

Abstract
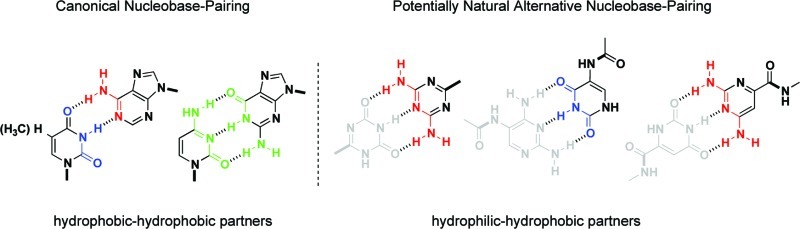
The formation of canonical base pairs through Watson–Crick hydrogen bonding sits at the heart of the genetic apparatus. The specificity of the base pairing of adenine with thymine/uracil and guanine with cytosine preserves accurate information for the biochemical blueprint and replicates the instructions necessary for carrying out biological function. The chemical evolution question of how these five canonical nucleobases were selected over various other possibilities remains intriguing. Since these and alternative nucleobases would have been available for chemical evolution, the reasons for the emergence of this system appear to be primarily functional.
While investigating the base-pairing properties of structural nucleic acid analogs, we encountered a relationship between the pKa of a series of nonstandard (and canonical) nucleobases and the pH of the aqueous medium. This relationship appeared to correspond with the propensity of these molecules to self-assemble via Watson–Crick-type base-pairing interactions. A simple correlation of the “magnitude of the difference between the pKa and pH” (pKa–pH correlation) enables a general prediction of which types of heterocyclic recognition elements form hydrogen-bonded base pairs in aqueous media. Using the pKa–pH relationship, we can rationalize why nature chose the canonical nucleobases in terms of hydrophobic and hydrophilic interactions, and further extrapolate its significance within the context of chemical evolution.
The connection between the physicochemical properties of bioorganic compounds and the interactions with their aqueous environment directly affects structure and function, at both a molecular and a supramolecular level. A general structure–function pattern emerges in biomolecules and biopolymers in aqueous media near neutral pH. A pKa – pH < 2 generally prompts catalytic functions, central to metabolism, but a difference in pKa – pH > 2 seems to result in the emergence of structure, central to replication. While this general trend is observed throughout extant biology, it could have also been an important factor in chemical evolution.
Introduction
The origins of chemical evolution are entangled with the events that led to the origins of life. Both physical and chemical processes must have played essential roles not only in prebiological and primitive biological processes but also during the emergence of the expanding and transforming sophisticated biochemical undertakings.1 The physicochemical properties of the molecules, as determined by their interaction with the geophysical and geochemical surroundings, expressed both at the individual and at the supramolecular level (and the gradation of complexes in between), would have dictated and selected what sort of constituents and assemblies would have been able to form, transform, adapt, and provide for chemical evolution to supervene.
Of these various geophysical and geochemical components, water, by far, would have had the most influence in determining the physicochemical properties of the molecules and in beginnings of chemical self-organization toward life’s origin. Not surprisingly, the presence of water is adjudged as one of the prerequisites for life to emerge. Two manifestations of the physicochemical properties of molecules, as dictated by their interaction with water, stand out: hydrophobicity and hydrophilicity. The interplay of these two characteristics plays a major role in determining the assemblage and, therefore, the function of (macro)molecules in extant biology. Such rudimentary interactions could have played a pivotal role also in the selection of molecules and in origins of chemical evolution.
This Account outlines the work that led to the realization of one such physicochemical property, the pKa (ionization or dissociation constant) of canonical and alternative nucleobases, which in turn dictates their hydrophobic and hydrophilic nature and thus their ability to function as base pairs in an aqueous medium. This work has its roots in (and is based on the fruits of) the seminal studies of Albert Eschenmoser and co-workers in the context of “chemical etiology of nucleic acid structure”,2 in which they examined the base-pairing properties of nucleic acids that were derived from various alternative sugar–phosphate backbones tagged with canonical nucleobases. The results of these extensive studies unambiguously demonstrated that Watson–Crick base pairing is not unique to the ribose–phosphate backbone of RNA/DNA but is also widespread among potentially natural alternative carbohydrate backbones. This conclusion, along with a host of other considerations, led Eschenmoser to expand this systematic investigation beyond the confines of the structural neighborhood of sugar backbone.3 The strategy entailed experimentally mapping the landscape of potentially primordial informational oligomers, without any restrictions on the structure types of backbones and recognition elements, provided these candidates satisfied the requirements of emerging under likely prebiotic conditions and entertained the potential for function (information).
Mapping the Landscape of Potentially Primordial Informational Oligomers
In this frame of reference, the base-pairing properties of a family of oligomers, derived from various dipeptides (e.g., Asp-Asp, Asp-Glu), deoxodipeptides, and dipeptoids tagged with the noncanonical recognition elements 2,4-diamino- and 2,4-dioxo-substituted triazines (1 and 2) and 5-aminopyrimidines (3 and 4), were investigated in the research group of Eschenmoser and the present author at Scripps (Figure 1).4
Figure 1.

The alternative recognition elements and backbones investigated in mapping the landscape of potentially primordial informational oligomers.
Base-pair-mediated duplex formation between these potentially natural oligomers and complementary RNA and DNA sequences was prevalent, but with an unexpected result: while 2,4-diaminotriazine-tagged backbones in all these series were found to be uniformly stronger base-pairing partners, the corresponding complementary dioxotriazine-tagged series exhibited (very) weak or no base pairing. Furthermore, the exact opposite behavior was observed in the series tagged with 2,4-disubstituted 5-aminopyrimidines. There, the 2,4-dioxo-5-aminopyrimidine tagged backbones formed duplexes that were stronger (base pairing) compared with the duplexes from corresponding complementary 2,4-diamino-5-aminopyrimidine tagged series. Thus, the 2,4-diamino substituent on a triazine scaffold was functioning as a good base-pairing partner, but the same 2,4-diamino substituent on a 5-aminopyrimidine chassis was inferior; this trend is the exact opposite for the 2,4-dioxo moiety. Such contrasting base-pairing behavior could not be rationalized by invoking strengths of hydrogen bonding, keto–enol tautomerism, stacking effects or change in base pairing or backbone axes, or the nature of the backbone to which these recognition elements were attached.
While searching for clues for this incongruity of the base-pairing behavior, the existence of a correlation between acid ionization/dissociation constants (pKa) of the complementary base-pairing partners and the base-pairing strength was noticed. The stronger base-pairing duplexes were formed whenever the difference between the pKa of the “acceptor” and the pKa of “donor” was at least 5 units or more; the smaller the ΔpKa between the donor and the acceptor was, the weaker was the strength of the duplex (Table 1). For example, the 2,4-diaminotriazine unit (pKa ≈ 4.5) paired strongly with deoxythymidine (pKa = 9.8), corresponding to ΔpKa of 5.3; however, the 2,4-diamino-5-aminopyrimidine unit (pKa ≈ 6.0) paired weakly with thymidine, where ΔpKa is 3.8. While the 2,4-dioxotriazine unit (pKa ≈ 7.2) paired weakly with deoxyadenosine (pKa 3.8), ΔpKa of 3.4, the 2,4-dioxo-5-aminopyrimidine unit (pKa ≈ 8.9) paired strongly with adenine; corresponding ΔpKa is 5.1.
Table 1. Comparison of Intra- and Intersystem Base-Pairing Strengths of Dodecameric Duplexes Containing Alternative Recognition Elements Tagged to Oligodipeptides as Determined by UV–Tm Melting Curvesa.
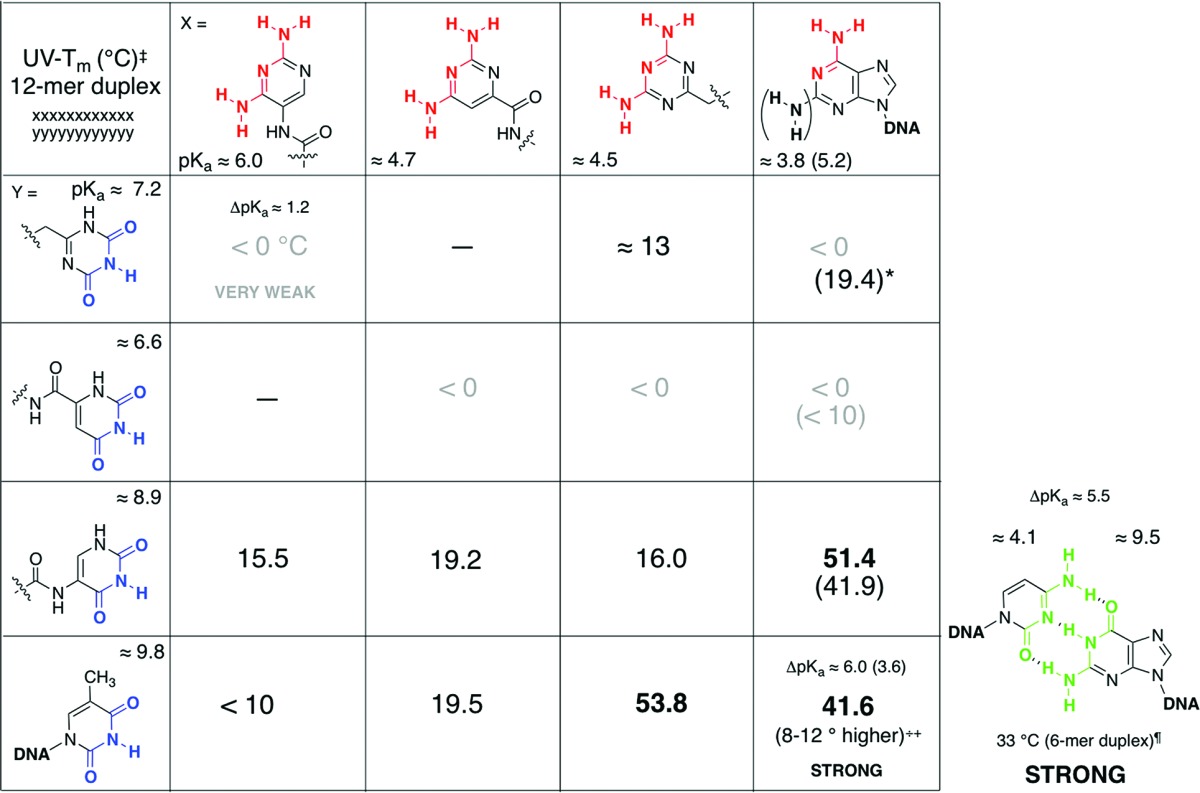
Taken from refs (4 and 5). ‡ indicates measured in 5 + 5 μM, 1 M NaCl, 10 mM Na2HPO4 buffer, pH 7.0, 0.1 mM Na2EDTA; * indicates triplex. ++ indicates as determined for poly-d(D):poly-d(T) versus poly-d(A):poly-d(T);13 ¶ indicates for a 6-mer homo-sequence duplex (25 + 25 μM) in 0.15 mM NaCl, 10 mM Na2HPO4 buffer, pH 7.0, 0.1 mM Na2EDTA;6 – indicates not measured.
We further investigated the base-pairing properties of orotic acid, 2,4-dioxopyrimidine-6-carboxylic acid (6), and its complementary base-pairing partner, 2,4-diaminopyrimidine-6-carboxylic acid (5), tagged to an oligodipeptide backbone derived from alternating units of l-aspartic acid and l-3-aminoalanine (asp-amAla, Figure 1).5 Oligodipeptide sequences tagged with 2,4-dioxo-derivative 6 were found to have extremely weak or no interactions with adenine-containing DNA/RNA sequences, while the corresponding complementary 2,4-diamino counterpart, 5, exhibited stronger duplex formation with thymine-tagged DNA/RNA sequences. In contrast to the 5-amino-pyrimidine (3 and 4) tagged series, here, the 2,4-dioxo moiety (as opposed to a 2,4-diamino unit) in a pyrimidine nucleus was the inferior pairing partner. As was the case with previous studies of alternative heterocycles,4 the property that is most consistent with the contradictory base-pairing behavior is the ionization constants; pKa of 2,4-dioxo derivative 6 is 6.6, and that of the corresponding 2,4-diamino derivative 5 is 4.7. The weak base-pairing behavior of orotamide 6 with adenine (ΔpKa ≈ 3) versus the stronger base pairing of 5 with thymine (ΔpKa ≈ 5) is consonant with the ΔpKa/base-pairing strength correlation (Table 1).
A comparison of the different pKa values of the alternative heterocycles with those of the complementary canonical nucleobases, with a side-by-side qualitative relationship with base-pairing strengths (Table 1) accentuates this correlation between the magnitude of ΔpKa of complementary partners with the base-pairing strength of the duplexes formed. While these correlations were largely consistent in their trends, they did not readily lend themselves to explanation.
While searching for explanations, the following sentence, “More precisely, the pK of an acid should be less than 4 and that of a base greater than 10 to ensure that only a small fraction of the compound remains in the un-ionized form at physiological pH. This general rule is not absolute”, from Frank Westheimer’s paper “Why Nature Chose Phosphates”,7 struck a chord. The pK values of 4 and 10 are exactly the same values around which the canonical nucleobases congregate; at physiological pH, the nucleobases are in the un-ionized form.8 The alternative nucleobases, whose pKa are more than 4 and less than 10, are probably in the “ionized form” under the measurement pH of 7.0.
Clues were obtained from the pH- and temperature-dependent UV-spectra behavior of orotamide derivatives. The pKa value of 6.6 of the orotamide unit in 6 correlated with the deprotonation of the N(1)-H proton (and not the N(3)-H proton) based on the shift in the λmax from 280 to 300 nm with increasing pH of the medium. Similar bathochromic shift was also observed in the temperature-dependent UV spectrum of the hexadecamer of 6 alone or in the presence of its pairing partner. All these facts indicated that the orotamide unit is deprotonated under the base-pairing measurement conditions and exists in its N(1)-anionic form. A similar λmax shift, from 232 to 255 nm pointing to a N(1 or 5)H deprotonation, was also observed in the temperature-dependent UV spectra of 2,4-dioxo-triazine tagged oligo-dipeptides; such a behavior was absent in the 2,4-dioxo-5-aminopyrimidine series suggesting the absence of deprotonation of N(1)-H in 4.
How exactly the deprotonation of the N(1 or 5)-H proton in 2 affects the base-pairing strength was not immediately clear, since it could be argued that it is the N(3)-H of 2,4-dioxo-triazine and -5-aminopyridmines that is involved in the hydrogen bonding. However, when the deprotonation and protonation (or the lack of it) of the recognition elements was linked via their corresponding “ionized” or “un-ionized” state in an aqueous environment with their hydrophilicity and hydrophobicity (and not solely with hydrogen-bonding capability), a qualitative understanding of the correlation between the base-pairing strength of the duplex, pKa of the heterocycle, and the pH of the aqueous medium emerged.
In the specific examples considered above, when the pKa of the heterocycles (2, 3, and 6) is close to the pH of the aqueous medium, they become deprotonated or protonated (ionized) and, therefore, hydrophilic (Figure 2). This, in turn, increases the solvation of the heterocycles by the aqueous medium (via polar interactions and hydrogen bonding), drastically hindering their ability to interact with their base-pairing partner (via stacking interactions and hydrogen bonding), weakening the duplex. With increasing number of such hydrophilic units, a “breaking point” is reached where no duplex formation is possible.9 On the other hand, when the pKa of the heterocycles (1, 4, and 5) is far removed from the pH of the aqueous medium, the heterocycles remain in their un-ionized form and, therefore, are hydrophobic (Figure 2). This coerces the heterocycles to minimize their interaction with the aqueous medium and congregate with their base-pairing partner (reinforced by stacking interactions10 and specific hydrogen bonding), leading to stronger base pairing. In other words, here, when the difference between the pKa of the heterocycle and the pH of the aqueous medium (pKa – pH < 2) is smaller, the base-pairing strength is expected to be weaker; conversely, when the difference between the pKa of the heterocycle and the pH of the aqueous medium is larger (pKa – pH > 2), the base-pairing strength is anticipated to be stronger, all other things being equal.
Figure 2.

Juxtaposition of the pKa values of the canonical nucleobases with those of the potentially natural alternative heterocycles, correlating with the degree of solvent interaction of the charged (hydrophilic) and uncharged (hydrophobic) nucleobases (in neutral aqueous conditions).
General Applicability of the pKa–pH Relationship and Implications
The relationship between pH of the medium, pKa of the heterocycle, and its base-pairing capacity is widely documented and has been exploited by various research groups largely in the context of mismatch discrimination and triplex formation, pertaining to diagnostic and antisense applications.11 The “pKa–pH rule” can be useful in understanding the base-pairing behavior of many of these nucleobase variations (Figure 3).12
Figure 3.

Selected nucleobase pairings illustrating the general applicability of the magnitude of the difference between pKa of the heterocycle and the pH of the medium (“pKa–pH rule”) in explaining the effect on thermal stability of the duplexes.
A classic example is the 2,6-diaminopurine–thymine base pair, which is weaker compared with guanine–cytosine (though both base pairs have three hydrogen bonds) and in some instances not as stable as A–T base pair.13 The higher basicity of 2,6-diaminopurine, pKa 5.2, leads to more protonation at neutral pH, increasing its hydrophilicity and interaction with water, thereby impeding its interaction with its complementary partner.14
Another instructive example is the strong base pairing between two purines, guanine (pKa 9.5) and isoguanine (pKa 9.2) in aqueous medium.15 Here ΔpKa < 1, which according to the original ΔpKa-correlation4 predicts weak or no base pairing;16 however, the experimental observation is in concordance with what would be expected from pKa–pH criterion (here >2). This case illustrates clearly that the ΔpKa criterion that was used before4 is actually a special manifestation of the pKa–pH correlation.17
This analysis is also valid for other noncanonical nucleobases and is exemplified here by one of the size-expanded guanine (dxG)–cytosine base pair, which is destabilizing compared with the inverse expanded cytosine (dxC)–guanine couplet (even though they both have three-hydrogen bonds and similar dimensions). When the pKa of dxG (7.2) is taken into consideration, this contrasting behavior falls in line with the pKa–pH/base pairing correlation.18
There are informative exceptions to the pKa–pH/base pairing correlation in aqueous medium, such as the nucleobases that do form self- and cross-base pairs upon protonation, for example, cytosine, adenine, and 8-aminoguanosine.19 Herein, protonation is needed for complementarity to be fulfilled, and therefore, the pKa of the nucleobase must be close to the pH of the medium. There are other cases of halogenated nucleobases20 that have pKa–pH < 2 but still base pair strongly, perhaps by maintaining their hydrophobicity (due to the nature of the substituents).
Among the majority of the alternative heterocycles and canonical nucleobases (investigated at neutral pH), in order to become ionized, the ones that have pKa values lower than pH of the medium are to be protonated, while the ones with pKa values higher are to be deprotonated; here, the smaller the difference between the pKa of the molecule and the pH of the medium, the greater would be the degree of ionization, and a weaker duplex formation is expected (and observed). However, there are instances where it is the other way around, with the exact opposite expectations and results. An example is xanthosine (pKa ≈ 5.5), which is present as a monoanion at neutral pH and has been shown to pair more strongly with adenine when the pH is lowered from 7.5 to 5.5;21 this is due to the increasing hydrophobicity of xanthosine when the pKa of nucleobase and the pH of medium become equal.16
There are examples that suggest there may be boundaries to the pKa–pH correlation (Figure 4): at one end of the spectrum there is 7-deazaguanine (pKa ≈ 10.3) and 3-deazaguanine (12.3), which exhibit decreased duplex stability although they have a pKa – pH > 2.22 At the other end, 5-aza- and 6-azacytosine (pKa of 2.6 and 2.8) are also known to decrease duplex stability.23 Thus, when the pKa of the nucleobase is below 3 or higher than 10, the base-pairing capability with complementary partner is compromised, perhaps, also due to an increase in hydrophilic character of the heterocycles. Such a suggestion finds support in the decreased lipophilic behavior of 3-deazaguanine and 5-aza- and 6-azacytosine nucleosides (compared with the parent canonical nucleosides).24 These limited examples indicate that there could be upper and lower limits to the pKa–pH correlation in this specific scenario, namely, 3.5 > pKa – pH > 2. Therefore, in an aqueous medium at near neutral pH, there appears to be a narrow window of pKa values of heterocycles, 3.5–4.5 and 9–10, wherein duplex formation mediated by base pairing seems to be optimal (Figure 4).
Figure 4.

A qualitative landscape associating the pKa of canonical nucleobases and their close structural analogs (in neutral aqueous medium) with their relative hydrophobicity and relative base-pairing strength (in oligomeric duplexes, as judged by thermal stability). In this context, the canonical nucleobases straddle a narrow pKa range and represent an “optimum” in terms of base-pairing capabilities.
In arguing as to “Why Nature Chose Phosphates”, Westheimer has emphasized the “importance of being ionized”, concluding that the ionization of phosphates has important consequences in an informational polymer as a linker under physiological conditions by (a) solubilizing the polymer in an aqueous medium, (b) stabilizing the backbone against hydrolysis, and (c) preventing the leakage of the charged polymer from the confines of the bilayer membrane.7 If one could reason, “why nature chose the canonical nucleobases”, then a significant part of the answer would be “the importance of being not ionized” under physiological conditions, the exact opposite of phosphate. The significance of the pKa values of the canonical nucleobases being less than 4 and greater than 9, correlating to their neutral forms and their ability to form Watson–Crick base pairing, has been pointed to by others in the context of structural studies.25
Among the whole range of canonical and noncanonical nucleobases, the complementary base-pairing sets as exemplified by A–T/U and G–C seem to be the most advantageous set of informational–recognition elements, based on their physicochemical properties as expressed and manifested in an aqueous environment at near neutral pH,26 satisfying the narrow window of pKa (3.5–4.5 and 9–10) where they are able to function as base pairs (Figure 4). Considering the potentially natural alternatives where one of the heterocycles is able to pair, the corresponding complementary partner seems to be inefficient. These two observations, more importantly, seem to be (largely) independent of the nature of the (plethora of) backbones to which the canonical and alternative nucleobases are attached, strengthening the implication that, in chemical evolution, the composition and structure of the recognition elements could have played a more influential role in Nature’s choice of an informational system than the backbone.4b
When pKa – pH < 2, It Leads to Catalytic Function Coupled with Structural Diversity
Among the major canonical RNA (DNA) nucleobases, only A and C are known to shift their pKa’s toward near neutrality by virtue of the unique environments created by RNA folding and strategic positioning of the nucleobases and the sugar–phosphate backbone, in conjunction with cations.27,28 This results in the protonation of A (shifted pKa 5.5–7.5) and C (shifted pKa 5.9–7.2) at near neutral pH;29 shifts in pKa of G and U (or T) do not seem to occur.28 For example, it is known that cytosine acquires a histidine-like pKa, which is optimal for general acid–base chemistry, implying that the catalytic sites in ribozymes may have a greater chance of possessing nucleobases that have shifted their pKa values; and not surprisingly most of the catalytic sites in smaller ribosomes seem to involve charged bases.30
Numerous examples of charged nucleobases and their contribution to (a) catalysis in ribozymes and (b) structural diversity stemming from non-Watson–Crick base-pairing modes are available from contemporary biology. Protonation is also known to increase the number of possible pairing modes leading to structural diversity.31 In Nature, the same nucleobases (A and C) that are used for both structural and informational purposes (Watson–Crick mode) in DNA and RNA are also used, in addition, for catalytic and functional purposes (non Watson–Crick mode) but in RNA.32
Thus, employing this phenomenon of perturbation/nonperturbation of pKa of nucleobases, Nature seems to have transformed a chameleonic set of “phenetic and genetic” alphabets (in RNA) to a pedantic and restricted set of “genetic only” alphabets (in DNA/RNA).33
pH of Early Oceans and Implications in the Context of pKa–pH Correlations
The pH of the early hydrosphere is thought to lie more on the acidic side (ranging from pH 4.5 to 6.0).34 If self-replicating oligomeric systems could have existed in their current canonical forms under acidic pH conditions, then the base-pairing rules would be expected to be different from what they are today, with the caveat that this acidic aqueous environment would have also been capable of harboring chemical evolution. Then, protonated base pairs might have been the rule, rather than the exception. It is widely accepted that the free energy minimum for folding in most RNAs occurs around pH 5.5. There are some ribozymes and aptamers that function at a pH as low as 4.0.29c,35 Considering these in the context of the RNA world, it is beguiling to consider whether RNA acted in these early stages as a better catalyst than replicator or followed a different set of base-pairing rules (e.g., Hoogsteen mode) for replication.
pKa (of Nucleobases) As a Driver of Congregation and As a Modulator of Emergent Properties
The magnitude of the pK–pH difference does not only influence the way nucleobases interact with themselves in aqueous surroundings;8 when amplified at a supramolecular level, it also modulates the expression of the emerging (chemical and physical) properties and the functioning of a bioassemblage.
In this context, the chemical constituents of RNA/DNA present an interesting contrast: while pKa – pH is greater than 2 for the canonical nucleobases rendering them hydrophobic at neutral pH, the phosphate diester backbone is ionized and exists as a polyanion. This creates a diverging hydrophobic and hydrophilic zone within the same polymer (akin to the phospholipids). In an aqueous environment, the hydrophobic zones tend to aggregate, sequestering themselves from interactions with water molecules, while the hydrophilic side is exposed to water (akin to lipid bilayer). Therefore, RNA/DNA duplexes could be viewed, superficially, as a “polynucleotide bilayer” (Figure 5) with a higher level of sophistication in terms of structure, information storage, and function (compared with the lipid bilayer).
Figure 5.

A balance between the magnitudes of hydrophobic interior versus hydrophilic exterior, regulating optimal base-pairing strength, may be a reason for the selection of a purine–pyrimidine (over a purine–purine or a pyrimidine–pyrimidine) base pair.
This hydrophobic interior, along with the well-known role played by water36 and a charged hydrophilic (versus uncharged hydrophobic37) backbone, enables the two polynucleotide strands to hybridize in a robust and dynamic manner. Such an assemblage is fine-tuned for optimal base pairing between complementary nucleobases and mismatch discrimination, sensed by weakening of the duplex (more so in DNA than in RNA38).
Changing the “internal hydrophobicity” of RNA/DNA (e.g., by varying the length of the bases) would affect not only the base-pairing strength but the also the sensitivity to base-pairing mismatch. This is exemplified by the studies of Kool and co-workers in their extended nucleobases, wherein a single mismatch in x-DNA leads to overall higher strength of the mismatched duplex of x-DNA compared with that of a mismatch in DNA; in the former case there is about a 25–44% drop in thermal stability compared with almost a 32–58% in the latter.39 A similar trend is also observed for the y-DNA series.40 Conversely, shortening of the bases (e.g., pyrimidine–pyrimidine base pairs) would be expected to weaken duplex strength. However, there may not be meaningful discrimination of base-pairing partners if the duplex strengths are compromised beyond a limit. Thus, the selection of a purine–pyrimidine base pair (over a purine–purine or pyrimidine–pyrimidine) may be a reflection of the optimization (and not maximization) of base-pairing strength,2 a result of the balancing act between strength of the assemblage and its sensitivity to mismatch or defects by control of the internal hydrophobicity versus external hydrophilicity (Figure 5). Such a cooperative effect and the tendency to minimize exposure of hydrophobic surfaces may also be at play in the selection of the Watson–Crick (over Hoogsteen and other) mode of pairing within the confines of a ribofuranosyl-phosphodiester backbone.41
The hydrophobic–hydrophilic interactions, arising from the fundamental physicochemical properties of molecules in an aqueous environment, are known to have important consequences in extant biology. The hydrophobic effect is a well-recognized driving force for assemblage42 and concentration43 in an aqueous medium. That these interactions would have also been consequential in the origins of chemical evolution has been illustrated, here, by considering the pKa of the canonical nucleobases, in the context of chemical etiology of nucleic acid structure.2
Acknowledgments
I am indebted to Professor Eschenmoser for his continuing inspiration, stimulation, guidance, support, generosity, and encouragement. This work was supported by the Skaggs Research Foundation and NASA Astrobiology: Exobiology & Evolutionary Biology (Grant NNX07AK18G). Continuing support is jointly provided by NSF and NASA Astrobiology Program, under the NSF Center for Chemical Evolution, Grant CHE-1004570.
Biography
Ramanarayanan Krishnamurthy received his B.Sc. in chemistry from Vivekananda College (University of Madras) in 1984 and M.Sc. in Chemistry (1986) from the Indian Institute of Technology, Bombay. He obtained his Ph.D. from The Ohio State University, Columbus, OH, in 1992 under the guidance of Professor David Hart. Captivated by a lecture given by Professor Albert Eschenmoser (OSU, 1990), he did his postdoctoral work at Swiss Federal Institute (ETH), Zürich, with Professor Eschenmoser. Following a NASA-NSCORT fellowship (1994–1996) with Professor Gustaf Arrhenius at Scripps Institution of Oceanography, UCSD, La Jolla, CA, he rejoined Professor Eschenmoser at the Skaggs Institute of Chemical Biology at The Scripps Research Institute, La Jolla, in 1996, spawning a nearly 13-year research collaboration. He is currently an Associate Professor of Chemistry at TSRI applying synthetic organic chemistry to understand the chemistry behind the origins of life and, in the process, developing molecular tools to probe biology and novel molecular leads for chemical therapeutics.
References
- a Budin I.; Szostak J. W. Expanding Roles for Diverse Physical Phenomena During the Origin of Life. Annu. Rev. Biophys. 2010, 39, 245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenmoser A. Chemical Etiology of Nucleic Acid Structure. Science 1999, 284, 2118–2124. [DOI] [PubMed] [Google Scholar]
- a Eschenmoser A. The TNA-Family of Nucleic Acid Systems: Properties and Prospects. Origins Life Evol. Biospheres 2004, 34, 277–306. [DOI] [PubMed] [Google Scholar]; b Eschenmoser A. The Search for the Chemistry of Life’s Origin. Tetrahedron 2007, 63, 12821–12844. [Google Scholar]
- a Mittapalli G. K.; Reddy K. R.; Xiong H.; Munoz O.; Han B.; De Riccardis F.; Krishnamurthy R.; Eschenmoser A. Mapping the Landscape of Potentially Primordial Informational Oligomers: Oligodipeptides and Oligodipeptoids Tagged with Triazines as Recognition Elements. Angew. Chem., Int. Ed. 2007, 46, 2470–2477. [DOI] [PubMed] [Google Scholar]; b Mittapalli G. K.; Osornio Y. M.; Guerreo M.; Reddy K. R.; Krishnamurthy R.; Eschenmoser A. Mapping the Landscape of Potentially Primordial Informational Oligomers: Oligodipeptides Tagged with 2,4-Disubstituted 5-Aminopyrimidines as Recognition Elements. Angew. Chem., Int. Ed. 2007, 46, 2478–2484. [DOI] [PubMed] [Google Scholar]
- Zhang X. J.; Krishnamurthy R. Mapping the Landscape of Potentially Primordial Informational Oligomers: Oligo-dipeptides Tagged with Orotic Acid Derivatives as Recognition Elements. Angew. Chem., Int. Ed. 2009, 48, 8124–8128. [DOI] [PubMed] [Google Scholar]
- Hunziker J.; Roth H.-J.; Böhringer M.; Giger A.; Diedrichsen U.; Göbel M.; Krishnan R.; Jaun B.; Leumann C.; Eschenmoser A. Warum Pentose- und nicht Hexose-Nucleinsäuren?. Helv. Chim. Acta 1993, 76, 259–352. [Google Scholar]
- Westheimer F. H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- Sanger W.Principles of Nucleic Acid Structure; Cantor C. R., Ed.; Springer-Verlag: New York, 1984; pp 105–140 and references cited therein. [Google Scholar]
- The ionized monomer or oligomer (at micromolar concentrations) has to compete with 55 M water (a factor of ∼108).
- Uncharged monomeric nucleobase derivatives associate, not by hydrogen bonding, but rather by stacking in aqueous medium:Ts’o P. O. P.; Melvin I. S.; Olson A. C. Interaction and Association of Bases and Nucleosides in Aqueous Solutions. J. Am. Chem. Soc. 1963, 85, 1289–1296. [DOI] [PubMed] [Google Scholar]
- Frank Seela’s group has methodically documented the effects of nucleobase modifications on duplex stability; for example, see:Peng A.; Li H.; Seela F. pH-Dependent Mismatch Discrimination of Oligonucleotide Duplexes Containing 2′-Deoxytubercidin and 2- or 7-Substituted Derivatives: Protonated Base Pairs Formed between 7-Deazapurines and Cytosine. Nucleic Acids Res. 2006, 34, 5987–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an in-depth review, see:Herdewijn P. Heterocyclic Modifications of Oligonucleotides and Antisense Technology. Antisense Nucleic Acid Drug Dev. 2000, 10, 297–310. [DOI] [PubMed] [Google Scholar]
- Caution must be exercised since a large majority of these examples have been investigated with only one insertion/replacement in a duplex.
- Cheong C.; Tinoco I. Jr.; Chollet A. Thermodynamic Studies of Base Pairing Involving 2,6-Diaminopurine. Nucleic Acids Res. 1988, 16, 5115–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “Charged” purines have less tendency to unstack compared with “charged” pyrimidines; see page 132 of ref (8).
- Groebke K.; Hunziker J.; Fraser W.; Peng L.; Diedrichsen U.; Zimmerman K.; Holzner A.; Leumann C.; Eschenmoser A. Warum Pentose- und nicht Hexose-Nucleinsäuren? (Purin-Purin)-Basenpaarung in der homo-DNS-Reihe: Guanin, Isoguanin, 2,6-Diaminopurin und Xanthin. Helv. Chim. Acta 1998, 81, 375–474. [Google Scholar]
- The well-studied hydrogen-bond mediated complex formation in non-polar organic medium has led to the “pKa-match” rule, where the difference between the pKa of the acceptor and the donor is small for strong hydrogen bonding to occur. Considering that in both aqueous and organic medium, the important factor seems to be the extent of the neutral character of the complementary partners that dictates the strength of hydrogen bond mediated association, there is no apparent contradiction. On the other hand, if ΔpKa between acceptor and donor is very large (≥7) (e.g., in amino acids and proteins), it leads to salt-bridge formation, which may also contribute to stability by electrostatic and hydrogen bond interactions:Donald J. E.; Kulp D. W.; DeGrado W. F. Salt Bridges: Geometrically Specific, Designable Interactions. Proteins: Struct., Funct., Bioinf. 2011, 79, 898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The “magnitude of ΔpKa/base-pairing strength correlation” rule would be applicable only if the pKa of the acceptor and donor are on opposite (and not on the same) side of the pH value of the medium.
- Liu H.; Lynch S. R.; Kool E. T. Solution Structure of xDNA: A Paired Genetic Helix with Increased Diameter. J. Am. Chem. Soc. 2004, 126, 6900–6905. [DOI] [PubMed] [Google Scholar]; This may be also the case for other extended purines such as 2,7-diamino-1,8-napthyridines (pKa 6.8):Minakawa N.; Kuramoto K.; Hikishima S.; Matsuda A. Four Hydrogen-Bonding Motifs in Oligonucleotides. ARKIVOC 2006, (vii), 326–337. [Google Scholar]
- a Gehring K.; Leroy J. L.; Gueron M. A Tetrameric DNA Structure with Protonated Cytosine-Cytosine Base Pairs. Nature 1993, 363, 561–565. [DOI] [PubMed] [Google Scholar]; b Hattori M.; Frazier J.; Miles H. T. Ordered Forms of 5′-8-Aminoguanylic Acid. Biopolymers 1975, 14, 2095–2106. [DOI] [PubMed] [Google Scholar]; c Engelhart A. E.; Morton T. H.; Hud N. V. Evidence of Strong Hydrogen Bonding by 8-Aminoguanine. Chem. Commun. 2009, 647–649. [DOI] [PubMed] [Google Scholar]
- Luyten I.; Herdewijn P. Hybridization Properties of Base-Modified Oligonucleotides within the Double and Triple Helix Motif. Eur. J. Med. 1998, 33, 515–576and references cited therein. [Google Scholar]
- Eritja R.; Horowitz D. M.; Walker P. A.; Ziehler-Martin J. P.; Boosalis M. S.; Goodman M. F.; Itakura K.; Kaplan B. E. Synthesis and Properties of Oligonucleotides Containing 2′-Deoxynebularine and 2′-Deoxyxanthosine. Nucleic Acids Res. 1986, 14, 8135–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Seela F.; Driller H. Solid-Phase Synthesis of the Self Complementary Hexamer d(C7GpCpc7GpCpc7GpC) via the O-3′-Phosphoramidite of 7-Deaza-2′-guanosine. Nucleic Acids Res. 1985, 13, 911–926. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seio K.; Sasami T.; Tawarada R.; Sekine M. Synthesis of 2′-O-Methyl-RNAs Incorporating a 3-Deazaguanine, and UV Melting and Computational Studies on Its Hybridization Properties. Nucleic Acids Res. 2006, 34, 4324–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Garcia R. G.; Brank A. S.; Christman J. K.; Marquez V. E.; Eritja R. Synthesis of Oligonucleotide Inhibitors of DNA (Cytosine-C5) Methyltransferase Containing 5-Azacytosine Residues at Specific Sites. Antisense Nucleic Acid Drug Dev. 2001, 11, 369–378. [DOI] [PubMed] [Google Scholar]; b Sanghvi Y. S.; Hoke G. D.; Freier S. M.; Zounes M. C.; Gonzalez C.; Cummins. L.; Samsor H.; Cook P. D. Antisense Oligodeoxynucleotides: Synthesis, Biophysical and Biological Evaluation of Oligodeoxynucleotides Containing Modified Pyrimidines. Nucleic Acids Res. 1993, 21, 3197–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Seela F.; Debelak H.; Andrews L.; Beigelman L. Synthesis and Enzymic Hydrolysis of Oligoribonucleotides Incorporating 3-Deazaguanosine: The Importance of the Nitrogen-3 Atom of Single Conserved Guanosine Residues on the Catalytic Activity of the Hammerhead Ribozyme. Helv. Chim. Acta 2003, 86, 2726–2740. [Google Scholar]; b Romanova D.; Novotny L. Chromatographic Properties of Cytosine, Cytidine and Their Synthetic Analogs. J. Chromatogr. B 1996, 675, 9–15. [DOI] [PubMed] [Google Scholar]
- a Ts’o P. O. P. The Hydrophobic-Stacking Properties of the Bases in Nucleic Acids. Ann. N.Y. Acad. Sci. 1969, 153, 785–804. [DOI] [PubMed] [Google Scholar]; b Ts’o P. O. P.Basic Principles in Nucleic Acid Chemistry Ts’o P. O. P., Ed.; Academic: New York, 1974; Vol. 45, Chapter 6. [Google Scholar]; c Pohorille A.; Burt S. K.; MacElroy R. D. Monte Carlo Simulation of the Influence of Solvent on Nucleic Acid Base Associations. J. Am. Chem. Soc. 1984, 106, 402–409. [DOI] [PubMed] [Google Scholar]; d Dang L. X.; Kollman P. A. Molecular Dynamics Simulations Study of the Free Energy of Association of 9-Methyladenine and 1-Methylthymine Bases in Water. J. Am. Chem. Soc. 1990, 112, 503–507. [Google Scholar]
- Strict control of pH is maintained in the nucleus compared with the cytoplasm. For example, see:Sesek O.; Bolard J. Nuclear pH Gradient in Mammalian Cells Revealed by Laser Microspectrofluorimetry. J. Cell Sci. 1996, 109, 257–262. [DOI] [PubMed] [Google Scholar]
- Lippert B. Ligand-pKa Shifts through Metals: Potential Relevance to Ribozyme Chemistry. Chem. Biodiversity 2008, 5, 1455–1474. [DOI] [PubMed] [Google Scholar]
- Wilcox J. L.; Ahluwalia A. K.; Bevilacqua P. C. Charged Nucleobases and Their Potential for RNA Catalysis. Acc. Chem. Res. 2011, 44, 1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nakano S.; Chadalavada D. M.; Bevilacqua P. C. General Acid-Base Catalysis in the Mechanism of a Hepatitis Delta Virus Ribozyme. Science 2000, 287, 1493–1497. [DOI] [PubMed] [Google Scholar]; b Wilson T. J.; Li N. S.; Lu J.; Frederiksen J. K.; Piccirilli J. A.; Lilley D. M. Nucleobase-Mediated General Acid-Base Catalysis in the Varkud Satellite Ribozyme. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burke D. H.; Hoffman D. C. A Novel Acidophilic RNA Motif That Recognizes Coenzyme A. Biochemistry 1998, 37, 4653–4663. [DOI] [PubMed] [Google Scholar]
- This leads to speculation whether (a) the selection of CCA sequence in tRNA (at the aminoacyl end) and (b) the central roles played by ATP, AMP, and cyclic-AMP could also have a connection to the malleable pKa’s of the adenine and cytosine. For example, increasing the positive charges on A or C (by protonation) increases their electron-withdrawing nature, which would make the electrophilic centers attached to these nucleosides more reactive towards nucleophiles.
- pH changes can also affect the percentage of keto–enol forms of the nucleobases, which in turn can affect the base-pairing modes: page 114 of ref (8).
- DNAzymes have been invented and are just as versatile; however, Nature seems not to have evolved one (so far):Breaker R. R.; Joyce G. F. A DNA Enzyme That Cleaves RNA. Chem. Biol. 1994, 4, 223–229. [DOI] [PubMed] [Google Scholar]
- a Geyer R. C.; Battersby T. R.; Benner S. A. Nucleobase Pairing in Expanded Watson-Crick-like Genetic Information Systems. Structure 2003, 11, 1485–1498. [DOI] [PubMed] [Google Scholar]
- a Walker J. C. G. Possible limits on the Composition of the Archean Ocean. Nature 1983, 302, 518–520. [Google Scholar]; b Russel M. J.; Hall A. J. The Emergence of Life from Iron Monosulphide Bubbles at a Submarine Hydrothermal Redox and pH Front. J. Geol. Soc. London 1997, 154, 377–402. [DOI] [PubMed] [Google Scholar]; c Kua J.; Bada J. L. Primordial Ocean Chemistry and its Compatibility with the RNA World. Origins Life Evol. Biospheres 2011, 41, 553–558. [DOI] [PubMed] [Google Scholar]
- Jayasena V.; Gold L. In Vitro Selection of Self-Cleaving RNAs with a Low pH Optimum. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10612–10617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See pages 368–384 of ref (8).
- As exemplified by PNA and sulfone DNA:Benner S. A. Understanding Nucleic Acids Using Synthetic Chemistry. Acc. Chem. Res. 2004, 37, 784–797. [DOI] [PubMed] [Google Scholar]
- Patra A.; Richert C. High Fidelity Base Pairing at the 3′-Terminus. J. Am. Chem. Soc. 2009, 131, 12671–12681. [DOI] [PubMed] [Google Scholar]
- Gao J.; Liu H.; Kool E. T. Assembly of the Complete Eight-Base Artificial Genetic Helix, xDNA, and Its Interaction with the Natural Genetic System. Angew. Chem., Int. Ed. 2005, 44, 3118–3122. [DOI] [PubMed] [Google Scholar]
- Lee A. H. F.; Kool E. T. A New Four-Base Genetic Helix, yDNA, Composed of Widened Benzopyrimidine–Purine Pairs. J. Am. Chem. Soc. 2005, 127, 3332–3338. [DOI] [PubMed] [Google Scholar]
- Eschenmoser has shown that the base-pairing modes can vary with a change in the nature of the sugar–phosphodiester backbone:Eschenmoser A.Toward a Chemical Etiology of the Natural Nucleic Acid’s Structure. 40 Years of DNA Double Helix. Proceedings of the R. A. Welch Foundation 37th Conference on Chemical Research; R. A. Welch Foundation: Houston, TX, 1993; pp 201–235. [Google Scholar]; Also see:; Bolli M.; Litten J. C.; Schültz R.; Leumann C. J. Bicyclo-DNA: A Hoogsteen-Selective Pairing System. Chem. Biol. 1996, 3, 197–206. [DOI] [PubMed] [Google Scholar]
- The hydrophobic effect has been described as a “unique organizing force” in the context of biological organization of membranes, protein folding, and membrane-bound proteins:; Tanford C. The Hydrophobic Effect and the Organization of Living Matter. Science 1978, 200, 1012–1018. [DOI] [PubMed] [Google Scholar]
- Two-chain lipids can associate in water at concentrations as low as 10–12 M.; Sackmann E. Physical Basis of Self-Organization and Function of Membranes: Physics of Vesicles. Handb. Biol. Phys. 1995, 1, 213–304. [Google Scholar]