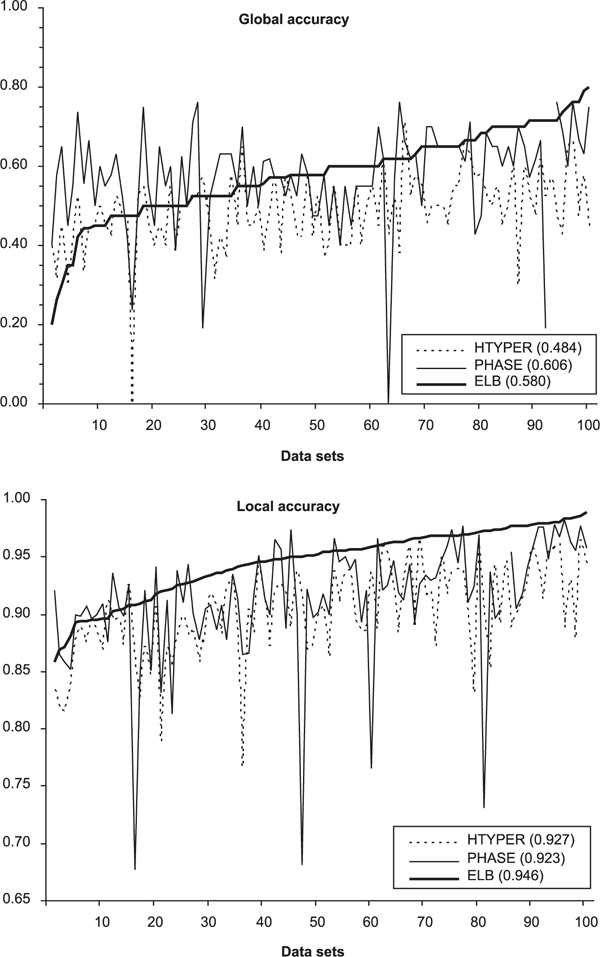
Global and local accuracies of ELB, PHASE (Ver. 1) and HTYPER in each of 100 datasets obtained by random pairings of 42 human male X chromosomes typed at 97 diallelic polymorphisms (predominantly single nucleotide polymorphisms). Datasets have been sorted by increasing values given by the ELB algorithm, separately for global and local accuracies. The mean values are reported within parentheses for each algorithm. ELB parameters: burn-in 300,000 steps; thinning interval 200; number of samples 10,000; α = 0.01; ε = 0.01; γ = 0.01. The one missing value for PHASE corresponds to an unexplained program crash. HTYPER results are those reported after 20 independent runs, as recommended by its authors.
Missing data
In handling missing data, the philosophy underpinning ELB is to ignore the affected loci rather than to impute missing data or to augment the space of possible genotypes. In the presence of missing data, the haplotype 'counts' nij and nij_1 are not necessarily integers: individuals with missing data at m loci within a current window of length L contribute 1 - m/L to nij (or nij_1) for each haplotype at which the remaining L - m loci match hij exactly (or with one mismatch).
Interpreting the output
After a burn-in period, the phase of each individual is recorded at fixed intervals, chosen so that the recorded phases are not too strongly correlated. For each individual, the most frequent among the recorded phases is chosen as the inferred phase. Its frequency in the output can be considered as an index of the quality of the inference. It is an approximate posterior probability under the stationary distribution of the ELB algorithm; however, because of the use of an adaptive window and the effects of the approximation inherent in (2), it does not seem possible to explicitly characterise this stationary distribution. The algorithm is designed so that its stationary distribution approximates one that can be characterised in terms of population frequencies of entire S-locus haplotypes, but the accuracy of this approximation is unknown and can only be assessed informally via simulation studies. The lack of an explicit likelihood formula means that the interpretation of output frequencies as posterior probabilities is less useful than in standard Bayesian settings. It is in this sense that ELB, like PHASE and HTYPER, is a pseudo-Gibbs sampler.
Data simulations for performance comparisons
Samples of SNP and STR genotypes were simulated under a coalescent model, which, in effect, assumes a large, random-mating population at demographic equilibrium. A modified version of the SIMCOAL program was used [26], allowing for arbitrary recombination between adjacent loci. For both SNP and STR markers, 100 datasets were generated for each of nine combinations of recombination and mutation parameters. Each dataset consisted of 100 simulated chromosome segments randomly paired into 50 genotypes. For both marker types, the total scaled recombination rates were R = 4Nr = 40, 100, and 200, where N is the population size. Assuming 1 cM = 1 Mb and N = 104 individuals, these values of R correspond to 100 kb, 250 kb and 500 kb.
For the SNP genotypes, mutation was simulated according to a finite-sites model, with a total scaled mutation rate of θ = 4Nu = 5, 10, or 20. Multiple mutations were thus allowed, but for compatibility with HTYPER, which only accepts diallelic data, all mutants were recorded as the same allele. The STR simulations employed 5, 10 and 20 equally-spaced loci and used the stepwise mutation model, with a scaled mutation rate of θ = 10 per locus, corresponding to a mutation rate of 2.5 × 10-4 per generation when N = 104 individuals. Details of the actual molecular diversity of the datasets can be found in Table 1.
Table 1.
Properties of simulated samples
| Case | Data type | θ 1 | R 2 | L 3 | π 4 |
|---|---|---|---|---|---|
| SNP | |||||
| 1 | 5 | 40 | 25 [14-39] | 4.8 | |
| 2 | 5 | 100 | 25 [13-44] | 4.8 | |
| 3 | 5 | 200 | 25 [10-38] | 4.9 | |
| 4 | 10 | 40 | 49 [33-69] | 9.9 | |
| 5 | 10 | 100 | 48 [31-70] | 9.6 | |
| 6 | 10 | 200 | 48 [30-61] | 9.6 | |
| 7 | 20 | 40 | 90 [65-127] | 18.3 | |
| 8 | 20 | 100 | 90 [65-109] | 18.7 | |
| 9 | 20 | 200 | 89 [65-119] | 18.5 | |
| STR | |||||
| 10 | 40 | 10 | 7.8 | ||
| 11 | 100 | 10 | 7.9 | ||
| 12 | 200 | 10 | 7.8 | ||
| 13 | 40 | 20 | 15.7 | ||
| 14 | 100 | 20 | 15.6 | ||
| 15 | 200 | 20 | 15.6 | ||
| 16 | 40 | 50 | 39.1 | ||
| 17 | 100 | 50 | 39.1 | ||
| 18 | 200 | 50 | 39.1 |
1. All simulations were performed in stationary random-mating populations. Samples consisted in 50 diploid individuals.
2. θ = 4Nu where N is the population size and u is the mutation rate per generation for the whole chromosomal segment.
3. R = 4Nr where r is the recombination rate for the whole chromosomal segment.
4. L is the number of polymorphic sites in the sample. For SNPs, we report the average number among 100 replicates, as well as the minimum and maximum numbers in brackets.
5. π is the average number of discordant sites between two gametes.
Two types of missing-data simulations were generated from the SNP datasets described above. In the first type of datasets, the proportion of missing data is kept low, but it is uniformly distributed: the genotype at any locus in any individual is missing with a common probability, which we set to 1, 2 or 4 per cent. In the second type of datasets, 40 of the 50 individuals were kept free of missing data and 5, 10 or 20 per cent of data were allowed to be missing in the remaining ten individuals.
Assessing the accuracy of phase reconstruction
Two statistics, one global and one local, were used to measure the accuracy of haplotype inference. The global accuracy statistic does not distinguish between a single phasing error and many such errors in an individual's reconstructed haplotype pair. It is defined as one minus the global error rate described by Stephens et al. [22], which is the proportion o ambiguous individuals whose haplotype pair is not recovered entirely correctly. The local accuracy index is the switch accuracy, introduced by Lin et al. [25], averaged over the ambiguous individuals in the sample. The switch accuracy for an individual is defined as 1 - W/(S - 1), where S denotes the number of heterozygous loci and W is the number of phase switches (equivalent to recombinations) required to obtain the correct haplotype pair from the reconstructed pair. For example, if the correct haplotypes at S = 3 heterozygous loci are ABC and abc, then the reconstructed haplotype pair AbC/aBc requires W = 2 phase switches to correct, and hence has a switch accuracy of 0, whereas Abc/aBC and ABc/abC both have a switch accuracy of 0.5
Results
The performance of ELB was compared, using both global and local accuracy measures, against that of HTYPER and PHASE (Ver. 1) in recovering the phases in the simulated SNP datasets described above. HTYPER was not included for the STR datasets, as it only handles diallelic markers. PHASE was not applied to the 50-locus STR datasets because of the prohibitive computation time. For the analysis of a sample of 50 genotypes typed at 10 STR loci, PHASE requires 7-8 hours on a 2.8 GHz Pentium 4 running Linux, and 20-21 hours for 20 STR genotypes. For the same analysis, ELB requires about 4 and 9 minutes, respectively.
SNP data
Figure 1 gives the means, and their standard errors, of the global and local accuracy statistics for each of the nine combinations of R and θ. In just 1 percent of datasets, PHASE stopped for unknown reasons and did not produce any results. HTYPER failed to find a legitimate solution for more than 6 per cent of datasets, the problem being inconsistencies between input and output files (ie heterozygous sites of some individuals in the input file were reported as homozygous in the output file). The results in Figure 1 are based, for each algorithm, only on the datasets for which the algorithm terminated successfully and produced a legitimate solution. Thus, for HTYPER, PHASE and ELB, respectively, the average number of datasets contributing to each mean in Figure 1 is 93, 99 and 100.
As expected, both the global and local accuracy of all algorithms is strongly and adversely affected by increasing the recombination rate, R. As the number of SNPs increases, with R fixed, there is higher LD and hence more accurate phase recovery between neighbouring SNP pairs, reflected in improved local accuracy for all algorithms. More phase calls must be correctly made in order to achieve an overall correct haplotype pair, however. The global accuracies of ELB and PHASE both decreased by about 5 to 10 per cent for each doubling of θ. For HTYPER, the global accuracy increases with θ in some cases.
PHASE has the highest global accuracy in four of the nine cases considered (not all differences are significant), including all of the low-recombination cases (R = 40). ELB has the highest accuracy in three cases, including two with high recombination (R = 200). HTYPER is best in two cases, both with high diversity (θ = 20). ELB has the highest local accuracy in eight of nine cases, while PHASE is best in the case with the lowest recombination and lowest diversity.
STR data
The mean global and local accuracy statistics of ELB and PHASE for the simulated STR datasets are shown in Figure 2. Once again, the overall performances of both PHASE and ELB algorithms are strongly affected by the amount of recombination between loci and, less markedly, by the number of loci. ELB is superior to PHASE in five of the six comparisons in terms of global accuracy and in all six comparisons in terms of local accuracy.
Figure 2.
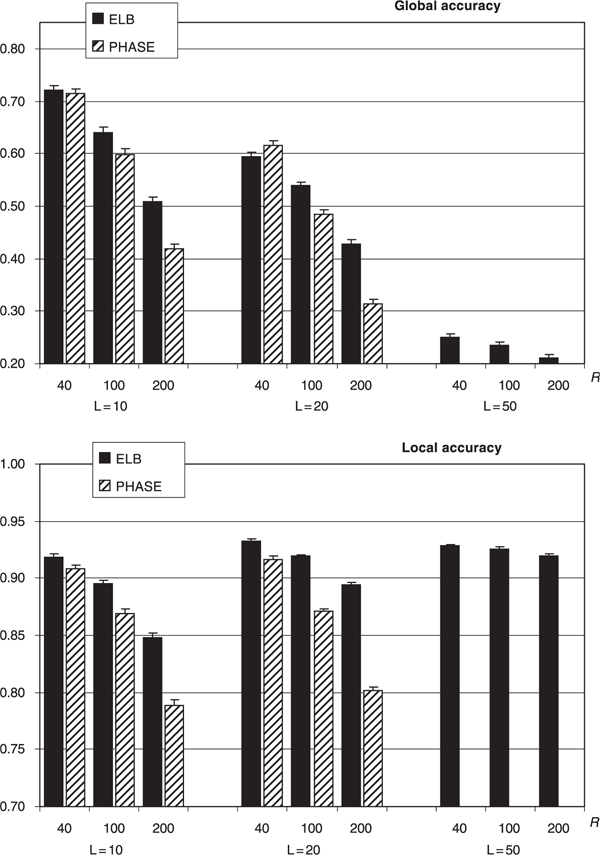
Mean (and standard error of the mean) global and local accuracy of ELB and PHASE (Ver. 1) algorithms when inferring gametic phase in 100 simulated short tandem repeat (microsatellite) datasets. PHASE was run with: burn-in 5,000 steps; thinning interval 100; number of samples 5,000. ELB was run with: burn-in 400,000 steps; thinning interval 1,000; number of samples 2,000; α = 0.01; ε = 0.1; γ = 0. HTYPER results are those reported after 20 independent runs, as recommended by its authors.
SNPs with missing data
In Figure 3A, the global and local accuracy of ELB is reported for datasets with a small fraction of missing data, uniformly distributed. Overall, there is a detectable, although small, degradation of performance with up to 2 per cent of missing data, this becomes more noticeable at 4 per cent, particularly when R = 100.
Figure 3.
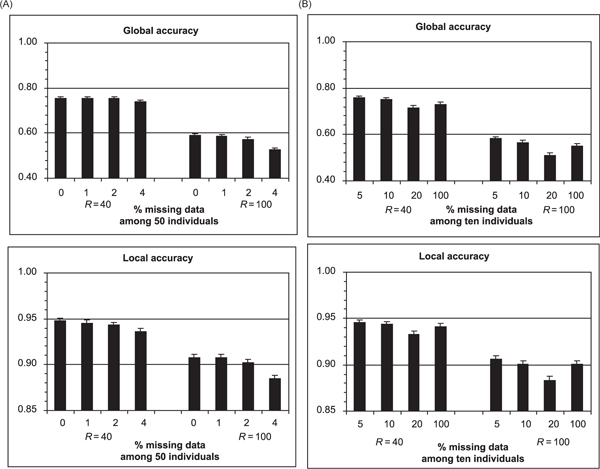
Mean (and standard error of the mean) global and local accuracy of ELB for single nucleotide polymorphism genotypes with varying amounts of missing data. (A) Data are missing at a uniform rate across all individuals. (B) Data are missing at a uniform rate among only ten individuals out of 50.
In Figure 3B, the estimation of gametic phase is compared in 40 individuals without missing data, when ten additional individuals having 5 per cent, 10 per cent and 20 per cent missing data are added. Figure 3B also reports results for the 'addition' of individuals having 100 per cent missing data, which corresponds to estimating the gametic phases only in the 40 individuals without missing data. We see here that adding individuals with up to 10 per cent missing data improves phase resolution in the 40 individuals without missing data, but when the proportion of missing data reaches 20 per cent, the additional individuals have a detrimental effect on phase resolution in the 40 individuals without missing data.
Application to real data
One hundred datasets were generated by randomly pairing 42 human male X chromosomes typed at 97 diallelic polymorphisms in a 193 kb low-recombination region. On average, the chromosome pairs differed at 31 sites. The chromosomes were drawn from 23 Afrikaner men, nine Ashkenazim, three British, three Swedes, three Greeks and one Italian. The polymorphisms were recorded as mismatches in comparisons with the Italian; they are overwhelmingly SNPs with occasional dinucleotide and small insertion/deletion polymorphisms [27].
In Figure 4, the global and local accuracy statistics are reported for each of the 100 datasets (corresponding to different random pairings of the same haplotypes). In agreement with the simulation results, ELB provides, on average, a slightly lower global accuracy than PHASE, although ELB is superior in almost half of the datasets (45 out of 100). Both of these algorithms are almost uniformly superior to HTYPER, which has the highest global accuracy in only two datasets. For local accuracy, it seems that ELB performs better than PHASE in 78 datasets, and better than HTYPER in 96 datasets.
Discussion
The ELB algorithm has been introduced for estimating gametic phase from multi-locus genotypes using a window that adapts to local levels of LD. ELB compares favourably with existing methods for reconstructing gametic phase, such as PHASE and HTYPER--especially for large genomic regions with a substantial total recombination rate. Like PHASE (but unlike HTYPER, which can only be applied to diallelic markers) ELB can be applied to either di- or multi-allelic markers.
Comparative performance of different algorithms
Analyses of simulated samples and of real data consisting of randomly-paired human X chromosome haplotypes, show that ELB leads generally to higher local accuracy than either PHASE or HTYPER, as measured by the switch index, for both SNP and STR data (Figures 1 and 2). It also shows higher global accuracy than PHASE for STR data. With high levels of recombination, corresponding to markers distributed over long chromosomal segments, global accuracy is often low (global accuracy lower than 50 per cent can be observed in Figures 1 and 2 when R = 200; corresponding to approximately 500 kb in humans) and local accuracy may be a more appropriate way to assess the success of phase estimation. The local accuracy of ELB is often the best among the three algorithms, even when its global accuracy is not the highest, which may be due to the local nature of the algorithm. This implies that when ELB fails to recover the entire haplotype pair of an individual, the reconstructed haplotypes tend to be closer to the correct pair than is the case for the other algorithms.
Another method (Stephens-Smith-Donnelly; SSD) recently introduced by Lin et al. [25], is a development of an algorithm first proposed by Stephens et al [22]. It was reported to produce better local accuracy than HTYPER, but SSD was especially optimised to perform well for this index, as it tries to predict the phase of a site mainly from the one immediately next to it. It was not possible to compare ELB with SSD, as no software was publicly available at the time of writing.
The choice of method to estimate gametic phase remains difficult, and no method appears to be uniformly superior in all scenarios. ELB is particularly well suited to large numbers of markers distributed over large chromosomal segments, both because of its local accuracy and its computational speed. Local accuracy is not affected by increasing the size of the genomic region, and the current implementation of ELB can readily handle several thousand SNPs in a single run. Thus, given sufficient marker density, regions of several Mb could be phased with high local accuracy, despite very low global accuracy.
Adaptive window size
A key feature of the authors' algorithm is that the phase of each heterozygous site in every individual is estimated based on information at surrounding sites, whose number varies depending on local levels of LD. For high levels of LD (low recombination), the size of windows will be on average larger than for low levels of LD, even though window size can vary greatly among sites for a given individual (results not shown). Therefore, the size of the window adapts automatically to the surrounding level of LD without the need to specify window size a priori. Even though the initial (minimum) size of a window depends on the number of sites separating the focal locus and neighbouring heterozygous sites, the size evolves towards a stationary value. Depending on the focal site and the level of polymorphism, the average stationary window size can vary from about five to more than 40 sites (results not shown). As mentioned previously, the authors' algorithm tracks two windows for each ambiguous locus, each one being initially limited at one extremity by the focal site and the other by one adjacent heterozygous site. In most cases, the size of these two chains converges towards the same value, but there are cases where they differ markedly over the whole estimation period. This may be due to poor mixing of the Gibbs chain, where adjacent heterozygous sites are very distant from the focal site.
Choice of parameters for ELB
ELB employs three parameters, α, ε and γ, which must be set by the user. The authors find (results not shown) that α = 0.01 works well for all data types. The parameter ε allows information to be incorporated about similar but not identical haplotypes in the phase inference process. The authors find that ε = 0.01 is suitable for SNP data, while ε = 0.1 is better for STRs, which is reasonable because two chromosomes differing by only one STR mutation step will tend to have a more recent common ancestor than two chromosomes discordant at a single nucleotide site. Large values of γ prevent the dynamic window from becoming too large, and consequently haplotype counts becoming too small, in regions with high LD. The authors find that it can be set to zero for STR data, irrespective of the overall level of recombination. For SNP data, γ = 0.01 was adopted for their simulation study and X chromosome data, but it should be noted that a larger value of γ may be required in low-recombination regions (and was used for the hot-spot simulation below).
While admittedly based on ad hoc features, ELB generally has better local accuracy than PHASE or HTYPER when the total recombination rate was large, and comparable performance for low or no recombination, while being fast and easy to implement and modify. For samples of 50 individuals, HTYPER typically takes seconds, ELB a few minutes, while PHASE takes several hours. ELB can thus analyse very large datasets in a reasonable amount of computing time, while maintaining a high degree of local phase accuracy.
Missing data and genotyping errors
Various methods have been adopted by other authors to address this problem [16,24]. Most approaches augment the space of possible genotypes using pseudo-data (inferred missing genotypes). ELB simply down-weights information from individuals having missing data within the current window. The present results (Figure 3) show that ELB is insensitive to low levels of missing data, but that including individuals with too much missing data (more than 10 per cent) can have detrimental effects on the reconstruction of the gametic phase in individuals without missing data.
Genotyping errors are another factor that can affect the performance of any algorithm aimed at reconstructing the gametic phase. Most genotyping errors, either due to allelic dropout or to the presence of null alleles, will result in an increased level of single-site homozygotes and will potentially introduce non-existing haplotypes in the sample. Even though all gametic phase inference procedures should be affected by the excess of single-site homozygotes due to genotyping errors, it is likely that algorithms allowing for the presence of haplotypes 'close' but not identical to those otherwise inferred in the rest of the sample (like PHASE or ELB) will be less sensitive to genotyping errors.
Blocks of linkage disequilibrium
The question remains open as to whether recombination can be safely ignored over small regions [28]. Recently, it has been suggested that the human genome is characterised by blocks within which there are high levels of LD and between which there is almost linkage equilibrium [21,29-32]. This pattern may be due to the presence of recombination hot-spots; however, randomly distributed crossovers can also lead to such a structure [33,34]. Moreover, these blocks are not observed in all populations and their size seems to be smaller in African populations, which may be linked to their different demographic history [21,35]. Also, the identification of the LD blocks boundaries is often made after gametic phases have been estimated [21,25]. It thus seems safer to assume that recombination can occur anywhere. The SSD algorithm [25] has been applied to LD blocks after an initial determination of their extent via an LD study. It was found that the accuracy in phase recovery was about 10 per cent higher when only considering SNPs within blocks, as compared to the whole SNP panel [25], (Table 5) possibly reflecting the importance of recombination hot-spots. Since the identification of these blocks cannot be fully dissociated from the estimation of the gametic phases, it seems desirable to allow for the presence of potential recombination hot-spots when estimating gametic phase.
In order to assess the performance of ELB in this setting, samples from a random-mating population were also simulated in the presence of a recombination hot-spot of infinite intensity. A series of 100 samples of 200 DNA sequences were simulated under a standard coalescent, each with mutation parameter θ = 2 and with no recombination within each sequence. Pairs of sequences were then ligated to produce 100 haplotypes that were then paired randomly into 50 genotypes. An average of 48 polymorphic sites were generated, and two haplotypes differed, on average, at about eight positions. Even though many sites are in complete LD for these datasets, the performance of the ELB algorithm (α = 0.01; ε = 0.01; γ = 0.5; global accuracy = 0.554, local accuracy = 0.886) is found here to be still slightly superior to PHASE (global accuracy = 0.520, local accuracy = 0.870), and, again, substantially better than HTYPER (global accuracy = 0.476, local accuracy = 0.846). It is likely that HTYPER and PHASE could be modified to allow explicitly for recombination. Since it appears that recombination levels may be quite heterogeneous along the chromosomes, this would seem to be a priority for future work on haplotype inference. It is likely that ELB can also be developed further, to estimate simultaneously the locations of recombination hot-spots and gametic phases.
Acknowledgements
We are grateful to Theo Anagnostopoulos, Pete Green and Francesco Giannelli of King's College London for kindly providing the human X haplotype data. Matthew Stephens kindly provided us with simulated samples and results on error rates from a previous study that helped us to calibrate our method. We are indebted to Jun Liu and Steve Qin for providing us with a version of their HTYPER software. We also thank two anonymous reviewers and Montgomery Slatkin for thoughtful comments on a previous version of the manuscript. GL was supported by a Swiss NSF grant No 31-56755.99 and LE by an EU grant No QLG2-CT-2001-00916. DB was supported in part by the UK Biotechnology and Biological Sciences (BBSRC) and Engineering and Physical Sciences (EPSRC) Research Councils grant E13397. The ELB procedure has been incorporated into version 3.0 of the Arlequin software, available from LE upon request.
References
- Beaudet L, Bedard J, Breton B. et al. 'Homogeneous assays for single-nucleotide polymorphism typing using AlphaScreen'. Genome Res. 2001;11:600–608. doi: 10.1101/gr.172501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman JI, Mindrinos MN, Oefner PJ. 'High-accuracy DNA sequence variation screening by DHPLC'. Biotechniques. 2000;29:1084–1090. doi: 10.2144/00295rr04. 1092. [DOI] [PubMed] [Google Scholar]
- Kidd KK, Morar B, Castiglione CM. et al. 'A global survey of haplotype frequencies and linkage disequilibrium at the DRD2 locus'. Hum Genet. 1998;103:211–227. doi: 10.1007/s004390050809. [DOI] [PubMed] [Google Scholar]
- Nicholson G, Smith AV, Jonsson F. et al. 'Assessing population differentiation and isolation from single-nucleotide polymorphism data'. J R Stat Soc B. 2002;64:695–715. doi: 10.1111/1467-9868.00357. [DOI] [Google Scholar]
- Stephens JC, Reich DE, Goldstein DB. et al. 'Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes'. Am J Hum Genet. 1998;62:1507–1515. doi: 10.1086/301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Varkonyi R, Cahinhinan N. et al. 'Haplotype diversity and linkage disequilibrium at human G6PD: Recent origin of alleles that confer malarial resistance'. Science. 2001;293:455–462. doi: 10.1126/science.1061573. [DOI] [PubMed] [Google Scholar]
- McGovern DP, van Heel DA, Ahmad T, Jewell DP. 'NOD2 (CARD15), the first susceptibility gene for Crohn's disease'. Gut. 2001;49:752–754. doi: 10.1136/gut.49.6.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian D, Thomas DC. 'Genome scan of complex traits by haplotype sharing correlation'. Genet Epidemiol. 2001;21(Suppl 1):S582–S587. doi: 10.1002/gepi.2001.21.s1.s582. [DOI] [PubMed] [Google Scholar]
- Rohde K, Fuerst R. 'Haplotyping and estimation of haplotype frequencies for closely linked biallelic multilocus genetic phenotypes including nuclear family information'. Hum Mutat. 2001;17:289–295. doi: 10.1002/humu.26. [DOI] [PubMed] [Google Scholar]
- Sabatti C, Risch N. 'Homozygosity and linkage disequilibrium'. Genetics. 2002;160:1707–1719. doi: 10.1093/genetics/160.4.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Daly MJ, Silverberg MS. et al. 'Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease'. Nat Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Taylor SL, Fullerton SM. et al. 'Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene'. Genome Res. 2000;10:1532–1545. doi: 10.1101/gr.146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas JA, Boehnke M, Gillanders E. et al. 'Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies'. Nat Genet. 2001;28:361–364. doi: 10.1038/ng582. [DOI] [PubMed] [Google Scholar]
- Clark A. 'Inference of haplotypes from PCR-amplified samples of diploid populations'. Mol Biol Evol. 1990;7:111–122. doi: 10.1093/oxfordjournals.molbev.a040591. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M. 'Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population'. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- Hawley ME, Kidd KK. 'HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes'. J Hered. 1995;86:409–411. doi: 10.1093/oxfordjournals.jhered.a111613. [DOI] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M. 'An E-M algorithm and testing strategy for multiple-locus haplotypes'. Am J Hum Genet. 1995;56:799–810. [PMC free article] [PubMed] [Google Scholar]
- Fallin D, Schork NJ. 'Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data'. Am J Hum Genet. 2000;67:947–959. doi: 10.1086/303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Ruano G, Kidd KK. 'The accuracy of statistical methods for estimation of haplotype frequencies: an example from the CD4 locus'. Am J Hum Genet. 2000;67:518–522. doi: 10.1086/303000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Pakstis AJ, Kidd KK, Zhao H. 'Comparisons of two methods for haplotype reconstruction and haplotype frequency estimation from population data'. Am J Hum Genet. 2001;69:906–914. doi: 10.1086/323622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H. et al. 'The structure of haplotype blocks in the human genome'. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. 'A new statistical method for haplotype reconstruction from population data'. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M. In: Handbook of Statistical Genetics. Cannings C, editor. John Wiley & Sons Ltd, New York, NY; 2001. pp. 179–212. [Google Scholar]
- Niu T, Qin ZS, Xu X, Liu JS. 'Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms'. Am J Hum Genet. 2002;70:157–169. doi: 10.1086/338446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Cutler DJ, Zwick ME, Chakravarti A. 'Haplotype inference in random population samples'. Am J Hum Genet. 2002;71:1129–1137. doi: 10.1086/344347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Novembre J, Schneider S. 'SIMCOAL: a general coalescent program for the simulation of molecular data in interconnected populations with arbitrary demography'. J Hered. 2000;91:506–510. doi: 10.1093/jhered/91.6.506. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos T, Green PM, Rowley G. et al. 'DNA vari ation in a 5-Mb region of the X chromosome and estimates of sex- specific/type-specific mutation rates'. Am J Hum Genet. 1999;64:508–517. doi: 10.1086/302250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Wall JD. 'Why is there so little intragenic linkage disequilibrium in humans?'. Genet Res. 2001;77:143–151. doi: 10.1017/s0016672301004967. [DOI] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF. et al. 'High-resolution hap-lotype structure in the human genome'. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Goldstein DB. 'Islands of linkage disequilibrium'. Nat Genet. 2001;29:109–111. doi: 10.1038/ng1001-109. [DOI] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M. 'Patterns of linkage disequilibrium in the human genome'. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- Stumpf MP. 'Haplotype diversity and the block structure of linkage disequilibrium'. Trends Genet. 2002;18:226–228. doi: 10.1016/S0168-9525(02)02641-0. [DOI] [PubMed] [Google Scholar]
- Phillips MS, Lawrence R, Sachidanandam R. et al. 'Chromosome-wide distribution of haplotype blocks and the role of recombination hot spots'. Nat Genet. 2003;33:382–387. doi: 10.1038/ng1100. [DOI] [PubMed] [Google Scholar]
- Zhang K, Akey JM, Wang N. et al. 'Randomly distributed crossovers may generate block-like patterns of linkage disequilibrium: An act of genetic drift'. Hum Genet. 2003;113:51–59. doi: 10.1007/s00439-003-0941-5. [DOI] [PubMed] [Google Scholar]
- Stumpf MP, Goldstein DB. 'Demography, recombination hotspot intensity, and the block structure of linkage disequilibrium'. Curr Biol. 2003;13:1–8. doi: 10.1016/S0960-9822(02)01404-5. [DOI] [PubMed] [Google Scholar]